Smad Phospho-Isoforms for Hepatocellular Carcinoma Risk Assessment in Patients with Nonalcoholic Steatohepatitis

 , ,
, ,
Abstract
:1. Introduction
2. Results
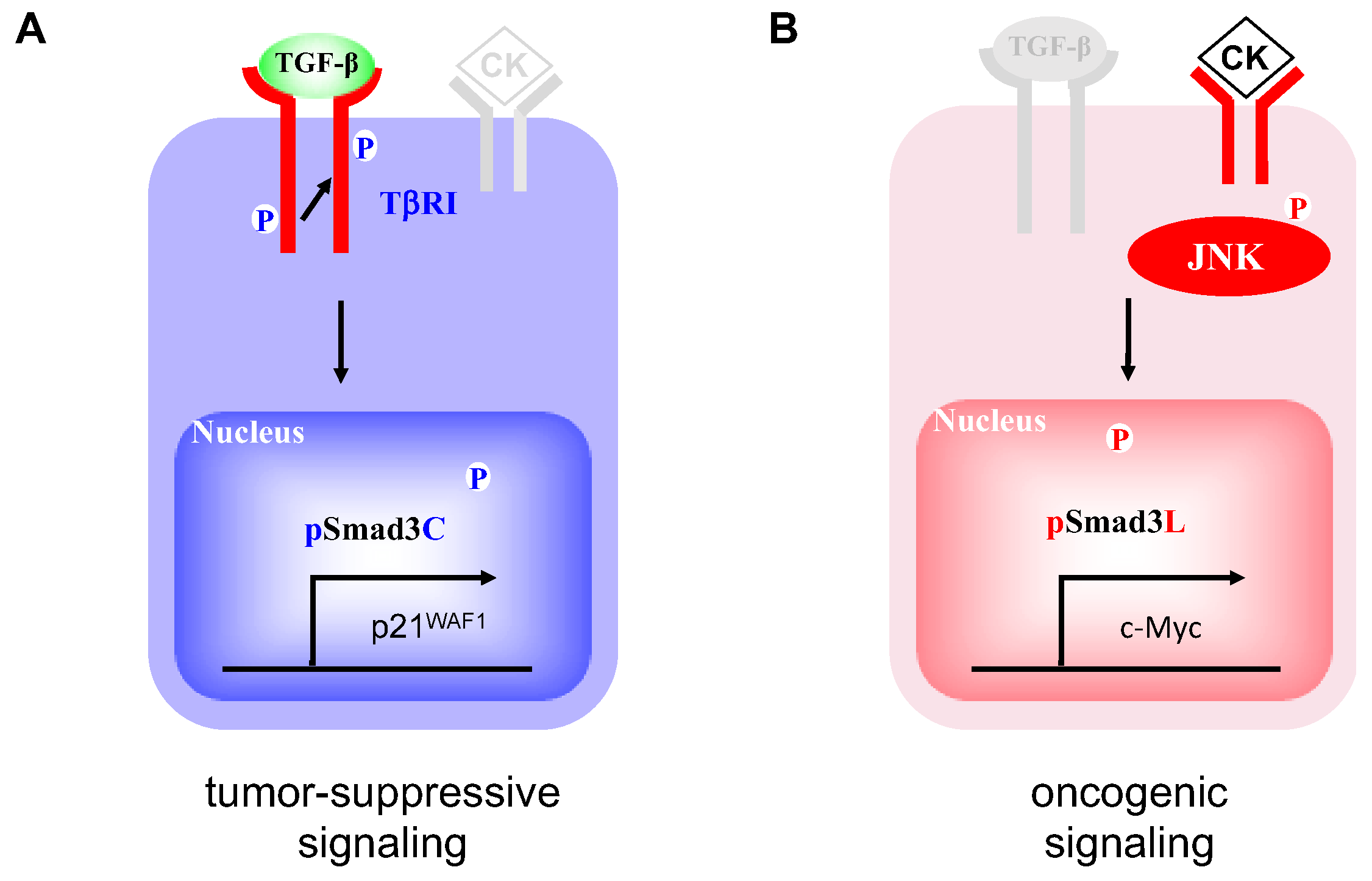
2.1. Two Distinct Hepatocytic Smad Phospho-Isoform Signaling Pathways in NASH Livers: pSmad3L- and pSmad3C-Dominant Types
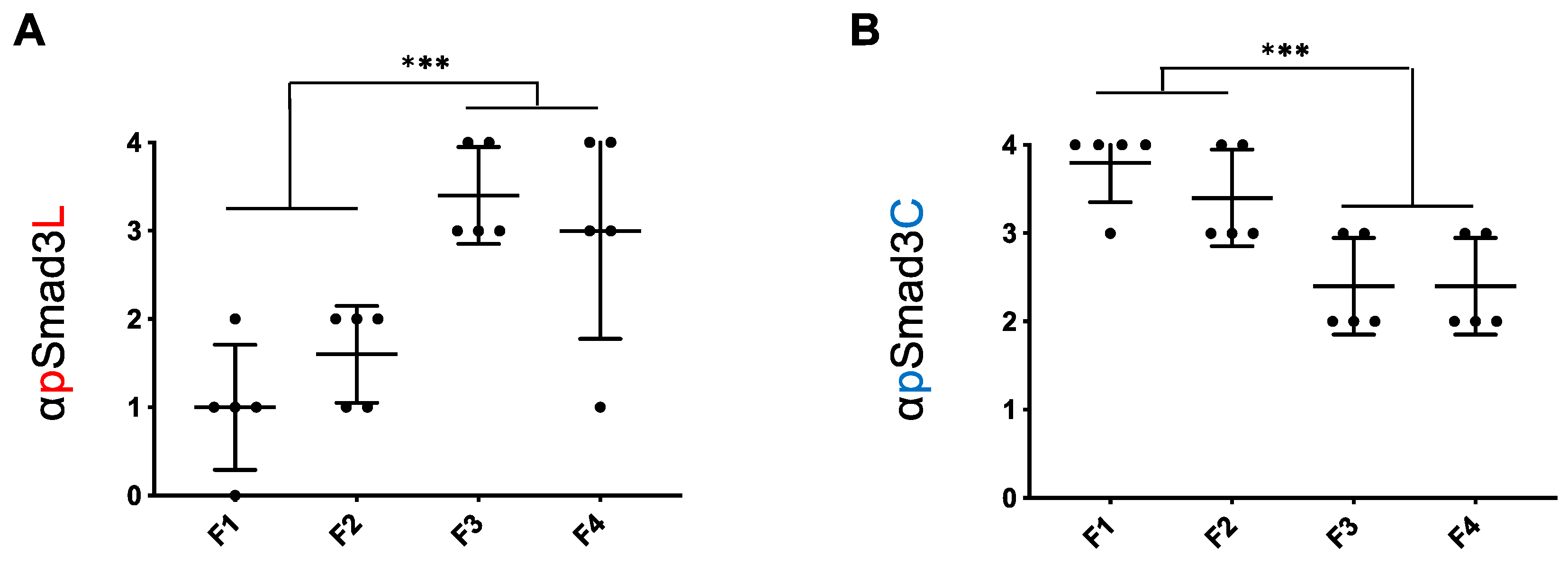
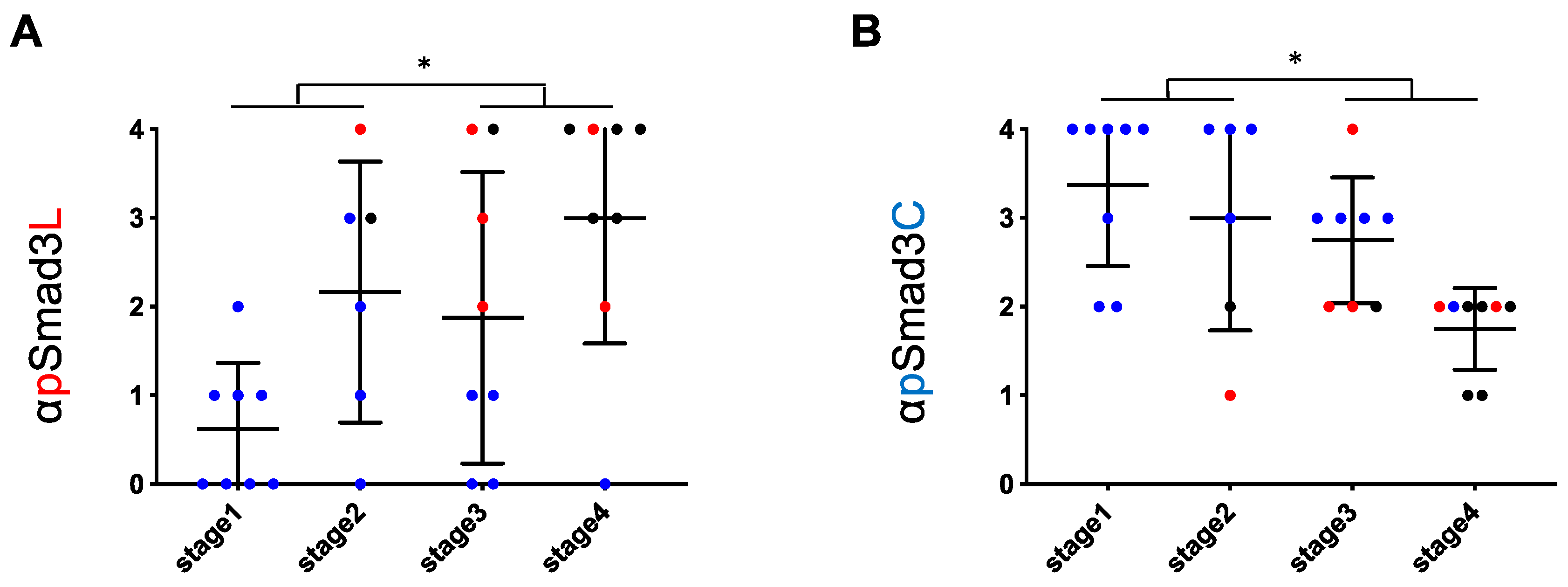
2.2. Although the pSmad3L Pathway in Hepatocytes Tended to Predominate While the pSmad3C Pathway Became Quiescent during Chronic Liver Disease Progression in both HCV and NASH, Smad3 Phospho-Isoforms in NASH Livers Varied more Widely than in HCV-Infected Livers at All Fibrotic Stages
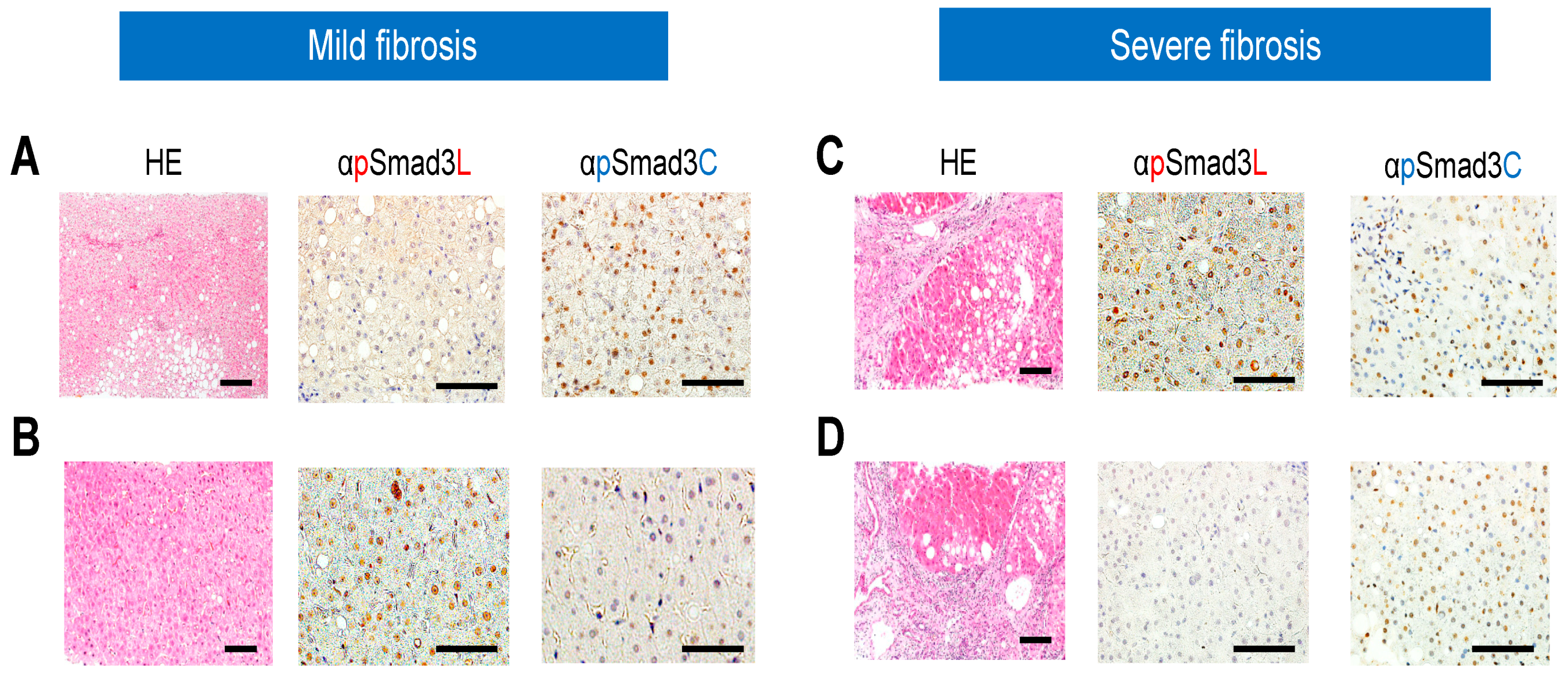
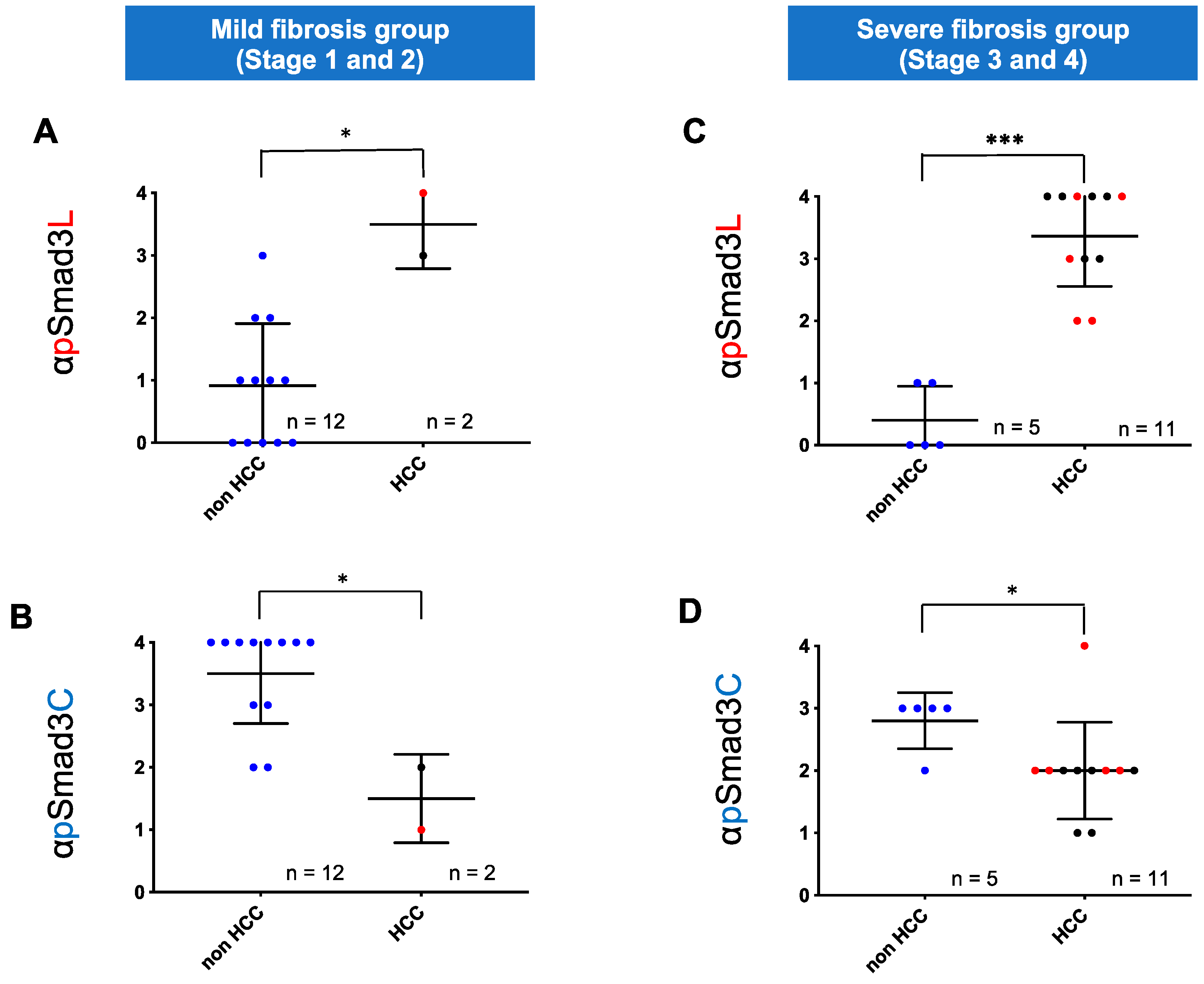
2.3. Regardless of Fibrotic Stage, HCC Developed from Hepatocytes in NASH Livers with Abundant Phosphorylation at Smad3L and Little Phosphorylation at Smad3C
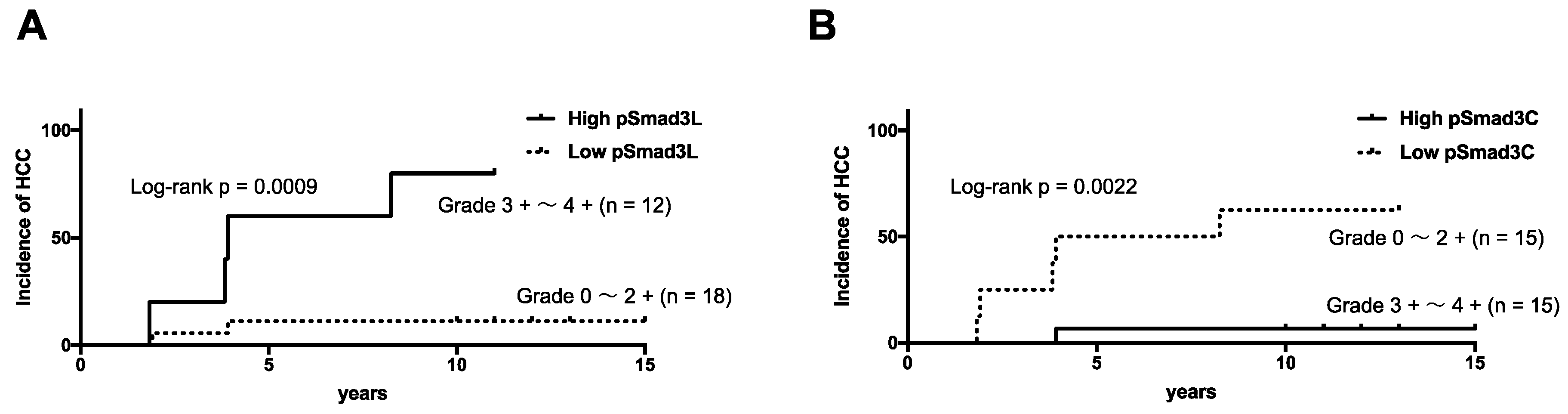
2.4. NASH Patients with Hepatocytes Positive for pSmad3L and Negative for pSmad3C Increased Risk of HCC Development
3. Discussion
4. Materials and Methods
4.1. Patient Enrollment and Clinical/Biochemical Evaluation
4.2. Evaluation of Liver Biopsy Specimens
4.3. Domain-Specific Antibodies (Abs) against Phosphorylated Smad2 and Smad3
4.4. Immunohistochemistry
4.5. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Parkin, D.M. Global cancer statistics in the year 2000. Lancet Oncol. 2001, 2, 533–543. [Google Scholar] [CrossRef]
- Augustine, M.M.; Fong, Y. Epidemiology and risk factors of biliary tract and primary liver tumors. Surg. Oncol. Clin. N. Am. 2014, 23, 171–188. [Google Scholar] [CrossRef] [PubMed]
- Liew, Z.H.; Goh, G.B.; Hao, Y.; Chang, P.E.; Tan, C.K. Comparison of Hepatocellular Carcinoma in Patients with Cryptogenic Versus Hepatitis B Etiology: A Study of 1079 Cases Over 3 Decades. Dig. Dis. Sci. 2019, 64, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.D.; Stengel, J.; Asike, M.I.; Torres, D.M.; Shaw, J.; Contreras, M.; Landt, C.L.; Harrison, S.A. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: A prospective study. Gastroenterology 2011, 140, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Torres, D.M.; Williams, C.D.; Harrison, S.A. Features, diagnosis, and treatment of nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2012, 10, 837–858. [Google Scholar] [CrossRef]
- Flegal, K.M.; Carroll, M.D.; Ogden, C.L.; Curtin, L.R. Prevalence and trends in obesity among US adults, 1999–2008. JAMA 2010, 303, 235–241. [Google Scholar] [CrossRef] [Green Version]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef]
- Singh, S.; Allen, A.M.; Wang, Z.; Prokop, L.J.; Murad, M.H.; Loomba, R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: A systematic review and meta-analysis of paired-biopsy studies. Clin. Gastroenterol. Hepatol. 2015, 13, 643–654. [Google Scholar] [CrossRef] [Green Version]
- Enomoto, H.; Ueno, Y.; Hiasa, Y.; Nishikawa, H.; Hige, S.; Takikawa, Y.; Taniai, M.; Ishikawa, T.; Yasui, K.; Takaki, A.; et al. Transition in the etiology of liver cirrhosis in Japan: A nationwide survey. J. Gastroenterol. 2019, 25, 1–10, (Epub). [Google Scholar] [CrossRef] [Green Version]
- Ertle, J.; Dechene, A.; Sowa, J.P.; Penndorf, V.; Herzer, K.; Kaiser, G.; Schlaak, J.F.; Gerken, G.; Syn, W.K.; Canbay, A. Non-alcoholic fatty liver disease progresses to hepatocellular carcinoma in the absence of apparent cirrhosis. Int. J. Cancer 2011, 128, 2436–2443. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Harrison, S.A.; Ratziu, V.; Abdelmalek, M.F.; Diehl, A.M.; Caldwell, S.; Shiffman, M.L.; Aguilar Schall, R.; Jia, C.; McColgan, B.; et al. The Natural History of Advanced Fibrosis Due to Nonalcoholic Steatohepatitis: Data From the Simtuzumab Trials. Hepatology 2019, 70, 1913–1927, (Epub). [Google Scholar] [CrossRef] [PubMed]
- Park, C.C.; Nguyen, P.; Hernandez, C.; Bettencourt, R.; Ramirez, K.; Fortney, L.; Hooker, J.; Sy, E.; Savides, M.T.; Alquiraish, M.H.; et al. Magnetic Resonance Elastography vs Transient Elastography in Detection of Fibrosis and Noninvasive Measurement of Steatosis in Patients With Biopsy-Proven Nonalcoholic Fatty Liver Disease. Gastroenterology 2017, 152, 598–607 e592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rockey, D.C. Noninvasive assessment of liver fibrosis and portal hypertension with transient elastography. Gastroenterology 2008, 134, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, Y.; Honda, Y.; Kessoku, T.; Tomeno, W.; Imajo, K.; Yoneda, M.; Kawanaka, M.; Kirikoshi, H.; Ono, M.; Taguri, M.; et al. Wisteria floribunda agglutinin-positive Mac-2-binding protein and type 4 collagen 7S: Useful markers for the diagnosis of significant fibrosis in patients with non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2018, 33, 1795–1803. [Google Scholar] [CrossRef]
- Uojima, H.; Hidaka, H.; Tanaka, Y.; Inoue, T.; Onoue, M.; Wada, N.; Kubota, K.; Nakazawa, T.; Shibuya, A.; Fujikawa, T.; et al. Wisteria floribunda agglutinin-positive human Mac-2 binding protein in decompensated cirrhosis. J. Gastroenterol. Hepatol. 2018, 33, 1889–1896. [Google Scholar] [CrossRef]
- Yamamura, S.; Kawaguchi, T.; Nakano, D.; Tomiyasu, Y.; Yoshinaga, S.; Doi, Y.; Takahashi, H.; Anzai, K.; Eguchi, Y.; Torimura, T.; et al. Profiles of advanced hepatic fibrosis evaluated by FIB-4 index and shear wave elastography in health checkup examinees. Hepatol. Res. 2019. [Google Scholar] [CrossRef]
- Mohamad, B.; Shah, V.; Onyshchenko, M.; Elshamy, M.; Aucejo, F.; Lopez, R.; Hanouneh, I.A.; Alhaddad, R.; Alkhouri, N. Characterization of hepatocellular carcinoma (HCC) in non-alcoholic fatty liver disease (NAFLD) patients without cirrhosis. Hepatol. Int. 2016, 10, 632–639. [Google Scholar] [CrossRef]
- Inagaki, Y.; Okazaki, I. Emerging insights into Transforming growth factor beta Smad signal in hepatic fibrogenesis. Gut 2007, 56, 284–292. [Google Scholar] [CrossRef] [Green Version]
- Dooley, S.; ten Dijke, P. TGF-beta in progression of liver disease. Cell Tissue Res. 2012, 347, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Moses, H.L.; Serra, R. Regulation of differentiation by TGF-beta. Curr. Opin. Genet. Dev. 1996, 6, 581–586. [Google Scholar] [CrossRef]
- Bellam, N.; Pasche, B. Tgf-beta signaling alterations and colon cancer. Cancer Treat Res. 2010, 155, 85–103. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Miyazono, K.; Ten Dijke, P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature 1997, 390, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Gordeeva, O. TGFbeta Family Signaling Pathways in Pluripotent and Teratocarcinoma Stem Cells’ Fate Decisions: Balancing Between Self-Renewal, Differentiation, and Cancer. Cells 2019, 8, 1500. [Google Scholar] [CrossRef] [Green Version]
- Matsuzaki, K. Smad phosphoisoform signals in acute and chronic liver injury: Similarities and differences between epithelial and mesenchymal cells. Cell Tissue Res. 2012, 347, 225–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wrighton, K.H.; Lin, X.; Feng, X.H. Phospho-control of TGF-beta superfamily signaling. Cell Res. 2009, 19, 8–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannon, G.J.; Beach, D. p15INK4B is a potential effector of TGF-beta-induced cell cycle arrest. Nature 1994, 371, 257–261. [Google Scholar] [CrossRef]
- Staller, P.; Peukert, K.; Kiermaier, A.; Seoane, J.; Lukas, J.; Karsunky, H.; Moroy, T.; Bartek, J.; Massague, J.; Hanel, F.; et al. Repression of p15INK4b expression by Myc through association with Miz-1. Nat. Cell Biol. 2001, 3, 392–399. [Google Scholar] [CrossRef]
- Lasorella, A.; Noseda, M.; Beyna, M.; Yokota, Y.; Iavarone, A. Id2 is a retinoblastoma protein target and mediates signalling by Myc oncoproteins. Nature 2000, 407, 592–598. [Google Scholar] [CrossRef]
- Feng, X.H.; Lin, X.; Derynck, R. Smad2, Smad3 and Smad4 cooperate with Sp1 to induce p15(Ink4B) transcription in response to TGF-beta. EMBO J. 2000, 19, 5178–5193. [Google Scholar] [CrossRef] [Green Version]
- Pardali, K.; Kurisaki, A.; Moren, A.; ten Dijke, P.; Kardassis, D.; Moustakas, A. Role of Smad proteins and transcription factor Sp1 in p21(Waf1/Cip1) regulation by transforming growth factor-beta. J. Biol. Chem. 2000, 275, 29244–29256. [Google Scholar] [CrossRef] [Green Version]
- Frederick, J.P.; Liberati, N.T.; Waddell, D.S.; Shi, Y.; Wang, X.F. Transforming growth factor beta-mediated transcriptional repression of c-myc is dependent on direct binding of Smad3 to a novel repressive Smad binding element. Mol. Cell Biol. 2004, 24, 2546–2559. [Google Scholar] [CrossRef] [Green Version]
- Hui, L.; Zatloukal, K.; Scheuch, H.; Stepniak, E.; Wagner, E.F. Proliferation of human HCC cells and chemically induced mouse liver cancers requires JNK1-dependent p21 downregulation. J. Clin. Investig. 2008, 118, 3943–3953. [Google Scholar] [CrossRef] [Green Version]
- Nagata, H.; Hatano, E.; Tada, M.; Murata, M.; Kitamura, K.; Asechi, H.; Narita, M.; Yanagida, A.; Tamaki, N.; Yagi, S.; et al. Inhibition of c-Jun NH2-terminal kinase switches Smad3 signaling from oncogenesis to tumor- suppression in rat hepatocellular carcinoma. Hepatology 2009, 49, 1944–1953. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Murata, M.; Yoshida, K.; Sekimoto, G.; Uemura, Y.; Sakaida, N.; Kaibori, M.; Kamiyama, Y.; Nishizawa, M.; Fujisawa, J.; et al. Chronic inflammation associated with hepatitis C virus infection perturbs hepatic transforming growth factor beta signaling, promoting cirrhosis and hepatocellular carcinoma. Hepatology 2007, 46, 48–57. [Google Scholar] [CrossRef]
- Murata, M.; Matsuzaki, K.; Yoshida, K.; Sekimoto, G.; Tahashi, Y.; Mori, S.; Uemura, Y.; Sakaida, N.; Fujisawa, J.; Seki, T.; et al. Hepatitis B virus X protein shifts human hepatic transforming growth factor (TGF)-beta signaling from tumor suppression to oncogenesis in early chronic hepatitis B. Hepatology 2009, 49, 1203–1217. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Matsuzaki, K.; Inokuchi, R.; Kawamura, R.; Yoshida, K.; Murata, M.; Fujisawa, J.; Fukushima, N.; Sata, M.; Kage, M.; et al. Phosphorylated Smad2 and Smad3 signaling: Shifting between tumor suppression and fibro-carcinogenesis in chronic hepatitis C. Hepatol. Res. 2013, 43, 1327–1342. [Google Scholar] [CrossRef]
- Deng, Y.R.; Yoshida, K.; Jin, Q.L.; Murata, M.; Yamaguchi, T.; Tsuneyama, K.; Moritoki, Y.; Niu, J.Q.; Matsuzaki, K.; Lian, Z.X. Reversible phospho-Smad3 signalling between tumour suppression and fibrocarcinogenesis in chronic hepatitis B infection. Clin. Exp. Immunol. 2014, 176, 102–111. [Google Scholar] [CrossRef]
- Furukawa, F.; Matsuzaki, K.; Mori, S.; Tahashi, Y.; Yoshida, K.; Sugano, Y.; Yamagata, H.; Matsushita, M.; Seki, T.; Inagaki, Y.; et al. p38 MAPK mediates fibrogenic signal through Smad3 phosphorylation in rat myofibroblasts. Hepatology 2003, 38, 879–889. [Google Scholar] [CrossRef]
- Ascha, M.S.; Hanouneh, I.A.; Lopez, R.; Tamimi, T.A.; Feldstein, A.F.; Zein, N.N. The incidence and risk factors of hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. Hepatology 2010, 51, 1972–1978. [Google Scholar] [CrossRef]
- Sanyal, A.; Poklepovic, A.; Moyneur, E.; Barghout, V. Population-based risk factors and resource utilization for HCC: US perspective. Curr. Med. Res. Opin. 2010, 26, 2183–2191. [Google Scholar] [CrossRef]
- McPherson, S.; Hardy, T.; Henderson, E.; Burt, A.D.; Day, C.P.; Anstee, Q.M. Evidence of NAFLD progression from steatosis to fibrosing-steatohepatitis using paired biopsies: Implications for prognosis and clinical management. J. Hepatol. 2015, 62, 1148–1155. [Google Scholar] [CrossRef]
- Arthur, M.J. Fibrogenesis II. Metalloproteinases and their inhibitors in liver fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 279, G245–G249. [Google Scholar] [CrossRef]
- Ha, H.; Oh, E.Y.; Lee, H.B. The role of plasminogen activator inhibitor 1 in renal and cardiovascular diseases. Nat. Rev. Nephrol. 2009, 5, 203–211. [Google Scholar] [CrossRef]
- Wells, R.G. The role of matrix stiffness in regulating cell behavior. Hepatology 2008, 47, 1394–1400. [Google Scholar] [CrossRef]
- Pinzani, M.; Macias-Barragan, J. Update on the pathophysiology of liver fibrosis. Expert Rev. Gastroenterol. Hepatol. 2010, 4, 459–472. [Google Scholar] [CrossRef]
- Kawamata, S.; Matsuzaki, K.; Murata, M.; Seki, T.; Matsuoka, K.; Iwao, Y.; Hibi, T.; Okazaki, K. Oncogenic Smad3 signaling induced by chronic inflammation is an early event in ulcerative colitis-associated carcinogenesis. Inflamm. Bowel. Dis. 2011, 17, 683–695. [Google Scholar] [CrossRef]
- Sekimoto, G.; Matsuzaki, K.; Yoshida, K.; Mori, S.; Murata, M.; Seki, T.; Matsui, H.; Fujisawa, J.; Okazaki, K. Reversible Smad-dependent signaling between tumor suppression and oncogenesis. Cancer Res. 2007, 67, 5090–5096. [Google Scholar] [CrossRef] [Green Version]
- Yamagata, H.; Matsuzaki, K.; Mori, S.; Yoshida, K.; Tahashi, Y.; Furukawa, F.; Sekimoto, G.; Watanabe, T.; Uemura, Y.; Sakaida, N.; et al. Acceleration of Smad2 and Smad3 phosphorylation via c-Jun NH(2)-terminal kinase during human colorectal carcinogenesis. Cancer Res. 2005, 65, 157–165. [Google Scholar]
- Kleiner, D.E.; Brunt, E.M.; Wilson, L.A.; Behling, C.; Guy, C.; Contos, M.; Cummings, O.; Yeh, M.; Gill, R.; Chalasani, N.; et al. Association of Histologic Disease Activity With Progression of Nonalcoholic Fatty Liver Disease. JAMA Netw. Open 2019, 2, e1912565. [Google Scholar] [CrossRef] [Green Version]
- Vilar-Gomez, E.; Martinez-Perez, Y.; Calzadilla-Bertot, L.; Torres-Gonzalez, A.; Gra-Oramas, B.; Gonzalez-Fabian, L.; Friedman, S.L.; Diago, M.; Romero-Gomez, M. Weight Loss Through Lifestyle Modification Significantly Reduces Features of Nonalcoholic Steatohepatitis. Gastroenterology 2015, 149, 367–378. [Google Scholar] [CrossRef]
- Khatun, M.; Ray, R.B. Mechanisms Underlying Hepatitis C Virus-Associated Hepatic Fibrosis. Cells 2019, 8, 1249. [Google Scholar] [CrossRef] [Green Version]
- Sabio, G.; Das, M.; Mora, A.; Zhang, Z.; Jun, J.Y.; Ko, H.J.; Barrett, T.; Kim, J.K.; Davis, R.J. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science 2008, 322, 1539–1543. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, H.; Maeda, S. Molecular mechanisms of liver injury and hepatocarcinogenesis: Focusing on the role of stress-activated MAPK. Pathol. Res. Int. 2012, 2012, 172894. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Kyo, S.; Takatura, M.; Sun, L. Autocrine transforming growth factor beta suppresses telomerase activity and transcription of human telomerase reverse transcriptase in human cancer cells. Cell Growth Differ. 2001, 12, 119–127. [Google Scholar]
- Glick, A.B.; Weinberg, W.C.; Wu, I.H.; Quan, W.; Yuspa, S.H. Transforming growth factor beta 1 suppresses genomic instability independent of a G1 arrest, p53, and Rb. Cancer Res. 1996, 56, 3645–3650. [Google Scholar]
- Tremain, R.; Marko, M.; Kinnimulki, V.; Ueno, H.; Bottinger, E.; Glick, A. Defects in TGF-beta signaling overcome senescence of mouse keratinocytes expressing v-Ha-ras. Oncogene 2000, 19, 1698–1709. [Google Scholar] [CrossRef] [Green Version]
- Brunt, E.M.; Janney, C.G.; Di Bisceglie, A.M.; Neuschwander-Tetri, B.A.; Bacon, B.R. Nonalcoholic steatohepatitis: A proposal for grading and staging the histological lesions. Am. J. Gastroenterol. 1999, 94, 2467–2474. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Age | Sex | AST | ALT | PLT | FiB4 | T-Bil | ALB | PT | F Stage | NI Grade | pSmad3L Staining | pSmad3C Staining | Folluw-up Period | Time to HCC * | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No HCC | 1 | 27 | M | 145 | 319 | 25.1 | 0.87 | 0.9 | 5.1 | 118 | 1 | 1 | 1 | 4 | 12 | |
| 2 | 29 | F | 150 | 248 | 28.0 | 0.99 | 0.7 | 4.3 | 75 | 1 | 1 | 1 | 4 | 12 | ||
| 3 | 38 | M | 59 | 178 | 21.1 | 0.8 | 0.8 | 4.9 | 101 | 1 | 1 | 0 | 4 | 10 | ||
| 4 | 60 | F | 90 | 118 | 26.3 | 1.89 | 0.5 | 4.0 | 102 | 1 | 1 | 1 | 3 | 11 | ||
| 5 | 63 | F | 38 | 43 | 28.4 | 1.29 | 0.5 | 4.9 | 110 | 1 | 1 | 0 | 4 | 15 | ||
| 6 | 68 | F | 84 | 82 | 28.3 | 2.23 | 1.1 | 4.4 | 1 | 1 | 0 | 2 | 13 | |||
| 7 | 72 | F | 49 | 45 | 20.1 | 2.62 | 0.6 | 4.2 | 127 | 1 | 1 | 0 | 2 | 13 | ||
| 8 | 70 | M | 168 | 214 | 16.9 | 4.76 | 1.2 | 4.4 | 74 | 1 | 2 | 2 | 4 | 11 | ||
| 9 | 44 | M | 93 | 169 | 23.9 | 1.32 | 1.4 | 4.5 | 118 | 2 | 1 | 2 | 4 | 13 | ||
| 10 | 45 | F | 84 | 145 | 23.1 | 1.36 | 0.7 | 4.9 | 119 | 2 | 1 | 1 | 3 | 10 | ||
| 11 | 66 | F | 29 | 29 | 19.2 | 1.85 | 0.5 | 4.2 | 115 | 2 | 1 | 0 | 4 | 13 | ||
| 12 | 65 | M | 98 | 35 | 19.3 | 5.58 | 0.4 | 4 | 88 | 2 | 2 | 3 | 4 | 11 | ||
| 13 | 46 | M | 27 | 14 | 11.3 | 2.94 | 0.8 | 3.9 | 88 | 3 | 1 | 1 | 3 | 11 | ||
| 14 | 54 | M | 40 | 86 | 21 | 1.11 | 1.1 | 4.4 | 89 | 3 | 2 | 0 | 3 | 11 | ||
| 15 | 56 | F | 155 | 192 | 12.5 | 5.01 | 1.1 | 4.3 | 3 | 2 | 1 | 3 | 12 | |||
| 16 | 57 | F | 30 | 24 | 16.9 | 2.07 | 0.7 | 4.3 | 107 | 3 | 2 | 0 | 3 | 10 | ||
| 17 | 54 | F | 80 | 99 | 12.8 | 3.39 | 1.2 | 4.4 | 101 | 4 | 2 | 0 | 2 | 13 | ||
| HCC | 18 | 50 | M | 51 | 70 | 9.1 | 3.35 | 1.2 | 4 | 38 | 2 | 1 | 4 | 1 | 8.25 | |
| 19 | 60 | M | 19 | 14 | 17.3 | 1.76 | 0.4 | 4.7 | 126 | 2 | 1 | 3 | 2 | 0 | ||
| 20 | 72 | M | 53 | 9.8 | 6.06 | 0.8 | 3.8 | 84 | 3 | 1 | 3 | 4 | 3.92 | |||
| 21 | 75 | M | 44 | 38 | 15.8 | 3.39 | 0.9 | 4 | 92 | 3 | 1 | 4 | 2 | 0 | ||
| 22 | 78 | F | 31 | 21 | 4.3 | 12.27 | 1.7 | 3.3 | 64 | 3 | 1 | 2 | 2 | 1.92 | ||
| 23 | 69 | M | 50 | 39 | 9.1 | 6.07 | 1.3 | 3.8 | 83 | 3 | 2 | 4 | 2 | 1.83 | ||
| 24 | 58 | M | 64 | 61 | 5 | 9.51 | 2.2 | 4 | 67 | 4 | 1 | 3 | 2 | 0 | ||
| 25 | 73 | M | 64 | 72 | 8.4 | 6.55 | 1 | 3.1 | 44 | 4 | 1 | 3 | 1 | 0 | ||
| 26 | 71 | M | 129 | 73 | 9.9 | 10.83 | 2.9 | 3.7 | 72 | 4 | 2 | 4 | 1 | 0 | ||
| 27 | 76 | F | 59 | 29 | 8.4 | 9.91 | 1.2 | 3.2 | 77 | 4 | 2 | 2 | 2 | 3.92 | ||
| 28 | 80 | F | 28 | 16 | 4.4 | 12.73 | 1.2 | 3.1 | 64 | 4 | 2 | 4 | 2 | 0 | ||
| 29 | 69 | F | 48 | 23 | 7 | 9.87 | 2.4 | 2.6 | 50 | 4 | 3 | 4 | 2 | 3.83 | ||
| 30 | 75 | M | 33 | 14 | 10.4 | 6.36 | 0.9 | 3.6 | 87 | 4 | 3 | 4 | 2 | 0 |
| No. | Age | Sex | AST | ALT | PLT | FiB4 | T-Bil | ALB | PT | F Stage | NI Grade | pSmad3L Staining | pSmad3C Staining |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 32 | F | 18 | 21 | 17.8 | 0.71 | 0.7 | 4.2 | 100 | 1 | 1 | 0 | 4 |
| 2 | 44 | M | 244 | 156 | 16.5 | 5.21 | 0.9 | 5.1 | 88 | 1 | 2 | 1 | 4 |
| 3 | 63 | F | 27 | 24 | 31.4 | 1.11 | 0.5 | 4.7 | 110 | 1 | 1 | 1 | 3 |
| 4 | 45 | M | 81 | 65 | 17.7 | 2.55 | 0.6 | 0.7 | 104 | 1 | 2 | 2 | 4 |
| 5 | 51 | M | 29 | 34 | 13.3 | 1.91 | 0.7 | 4.3 | 92 | 1 | 2 | 1 | 4 |
| 6 | 70 | F | 22 | 31 | 13.9 | 1.99 | 0.6 | 3.9 | 105 | 2 | 2 | 1 | 4 |
| 7 | 45 | M | 57 | 101 | 12.7 | 2.01 | 0.7 | 4.4 | 88 | 2 | 3 | 2 | 3 |
| 8 | 72 | F | 297 | 197 | 11.6 | 13.13 | 1.2 | 3.8 | 102 | 2 | 2 | 2 | 4 |
| 9 | 64 | M | 105 | 83 | 16.8 | 4.39 | 0.7 | 4.1 | 77 | 2 | 2 | 1 | 3 |
| 10 | 69 | F | 94 | 75 | 23.4 | 3.2 | 1.2 | 3.9 | 81 | 2 | 2 | 2 | 3 |
| 11 | 57 | M | 41 | 43 | 15.0 | 2.38 | 0.7 | 4.6 | 107 | 3 | 2 | 3 | 3 |
| 12 | 59 | M | 161 | 169 | 11.4 | 6.41 | 1.0 | 4.2 | 89 | 3 | 2 | 4 | 2 |
| 13 | 68 | M | 83 | 93 | 7.8 | 7.5 | 1.7 | 3.4 | 71 | 3 | 3 | 3 | 2 |
| 14 | 55 | M | 30 | 26 | 15.7 | 2.06 | 1.2 | 3.9 | 72 | 3 | 2 | 3 | 2 |
| 15 | 64 | F | 43 | 31 | 14.4 | 3.43 | 1.1 | 4.5 | 63 | 3 | 2 | 4 | 3 |
| 16 | 69 | F | 26 | 19 | 13.7 | 3.00 | 1.2 | 4.7 | 88 | 4 | 2 | 4 | 2 |
| 17 | 58 | M | 64 | 69 | 11.0 | 4.06 | 1.2 | 4.2 | 96 | 4 | 3 | 4 | 3 |
| 18 | 48 | M | 62 | 103 | 13.2 | 2.22 | 0.6 | 4.3 | 108 | 4 | 2 | 1 | 2 |
| 19 | 34 | F | 96 | 79 | 15.3 | 2.40 | 1.0 | 4.1 | 83 | 4 | 2 | 3 | 3 |
| 20 | 61 | F | 87 | 56 | 9.0 | 7.88 | 0.4 | 3.9 | 79 | 4 | 1 | 3 | 2 |
| Variables | Category | Univariate Analysis | Multivariate Analysis | ||
|---|---|---|---|---|---|
| Hazard Ratio (95% CI) | p Value | Hazard Ratio (95% CI) | p Value | ||
| Fibrosis stage | Low (1 and 2) | ||||
| High (3 and 4) | 8.823 (1.023–76.12) | 0.04766 | 22.36 (1.269–394.1) | 0.0338 | |
| Necroinflammatory grade | Low (0 and 1) | ||||
| High (2 and 3) | 1.768 (0.3563–8.771) | 0.4857 | |||
| NAFLD activity score | <5 | ||||
| ≥5 | 0 | 0.999 | |||
| pSmad3C positivity | High (3 and 4) | ||||
| Low (1 and 2) | 13.25 (1.538–114.2) | 0.01868 | 15.61 (1.254–194.3) | 0.0327 | |
| pSmad3L positivity | Low (1 and 2) | ||||
| High (3 and 4) | 10.32 (1.866–57.05) | 0.007479 | 18.53 (1.712–200.6) | 0.0163 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suwa, K.; Yamaguchi, T.; Yoshida, K.; Murata, M.; Ichimura, M.; Tsuneyama, K.; Seki, T.; Okazaki, K. Smad Phospho-Isoforms for Hepatocellular Carcinoma Risk Assessment in Patients with Nonalcoholic Steatohepatitis. Cancers 2020, 12, 286. https://doi.org/10.3390/cancers12020286
Suwa K, Yamaguchi T, Yoshida K, Murata M, Ichimura M, Tsuneyama K, Seki T, Okazaki K. Smad Phospho-Isoforms for Hepatocellular Carcinoma Risk Assessment in Patients with Nonalcoholic Steatohepatitis. Cancers. 2020; 12(2):286. https://doi.org/10.3390/cancers12020286
Chicago/Turabian StyleSuwa, Kanehiko, Takashi Yamaguchi, Katsunori Yoshida, Miki Murata, Mayuko Ichimura, Koichi Tsuneyama, Toshihito Seki, and Kazuichi Okazaki. 2020. "Smad Phospho-Isoforms for Hepatocellular Carcinoma Risk Assessment in Patients with Nonalcoholic Steatohepatitis" Cancers 12, no. 2: 286. https://doi.org/10.3390/cancers12020286
APA StyleSuwa, K., Yamaguchi, T., Yoshida, K., Murata, M., Ichimura, M., Tsuneyama, K., Seki, T., & Okazaki, K. (2020). Smad Phospho-Isoforms for Hepatocellular Carcinoma Risk Assessment in Patients with Nonalcoholic Steatohepatitis. Cancers, 12(2), 286. https://doi.org/10.3390/cancers12020286