Toll-like Receptors from the Perspective of Cancer Treatment



Abstract
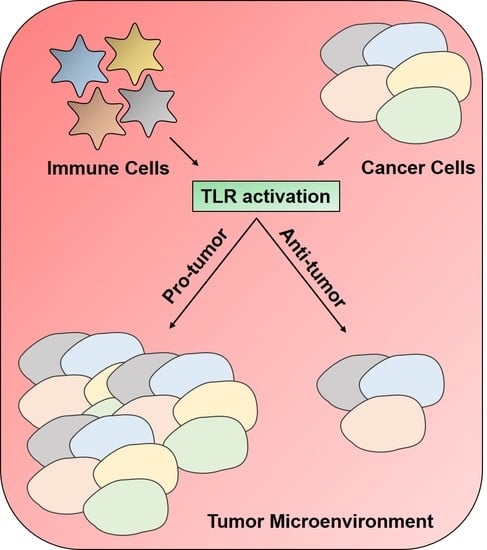
1. Introduction
2. The NF-κB Pathway
3. The MAPK Pathway
3.1. ERKs
3.2. JNKs
3.3. p38
4. The Type I IFN Pathway
5. TLR Signaling in Immune and Cancer Cells
5.1. TLR Signaling in DC Subsets
5.2. TLR Signaling in T-Cell Subsets
5.2.1. TLR1/2 and TLR2/6
5.2.2. TLR3
5.2.3. TLR4
5.2.4. TLR5
5.2.5. TLR7/8
5.2.6. TLR9
5.3. TLR Signaling in the Cancer Cell
6. TLRs as Therapeutic Targets in Cancers
6.1. TLR Agonism for Cancer Prevention or Treatment
6.1.1. TLR2/TLR4
6.1.2. TLR3
6.1.3. TLR5
6.1.4. TLR7/8
6.1.5. TLR9
6.2. TLR Antagonism for Cancer Treatment or Prevention
6.2.1. Manipulation of the Gut Microbiota
6.2.2. Inhibition of TLR2 and TLR4
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A.; Medzhitov, R. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 2004, 5, 987. [Google Scholar] [CrossRef]
- Bowie, A.; O’Neill, L.A. The interleukin-1 receptor/Toll-like receptor superfamily: signal generators for pro-inflammatory interleukins and microbial products. J. Leuko. Biol. 2000, 67, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Choe, J.; Kelker, M.S.; Wilson, I.A. Crystal structure of human toll-like receptor 3 (TLR3) ectodomain. Science 2005, 309, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499. [Google Scholar] [CrossRef] [PubMed]
- Javaid, N.; Yasmeen, F.; Choi, S. Toll-Like Receptors and Relevant Emerging Therapeutics with Reference to Delivery Methods. Pharmaceutics 2019, 11, 441. [Google Scholar] [CrossRef]
- Sato, S.; Sanjo, H.; Takeda, K.; Ninomiya-Tsuji, J.; Yamamoto, M.; Kawai, T.; Matsumoto, K.; Takeuchi, O.; Akira, S. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat. Immunol. 2005, 6, 1087. [Google Scholar] [CrossRef]
- Campisi, J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell 2005, 120, 513–522. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Bishop, J.M. Cancer: what should be done? American Association for the Advancement of Science, 1997. [Google Scholar]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: from immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991. [Google Scholar] [CrossRef]
- Pardoll, D. Does the immune system see tumors as foreign or self? Annu/ Rev. Immunol. 2003, 21, 807–839. [Google Scholar] [CrossRef] [PubMed]
- Boudreau, J.E.; Bonehill, A.; Thielemans, K.; Wan, Y. Engineering dendritic cells to enhance cancer immunotherapy. Mol. Ther. 2011, 19, 841–853. [Google Scholar] [CrossRef] [PubMed]
- Fukata, M.; Abreu, M.T. Role of Toll-like receptors in gastrointestinal malignancies. Oncogene 2008, 27, 234. [Google Scholar] [CrossRef] [PubMed]
- Spencer, E.; Jiang, J.; Chen, Z.J. Signal-induced ubiquitination of IκBα by the F-box protein Slimb/β-TrCP. Genes Dev. 1999, 13, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.J.; Parent, L.; Maniatis, T. Site-specific phosphorylation of IκBα by a novel ubiquitination-dependent protein kinase activity. Cell 1996, 84, 853–862. [Google Scholar] [CrossRef]
- Chen, Z.; Hagler, J.; Palombella, V.J.; Melandri, F.; Scherer, D.; Ballard, D.; Maniatis, T. Signal-induced site-specific phosphorylation targets I kappa B alpha to the ubiquitin-proteasome pathway. Genes Dev. 1995, 9, 1586–1597. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Signaling to NF-κB. Genes Dev. 2004, 18, 2195–2224. [Google Scholar] [CrossRef]
- Janssens, S.; Tinel, A.; Lippens, S.; Tschopp, J. PIDD mediates NF-κB activation in response to DNA damage. Cell 2005, 123, 1079–1092. [Google Scholar] [CrossRef]
- Festjens, N.; Berghe, T.V.; Cornelis, S.; Vandenabeele, P. RIP1, a kinase on the crossroads of a cell’s decision to live or die. Cell Death Differ. 2007, 14, 400. [Google Scholar] [CrossRef]
- Wu, Z.-H.; Mabb, A.; Miyamoto, S. PIDD: a switch hitter. Cell 2005, 123, 980–982. [Google Scholar] [CrossRef]
- Wu, C.-J.; Conze, D.B.; Li, T.; Srinivasula, S.M.; Ashwell, J.D. Sensing of Lys 63-linked polyubiquitination by NEMO is a key event in NF-κB activation. Nat. Cell Biol. 2006, 8, 398. [Google Scholar] [CrossRef] [PubMed]
- Zarnegar, B.J.; Wang, Y.; Mahoney, D.J.; Dempsey, P.W.; Cheung, H.H.; He, J.; Shiba, T.; Yang, X.; Yeh, W.-c.; Mak, T.W. Noncanonical NF-κB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat. Immunol. 2008, 9, 1371. [Google Scholar] [CrossRef] [PubMed]
- Kato, T., Jr.; Delhase, M.; Hoffmann, A.; Karin, M. CK2 is a C-terminal IκB kinase responsible for NF-κB activation during the UV response. Mol. Cell 2003, 12, 829–839. [Google Scholar] [CrossRef]
- Tergaonkar, V.; Bottero, V.; Ikawa, M.; Li, Q.; Verma, I.M. IκB kinase-independent IκBα degradation pathway: functional NF-κB activity and implications for cancer therapy. Mol. Cell. Biol. 2003, 23, 8070–8083. [Google Scholar] [CrossRef] [PubMed]
- Viatour, P.; Merville, M.-P.; Bours, V.; Chariot, A. Phosphorylation of NF-κB and IκB proteins: implications in cancer and inflammation. Trends Biochem. Sci. 2005, 30, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B. Nuclear factor-κB: the enemy within. Cancer Cell 2004, 6, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.J.; Rocha, S.; Perkins, N.D. Active repression of antiapoptotic gene expression by RelA (p65) NF-κB. Mol. Cell 2004, 13, 853–865. [Google Scholar] [CrossRef]
- Janssens, S.; Tschopp, J. Signals from within: the DNA-damage-induced NF-κB response. Cell Death Differ. 2006, 13, 773. [Google Scholar] [CrossRef]
- Hur, G.M.; Lewis, J.; Yang, Q.; Lin, Y.; Nakano, H.; Nedospasov, S.; Liu, Z.-g. The death domain kinase RIP has an essential role in DNA damage-induced NF-κB activation. Genes Dev. 2003, 17, 873–882. [Google Scholar] [CrossRef]
- Karin, M.; Greten, F.R. NF-κB: linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5, 749. [Google Scholar] [CrossRef]
- Singh, N.P.; Nagarkatti, M.; Nagarkatti, P.S. Role of dioxin response element and nuclear factor-κB motifs in 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin-mediated regulation of Fas and Fas ligand expression. Mol. Pharmacol. 2007, 71, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Shou, Y.; Li, N.; Li, L.; Borowitz, J.L.; Isom, G.E. NF-κB-mediated up-regulation of Bcl-XS and Bax contributes to cytochrome c release in cyanide-induced apoptosis. J. Neurochem. 2002, 81, 842–852. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Qiu, W.; Dudgeon, C.; Liu, H.; Huang, C.; Zambetti, G.; Yu, J.; Zhang, L. PUMA is directly activated by NF-κB and contributes to TNF-α-induced apoptosis. Cell Death Differ. 2009, 16, 1192. [Google Scholar] [CrossRef] [PubMed]
- Sen, R.; Baltimore, D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell 1986, 46, 705–716. [Google Scholar] [CrossRef]
- Prasad, S.; Ravindran, J.; Aggarwal, B.B. NF-κB and cancer: how intimate is this relationship. Mol. Cell. Biochem. 2010, 336, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.; Ihekwaba, A.; Elliott, M.; Johnson, J.; Gibney, C.; Foreman, B.; Nelson, G.; See, V.; Horton, C.; Spiller, D. Oscillations in NF-κB signaling control the dynamics of gene expression. Science 2004, 306, 704–708. [Google Scholar] [CrossRef]
- Baldwin, A.S. Regulation of cell death and autophagy by IKK and NF-κB: critical mechanisms in immune function and cancer. Immunol. Rev. 2012, 246, 327–345. [Google Scholar] [CrossRef]
- Chu, Z.-L.; McKinsey, T.A.; Liu, L.; Gentry, J.J.; Malim, M.H.; Ballard, D.W. Suppression of tumor necrosis factor-induced cell death by inhibitor of apoptosis c-IAP2 is under NF-κB control. Proc. Natl. Acad. Sci. USA 1997, 94, 10057–10062. [Google Scholar] [CrossRef]
- Wang, C.-Y.; Mayo, M.W.; Korneluk, R.G.; Goeddel, D.V.; Baldwin, A.S. NF-κB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science 1998, 281, 1680–1683. [Google Scholar] [CrossRef]
- Basseres, D.; Baldwin, A. Nuclear factor-κB and inhibitor of κB kinase pathways in oncogenic initiation and progression. Oncogene 2006, 25, 6817. [Google Scholar] [CrossRef]
- Hinz, M.; Krappmann, D.; Eichten, A.; Heder, A.; Scheidereit, C.; Strauss, M. NF-κB function in growth control: regulation of cyclin D1 expression and G0/G1-to-S-phase transition. Mol. Cell. Biol. 1999, 19, 2690–2698. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Bonizzi, G.; Seagroves, T.N.; Greten, F.R.; Johnson, R.; Schmidt, E.V.; Karin, M. IKKα provides an essential link between RANK signaling and cyclin D1 expression during mammary gland development. Cell 2001, 107, 763–775. [Google Scholar] [CrossRef]
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37. [Google Scholar] [CrossRef] [PubMed]
- Krens, S.G.; Spaink, H.P.; Snaar-Jagalska, B.E. Functions of the MAPK family in vertebrate-development. FEBS Lett. 2006, 580, 4984–4990. [Google Scholar] [CrossRef] [PubMed]
- Kyriakis, J.M.; Avruch, J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 2001, 81, 807–869. [Google Scholar] [CrossRef]
- McKay, M.; Morrison, D. Integrating signals from RTKs to ERK/MAPK. Oncogene 2007, 26, 3113. [Google Scholar] [CrossRef]
- Raman, M.; Chen, W.; Cobb, M. Differential regulation and properties of MAPKs. Oncogene 2007, 26, 3100. [Google Scholar] [CrossRef]
- Yoon, S.; Seger, R. The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors 2006, 24, 21–44. [Google Scholar] [CrossRef]
- Dunn, K.L.; Espino, P.S.; Drobic, B.; He, S.; Davie, J.R. The Ras-MAPK signal transduction pathway, cancer and chromatin remodeling. Biochem. Cell Biol. 2005, 83, 1–14. [Google Scholar] [CrossRef]
- Murphy, L.O.; MacKeigan, J.P.; Blenis, J. A network of immediate early gene products propagates subtle differences in mitogen-activated protein kinase signal amplitude and duration. Mol. Cell. Biol. 2004, 24, 144–153. [Google Scholar] [CrossRef]
- Yamamoto, T.; Ebisuya, M.; Ashida, F.; Okamoto, K.; Yonehara, S.; Nishida, E. Continuous ERK activation downregulates antiproliferative genes throughout G1 phase to allow cell-cycle progression. Curr. Biol. 2006, 16, 1171–1182. [Google Scholar] [CrossRef] [PubMed]
- Mirza, A.M.; Gysin, S.; Malek, N.; Nakayama, K.-i.; Roberts, J.M.; McMahon, M. Cooperative regulation of the cell division cycle by the protein kinases RAF and AKT. Mol. and cellular Biol. 2004, 24, 10868–10881. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Coleman, M.L.; Marshall, C.J.; Olson, M.F. RAS and RHO GTPases in G1-phase cell-cycle regulation. Nat. Rev. Mol. Cell Biol. 2004, 5, 355. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, K.; Wakioka, T.; Nishinakamura, H.; Kamio, M.; Yang, L.; Inoue, M.; Hasegawa, M.; Yonemitsu, Y.; Komiya, S.; Yoshimura, A. The Sprouty-related protein, Spred, inhibits cell motility, metastasis, and Rho-mediated actin reorganization. Oncogene 2004, 23, 5567. [Google Scholar] [CrossRef] [PubMed]
- Bloethner, S.; Chen, B.; Hemminki, K.; Müller-Berghaus, J.; Ugurel, S.; Schadendorf, D.; Kumar, R. Effect of common B-RAF and N-RAS mutations on global gene expression in melanoma cell lines. Carcinogenesis 2005, 26, 1224–1232. [Google Scholar] [CrossRef]
- Saitoh, M.; Nishitoh, H.; Fujii, M.; Takeda, K.; Tobiume, K.; Sawada, Y.; Kawabata, M.; Miyazono, K.; Ichijo, H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998, 17, 2596–2606. [Google Scholar] [CrossRef]
- Dérijard, B.; Hibi, M.; Wu, I.-H.; Barrett, T.; Su, B.; Deng, T.; Karin, M.; Davis, R.J. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell 1994, 76, 1025–1037. [Google Scholar] [CrossRef]
- Kyriakis, J.M.; Banerjee, P.; Nikolakaki, E.; Dai, T.; Rubie, E.A.; Ahmad, M.F.; Avruch, J.; Woodgett, J.R. The stress-activated protein kinase subfamily of c-Jun kinases. Nature 1994, 369, 156. [Google Scholar] [CrossRef]
- Weston, C.R.; Davis, R.J. The JNK signal transduction pathway. Curr. Opin. Cell Biol. 2007, 19, 142–149. [Google Scholar] [CrossRef]
- Kyriakis, J.; Brautigan, D.; Ingebritsen, T.; Avruch, J. pp54 microtubule-associated protein-2 kinase requires both tyrosine and serine/threonine phosphorylation for activity. J. Biol. Chem. 1991, 266, 10043–10046. [Google Scholar]
- Dai, T.; Rubie, E.; Franklin, C.; Kraft, A.; Gillespie, D.; Avruch, J.; Kyriakis, J.; Woodgett, J. Stress-activated protein kinases bind directly to the delta domain of c-Jun in resting cells: implications for repression of c-Jun function. Oncogene 1995, 10, 849–855. [Google Scholar] [PubMed]
- Kallunki, T.; Su, B.; Tsigelny, I.; Sluss, H.K.; Dérijard, B.; Moore, G.; Davis, R.; Karin, M. JNK2 contains a specificity-determining region responsible for efficient c-Jun binding and phosphorylation. Genes Dev. 1994, 8, 2996–3007. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, N.J.; Davis, R.J. Role of JNK in tumor development. Cell Cycle 2003, 2, 199–201. [Google Scholar] [PubMed]
- Johnson, R.; Spiegelman, B.; Hanahan, D.; Wisdom, R. Cellular transformation and malignancy induced by ras require c-jun. Mol. Cell. Biol. 1996, 16, 4504–4511. [Google Scholar] [CrossRef]
- Eferl, R.; Ricci, R.; Kenner, L.; Zenz, R.; David, J.-P.; Rath, M.; Wagner, E.F. Liver tumor development: c-Jun antagonizes the proapoptotic activity of p53. Cell 2003, 112, 181–192. [Google Scholar] [CrossRef]
- Kennedy, N.J.; Sluss, H.K.; Jones, S.N.; Bar-Sagi, D.; Flavell, R.A.; Davis, R.J. Suppression of Ras-stimulated transformation by the JNK signal transduction pathway. Genes & development 2003, 17, 629–637. [Google Scholar]
- Vasilevskaya, I.; O’Dwyer, P.J. Role of Jun and Jun kinase in resistance of cancer cells to therapy. Drug Resist. Updat. 2003, 6, 147–156. [Google Scholar] [CrossRef]
- Bubici, C.; Papa, S.; Pham, C.G.; Zazzeroni, F.; Franzoso, G. NF-κB and JNK: an intricate affair. Cell Cycle 2004, 3, 1524–1529. [Google Scholar] [CrossRef]
- Javelaud, D.; Besançon, F. NF-κB activation results in rapid inactivation of JNK in TNFα-treated Ewing sarcoma cells: a mechanism for the anti-apoptotic effect of NF-κB. Oncogene 2001, 20, 4365. [Google Scholar] [CrossRef]
- Han, J.; Lee, J.; Bibbs, L.; Ulevitch, R. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science 1994, 265, 808–811. [Google Scholar] [CrossRef]
- Herskowitz, I. MAP kinase pathways in yeast: for mating and more. Cell 1995, 80, 187–197. [Google Scholar] [CrossRef]
- Freshney, N.W.; Rawlinson, L.; Guesdon, F.; Jones, E.; Cowley, S.; Hsuan, J.; Saklatvala, J. Interleukin-1 activates a novel protein kinase cascade that results in the phosphorylation of Hsp27. Cell 1994, 78, 1039–1049. [Google Scholar] [CrossRef]
- Rouse, J.; Cohen, P.; Trigon, S.; Morange, M.; Alonso-Llamazares, A.; Zamanillo, D.; Hunt, T.; Nebreda, A.R. A novel kinase cascade triggered by stress and heat shock that stimulates MAPKAP kinase-2 and phosphorylation of the small heat shock proteins. Cell 1994, 78, 1027–1037. [Google Scholar] [CrossRef]
- Goedert, M.; Cuenda, A.; Craxton, M.; Jakes, R.; Cohen, P. Activation of the novel stress-activated protein kinase SAPK4 by cytokines and cellular stresses is mediated by SKK3 (MKK6); comparison of its substrate specificity with that of other SAP kinases. EMBO J. 1997, 16, 3563–3571. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Chen, C.; Li, Z.; Guo, W.; Gegner, J.A.; Lin, S.; Han, J. Characterization of the structure and function of a new mitogen-activated protein kinase (p38β). J. Biol. Chem. 1996, 271, 17920–17926. [Google Scholar] [CrossRef]
- Jiang, Y.; Gram, H.; Zhao, M.; New, L.; Gu, J.; Feng, L.; Di Padova, F.; Ulevitch, R.J.; Han, J. Characterization of the structure and function of the fourth member of p38 group mitogen-activated protein kinases, p38δ. J. Biol. Chem. 1997, 272, 30122–30128. [Google Scholar] [CrossRef]
- Lee, J.C.; Laydon, J.T.; McDonnell, P.C.; Gallagher, T.F.; Kumar, S.; Green, D.; McNulty, D.; Blumenthal, M.J.; Keys, J.R.; Strickler, J.E. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature 1994, 372, 739. [Google Scholar] [CrossRef]
- Eyers, P.A.; Craxton, M.; Morricel, N.; Cohen, P.; Goedert, M. Conversion of SB 203580-insensitive MAP kinase family members to drug-sensitive forms by a single amino-acid substitution. Chem. Biol. 1998, 5, 321–328. [Google Scholar] [CrossRef]
- Saklatvala, J. The p38 MAP kinase pathway as a therapeutic target in inflammatory disease. Curr. Opin. Pharmacol. 2004, 4, 372–377. [Google Scholar] [CrossRef]
- Hirai, S.-I.; Noda, K.; Moriguchi, T.; Nishida, E.; Yamashita, A.; Deyama, T.; Fukuyama, K.; Ohno, S. Differential activation of two JNK activators, MKK7 and SEK1, by MKN28-derived nonreceptor serine/threonine kinase/mixed lineage kinase 2. J. Biol. Chem. 1998, 273, 7406–7412. [Google Scholar] [CrossRef]
- Zarubin, T.; Jiahuai, H. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005, 15, 11. [Google Scholar] [CrossRef] [PubMed]
- Bulavin, D.V.; Fornace, A.J. p38 MAP kinase’s emerging role as a tumor suppressor. Advances Cancer Res. 2004, 92, 95–118. [Google Scholar]
- Timofeev, O.; Lee, T.Y.; Bulavin, D.V. A subtle change in p38 MAPK activity is sufficient to suppress in vivo tumorigenesis. Cell Cycle 2005, 4, 118–120. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, K.; Pruitt, W.M.; Bilter, G.K.; Westwick, J.K.; Der, C.J. Raf-independent deregulation of p38 and JNK mitogen-activated protein kinases are critical for Ras transformation. J. Biol. Chem. 2002, 277, 31808–31817. [Google Scholar] [CrossRef]
- She, Q.-B.; Bode, A.M.; Ma, W.-Y.; Chen, N.-Y.; Dong, Z. Resveratrol-induced activation of p53 and apoptosis is mediated by extracellular-signal-regulated protein kinases and p38 kinase. Cancer Res. 2001, 61, 1604–1610. [Google Scholar]
- Bulavin, D.V.; Kovalsky, O.; Hollander, M.C.; Fornace Jr, A.J. Loss of oncogenic H-ras-induced cell cycle arrest and p38 mitogen-activated protein kinase activation by disruption of Gadd45a. Mol. and cellular Biol. 2003, 23, 3859–3871. [Google Scholar] [CrossRef]
- Iyoda, K.; Sasaki, Y.; Horimoto, M.; Toyama, T.; Yakushijin, T.; Sakakibara, M.; Takehara, T.; Fujimoto, J.; Hori, M.; Wands, J.R. Involvement of the p38 mitogen-activated protein kinase cascade in hepatocellular carcinoma. Cancer 2003, 97, 3017–3026. [Google Scholar] [CrossRef]
- Olson, J.M.; Hallahan, A.R. p38 MAP kinase: a convergence point in cancer therapy. Trends Mol. Med. 2004, 10, 125–129. [Google Scholar] [CrossRef]
- Deacon, K.; Mistry, P.; Chernoff, J.; Blank, J.L.; Patel, R. p38 Mitogen-activated protein kinase mediates cell death and p21-activated kinase mediates cell survival during chemotherapeutic drug-induced mitotic arrest. Mol. Biol. Cell 2003, 14, 2071–2087. [Google Scholar] [CrossRef]
- Losa, J.H.; Cobo, C.P.; Viniegra, J.G.; Lobo, V.J.S.-A.; y Cajal, S.R.; Sanchez-Prieto, R. Role of the p38 MAPK pathway in cisplatin-based therapy. Oncogene 2003, 22, 3998. [Google Scholar] [CrossRef]
- Lee, E.-R.; Kim, J.-Y.; Kang, Y.-J.; Ahn, J.-Y.; Kim, J.-H.; Kim, B.-W.; Choi, H.-Y.; Jeong, M.-Y.; Cho, S.-G. Interplay between PI3K/Akt and MAPK signaling pathways in DNA-damaging drug-induced apoptosis. Biochimica et Biophysica Acta (BBA)-Mol. Cell Res. 2006, 1763, 958–968. [Google Scholar] [CrossRef] [PubMed]
- Hardy, M.P.; Owczarek, C.M.; Jermiin, L.S.; Ejdebäck, M.; Hertzog, P.J. Characterization of the type I interferon locus and identification of novel genes. Genomics 2004, 84, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Van Pesch, V.; Lanaya, H.; Renauld, J.-C.; Michiels, T. Characterization of the murine alpha interferon gene family. J. Virol. 2004, 78, 8219–8228. [Google Scholar] [CrossRef] [PubMed]
- Pestka, S. The human interferon α species and receptors. Peptide Sci. 2000, 55, 254–287. [Google Scholar] [CrossRef]
- Zhou, A.; Hassel, B.A.; Silverman, R.H. Expression cloning of 2-5A-dependent RNAase: a uniquely regulated mediator of interferon action. Cell 1993, 72, 753–765. [Google Scholar] [CrossRef]
- Lu, J.; O’Hara, E.B.; Trieselmann, B.A.; Romano, P.R.; Dever, T.E. The interferon-induced double-stranded RNA-activated protein kinase PKR will phosphorylate serine, threonine, or tyrosine at residue 51 in eukaryotic initiation factor 2α. J. Biol. Chem. 1999, 274, 32198–32203. [Google Scholar] [CrossRef]
- Stranden, A.M.; Staeheli, P.; Pavlovic, J. Function of the mouse Mx1 protein is inhibited by overexpression of the PB2 protein of influenza virus. Virology 1993, 197, 642–651. [Google Scholar] [CrossRef]
- Hu, X.; Bies, J.; Wolff, L. Interferon beta increases c-Myc proteolysis in mouse monocyte/macrophage leukemia cells. Leukemia Res. 2005, 29, 1307–1314. [Google Scholar] [CrossRef]
- Iacopino, F.; Ferrandina, G.; Scambia, G.; Benedetti-Panici, P.; Mancuso, S.; Sica, G. Interferons inhibit EGF-stimulated cell growth and reduce EGF binding in human breast cancer cells. Anticancer Res. 1996, 16, 1919–1924. [Google Scholar]
- Hamilton, J.A.; Whitty, G.A.; Kola, I.; Hertzog, P.J. Endogenous IFN-alpha beta suppresses colony-stimulating factor (CSF)-1-stimulated macrophage DNA synthesis and mediates inhibitory effects of lipopolysaccharide and TNF-alpha. J. Immunol. 1996, 156, 2553–2557. [Google Scholar]
- Hwang, S.Y.; Hertzog, P.J.; Holland, K.A.; Sumarsono, S.H.; Tymms, M.J.; Hamilton, J.A.; Whitty, G.; Bertoncello, I.; Kola, I. A null mutation in the gene encoding a type I interferon receptor component eliminates antiproliferative and antiviral responses to interferons alpha and beta and alters macrophage responses. Proc. Natl. Acad. Sci. USA 1995, 92, 11284–11288. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Zhang, X.; Tough, D.; Sprent, J. Multiple effects of immunostimulatory DNA on T cells and the role of type I interferons. Immunostimulatory DNA Seq. 2001, 22, 77–84. [Google Scholar]
- Chawla-Sarkar, M.; Lindner, D.J.; Liu, Y.-F.; Williams, B.; Sen, G.C.; Silverman, R.H.; Borden, E.C. Apoptosis and interferons: role of interferon-stimulated genes as mediators of apoptosis. Apoptosis 2003, 8, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Juang, S.-H.; Wei, S.-J.; Hung, Y.-M.; Hsu, C.-Y.; Yang, D.-M.; Liu, K.-J.; Chen, W.-S.; Yang, W.K. IFN-β induces caspase-mediated apoptosis by disrupting mitochondria in human advanced stage colon cancer cell lines. J. Interferon Cytokine Res. 2004, 24, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Kirou, K.A.; Krishna, R.; Maria, V.; Butler, J.; Crow, M.K. Induction of Fas ligand-mediated apoptosis by interferon-α. Clin. Immunol. 2000, 95, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Oshima, K.; Yanase, N.; Ibukiyama, C.; Yamashina, A.; Kayagaki, N.; Yagita, H.; Mizuguchi, J. Involvement of TRAIL/TRAIL-R interaction in IFN-α-induced apoptosis of Daudi B lymphoma cells. Cytokine 2001, 14, 193–201. [Google Scholar] [CrossRef]
- Sanceau, J.; Hiscott, J.; Delattre, O.; Wietzerbin, J. IFN-β induces serine phosphorylation of Stat-1 in Ewing’s sarcoma cells and mediates apoptosis via induction of IRF-1 and activation of caspase-7. Oncogene 2000, 19, 3372. [Google Scholar] [CrossRef]
- Zhou, A.; Paranjape, J.; Brown, T.L.; Nie, H.; Naik, S.; Dong, B.; Chang, A.; Trapp, B.; Fairchild, R.; Colmenares, C. Interferon action and apoptosis are defective in mice devoid of 2′,5′-oligoadenylate-dependent RNase L. EMBO J. 1997, 16, 6355–6363. [Google Scholar] [CrossRef]
- Rogge, L.; D’Ambrosio, D.; Biffi, M.; Penna, G.; Minetti, L.J.; Presky, D.H.; Adorini, L.; Sinigaglia, F. The role of Stat4 in species-specific regulation of Th cell development by type I IFNs. J. Immunol. 1998, 161, 6567–6574. [Google Scholar]
- Blanco, P.; Palucka, A.K.; Gill, M.; Pascual, V.; Banchereau, J. Induction of dendritic cell differentiation by IFN-α in systemic lupus erythematosus. Science 2001, 294, 1540–1543. [Google Scholar] [CrossRef]
- Theofilopoulos, A.N.; Baccala, R.; Beutler, B.; Kono, D.H. Type I interferons (α/β) in immunity and autoimmunity. Annu. Rev. Immunol. 2005, 23, 307–335. [Google Scholar] [CrossRef] [PubMed]
- Biron, C.A.; Nguyen, K.B.; Pien, G.C.; Cousens, L.P.; Salazar-Mather, T.P. Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annu. Rev. Immunol. 1999, 17, 189–220. [Google Scholar] [CrossRef] [PubMed]
- Salazar-Mather, T.P.; Lewis, C.A.; Biron, C.A. Type I interferons regulate inflammatory cell trafficking and macrophage inflammatory protein 1α delivery to the liver. J. Clin. Invest. 2002, 110, 321–330. [Google Scholar] [CrossRef]
- Thomas, K.E.; Galligan, C.L.; Newman, R.D.; Fish, E.N.; Vogel, S.N. Contribution of interferon-β to the murine macrophage response to the Toll-like receptor 4 agonist, lipopolysaccharide. J. Biol. Chem. 2006, 281, 31119–31130. [Google Scholar] [CrossRef]
- Hennessy, E.J.; Parker, A.E.; O’neill, L.A. Targeting Toll-like receptors: emerging therapeutics? Nat. Rev. Drug Discov. 2010, 9, 293. [Google Scholar] [CrossRef]
- So, E.Y.; Ouchi, T. The application of Toll like receptors for cancer therapy. Int. J. Biol. Sci. 2010, 6, 675. [Google Scholar] [CrossRef]
- Rudilla, F.; Fayolle, C.; Casares, N.; Durantez, M.; Arribillaga, L.; Lozano, T.; Villanueva, L.; Pio, R.; Sarobe, P.; Leclerc, C. Combination of a TLR4 ligand and anaphylatoxin C5a for the induction of antigen-specific cytotoxic T cell responses. Vaccine 2012, 30, 2848–2858. [Google Scholar] [CrossRef]
- Stone, G.W.; Barzee, S.; Snarsky, V.; Santucci, C.; Tran, B.; Langer, R.; Zugates, G.T.; Anderson, D.G.; Kornbluth, R.S. Nanoparticle-delivered multimeric soluble CD40L DNA combined with Toll-Like Receptor agonists as a treatment for melanoma. PLoS One 2009, 4, e7334. [Google Scholar] [CrossRef]
- Schneider, C.; Schmidt, T.; Ziske, C.; Tiemann, K.; Lee, K.; Uhlinsky, V.; Behrens, P.; Sauerbruch, T.; Schmidt-Wolf, I.; Mühlradt, P. Tumour suppression induced by the macrophage activating lipopeptide MALP-2 in an ultrasound guided pancreatic carcinoma mouse model. Gut 2004, 53, 355–361. [Google Scholar] [CrossRef]
- Triozzi, P.L.; Aldrich, W.; Ponnazhagan, S. Inhibition and promotion of tumor growth with adeno-associated virus carcinoembryonic antigen vaccine and Toll-like receptor agonists. Cancer Gene Ther. 2011, 18, 850. [Google Scholar] [CrossRef]
- Broomfield, S.A.; Van Der Most, R.G.; Prosser, A.C.; Mahendran, S.; Tovey, M.G.; Smyth, M.J.; Robinson, B.W.; Currie, A.J. Locally administered TLR7 agonists drive systemic antitumor immune responses that are enhanced by anti-CD40 immunotherapy. J. Immunol. 2009, 182, 5217–5224. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.B.; Vasquez-Dunddel, D.; Fu, J.; Albesiano, E.; Pardoll, D.; Kim, Y.J. Intratumoral administration of TLR4 agonist absorbed into a cellular vector improves antitumor responses. Clin. Cancer Res. 2011, 17, 3984–3992. [Google Scholar] [CrossRef] [PubMed]
- Sobek, V.; Birkner, N.; Falk, I.; Würch, A.; Kirschning, C.J.; Wagner, H.; Wallich, R.; Lamers, M.C.; Simon, M.M. Direct Toll-like receptor 2 mediated co-stimulation of T cells in the mouse system as a basis for chronic inflammatory joint disease. Arthritis Res. Ther. 2004, 6, R433. [Google Scholar] [CrossRef] [PubMed]
- Asprodites, N.; Zheng, L.; Geng, D.; Velasco-Gonzalez, C.; Sanchez-Perez, L.; Davila, E. Engagement of Toll-like receptor-2 on cytotoxic T-lymphocytes occurs in vivo and augments antitumor activity. FASEB J. 2008, 22, 3628–3637. [Google Scholar] [CrossRef] [PubMed]
- Cottalorda, A.; Verschelde, C.; Marçais, A.; Tomkowiak, M.; Musette, P.; Uematsu, S.; Akira, S.; Marvel, J.; Bonnefoy-Berard, N. TLR2 engagement on CD8 T cells lowers the thresholdfor optimal antigen-induced T cell activation. Eur. J. Immunol. 2006, 36, 1684–1693. [Google Scholar] [CrossRef] [PubMed]
- Morrison, C.; Baer, M.R.; Zandberg, D.P.; Kimball, A.; Davila, E. Effects of Toll-like receptor signals in T-cell neoplasms. Future Oncol. 2011, 7, 309–320. [Google Scholar] [CrossRef]
- Babu, S.; Blauvelt, C.P.; Kumaraswami, V.; Nutman, T.B. Cutting edge: diminished T cell TLR expression and function modulates the immune response in human filarial infection. J. Immunol. 2006, 176, 3885–3889. [Google Scholar] [CrossRef]
- Hervas-Stubbs, S.; Olivier, A.; Boisgerault, F.; Thieblemont, N.; Leclerc, C. TLR3 ligand stimulates fully functional memory CD8+ T cells in the absence of CD4+ T-cell help. Blood 2007, 109, 5318–5326. [Google Scholar] [CrossRef]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245. [Google Scholar] [CrossRef]
- Shortman, K.; Liu, Y.-J. Mouse and human dendritic cell subtypes. Nat. Rev. Immunol. 2002, 2, 151. [Google Scholar] [CrossRef]
- Piccioli, D.; Tavarini, S.; Borgogni, E.; Steri, V.; Nuti, S.; Sammicheli, C.; Bardelli, M.; Montagna, D.; Locatelli, F.; Wack, A. Functional specialization of human circulating CD16 and CD1c myeloid dendritic-cell subsets. Blood 2007, 109, 5371–5379. [Google Scholar] [CrossRef] [PubMed]
- Hovanessian, A.G. On the discovery of interferon-inducible, double-stranded RNA activated enzymes: the 2′–5′ oligoadenylate synthetases and the protein kinase PKR. Cytokine Growth Factor Rev. 2007, 18, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Jarrossay, D.; Napolitani, G.; Colonna, M.; Sallusto, F.; Lanzavecchia, A. Specialization and complementarity in microbial molecule recognition by human myeloid and plasmacytoid dendritic cells. Eur. J. Immunol. 2001, 31, 3388–3393. [Google Scholar] [CrossRef]
- Kadowaki, N.; Ho, S.; Antonenko, S.; de Waal Malefyt, R.; Kastelein, R.A.; Bazan, F.; Liu, Y.-J. Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens. J. Exp. Med. 2001, 194, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Amakawa, R.; Kaisho, T.; Hemmi, H.; Tajima, K.; Uehira, K.; Ozaki, Y.; Tomizawa, H.; Akira, S.; Fukuhara, S. Interferon-α and interleukin-12 are induced differentially by Toll-like receptor 7 ligands in human blood dendritic cell subsets. J. Exp. Med. 2002, 195, 1507–1512. [Google Scholar] [CrossRef] [PubMed]
- Krug, A.; Towarowski, A.; Britsch, S.; Rothenfusser, S.; Hornung, V.; Bals, R.; Giese, T.; Engelmann, H.; Endres, S.; Krieg, A.M. Toll-like receptor expression reveals CpG DNA as a unique microbial stimulus for plasmacytoid dendritic cells which synergizes with CD40 ligand to induce high amounts of IL-12. Eur. J. Immunol. 2001, 31, 3026–3037. [Google Scholar] [CrossRef]
- Hasan, U.; Chaffois, C.; Gaillard, C.; Saulnier, V.; Merck, E.; Tancredi, S.; Guiet, C.; Brière, F.; Vlach, J.; Lebecque, S. Human TLR10 is a functional receptor, expressed by B cells and plasmacytoid dendritic cells, which activates gene transcription through MyD88. J. Immunol. 2005, 174, 2942–2950. [Google Scholar] [CrossRef]
- Boonstra, A.; Asselin-Paturel, C.; Gilliet, M.; Crain, C.; Trinchieri, G.; Liu, Y.-J.; O’Garra, A. Flexibility of mouse classical and plasmacytoid-derived dendritic cells in directing T helper type 1 and 2 cell development: dependency on antigen dose and differential toll-like receptor ligation. J. Exp. Med. 2003, 197, 101–109. [Google Scholar] [CrossRef]
- Salio, M.; Cella, M.; Vermi, W.; Facchetti, F.; Palmowski, M.J.; Smith, C.L.; Shepherd, D.; Colonna, M.; Cerundolo, V. Plasmacytoid dendritic cells prime IFN-γ-secreting melanoma-specific CD8 lymphocytes and are found in primary melanoma lesions. Eur. J. Immunol. 2003, 33, 1052–1062. [Google Scholar] [CrossRef]
- Zou, W.; Machelon, V.; Coulomb-L’Hermin, A.; Borvak, J.; Nome, F.; Isaeva, T.; Wei, S.; Krzysiek, R.; Durand-Gasselin, I.; Gordon, A. Stromal-derived factor-1 in human tumors recruits and alters the function of plasmacytoid precursor dendritic cells. Nat. Med. 2001, 7, 1339. [Google Scholar] [CrossRef]
- Hartmann, E.; Wollenberg, B.; Rothenfusser, S.; Wagner, M.; Wellisch, D.; Mack, B.; Giese, T.; Gires, O.; Endres, S.; Hartmann, G. Identification and functional analysis of tumor-infiltrating plasmacytoid dendritic cells in head and neck cancer. Cancer Res. 2003, 63, 6478–6487. [Google Scholar] [PubMed]
- Treilleux, I.; Blay, J.-Y.; Bendriss-Vermare, N.; Ray-Coquard, I.; Bachelot, T.; Guastalla, J.-P.; Bremond, A.; Goddard, S.; Pin, J.-J.; Barthelemy-Dubois, C. Dendritic cell infiltration and prognosis of early stage breast cancer. Clin. Cancer Res. 2004, 10, 7466–7474. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Corak, J.; Ciernik, I.F.; Kavanaugh, D.; Carbone, D.P. Decreased antigen presentation by dendritic cells in patients with breast cancer. Clin. Cancer Res. 1997, 3, 483–490. [Google Scholar] [PubMed]
- Bell, D.; Chomarat, P.; Broyles, D.; Netto, G.; Harb, G.M.; Lebecque, S.; Valladeau, J.; Davoust, J.; Palucka, K.A.; Banchereau, J. In breast carcinoma tissue, immature dendritic cells reside within the tumor, whereas mature dendritic cells are located in peritumoral areas. J. Exp. Med. 1999, 190, 1417–1426. [Google Scholar] [CrossRef]
- Menetrier-Caux, C.; Montmain, G.; Dieu, M.; Bain, C.; Favrot, M.; Caux, C.; Blay, J. Inhibition of the differentiation of dendritic cells from CD34+ progenitors by tumor cells: role of interleukin-6 and macrophage colony-stimulating factor. Blood 1998, 92, 4778–4791. [Google Scholar] [CrossRef]
- Wei, S.; Kryczek, I.; Zou, L.; Daniel, B.; Cheng, P.; Mottram, P.; Curiel, T.; Lange, A.; Zou, W. Plasmacytoid dendritic cells induce CD8+ regulatory T cells in human ovarian carcinoma. Cancer Res. 2005, 65, 5020–5026. [Google Scholar] [CrossRef]
- Dummer, R.; Urosevic, M.; Kempf, W.; Hoek, K.; Hafner, J.; Burg, G. Imiquimod in basal cell carcinoma: how does it work? Br. J. Dermatol. 2003, 149, 57–58. [Google Scholar] [CrossRef]
- Tyring, S. Imiquimod applied topically: a novel immune response modifier. Skin Ther. Lett. 2001, 6, 1–4. [Google Scholar]
- Hofmann, M.A.; Kors, C.; Audring, H.; Walden, P.; Sterry, W.; Trefzer, U. Phase 1 evaluation of intralesionally injected TLR9-agonist PF-3512676 in patients with basal cell carcinoma or metastatic melanoma. J. Immunother. 2008, 31, 520–527. [Google Scholar] [CrossRef]
- Lou, Y.; Liu, C.; Kim, G.J.; Liu, Y.-J.; Hwu, P.; Wang, G. Plasmacytoid dendritic cells synergize with myeloid dendritic cells in the induction of antigen-specific antitumor immune responses. J. Immunol. 2007, 178, 1534–1541. [Google Scholar] [CrossRef]
- Piccioli, D.; Sammicheli, C.; Tavarini, S.; Nuti, S.; Frigimelica, E.; Manetti, A.G.; Nuccitelli, A.; Aprea, S.; Valentini, S.; Borgogni, E. Human plasmacytoid dendritic cells are unresponsive to bacterial stimulation and require a novel type of cooperation with myeloid dendritic cells for maturation. Blood 2009, 113, 4232–4239. [Google Scholar] [CrossRef] [PubMed]
- Komai-Koma, M.; Jones, L.; Ogg, G.S.; Xu, D.; Liew, F.Y. TLR2 is expressed on activated T cells as a costimulatory receptor. Proc. Natl. Acad. Sci. USA 2004, 101, 3029–3034. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Yang, Y.; Gad, E.; Wenner, C.A.; Chang, A.; Larson, E.R.; Dang, Y.; Martzen, M.; Standish, L.J.; Disis, M.L. Polysaccharide krestin is a novel TLR2 agonist that mediates inhibition of tumor growth via stimulation of CD8 T cells and NK cells. Clin. Cancer Res. 2010, 17, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Luo, F.; Cai, Y.; Liu, N.; Wang, L.; Xu, D.; Chu, Y. TLR1/TLR2 agonist induces tumor regression by reciprocal modulation of effector and regulatory T cells. J. Immunol. 2011, 186, 1963–1969. [Google Scholar] [CrossRef] [PubMed]
- Geng, D.; Zheng, L.; Srivastava, R.; Asprodites, N.; Velasco-Gonzalez, C.; Davila, E. When toll-like receptor and T-cell receptor signals collide: a mechanism for enhanced CD8 T-cell effectors function. Blood 2010, 116, 3494–3504. [Google Scholar] [CrossRef]
- Geng, D.; Zheng, L.; Srivastava, R.; Riker, A.I.; Velasco-Gonzales, C.; Markovic, S.N.; Davila, E. Amplifying TLR-MyD88 signals within tumor-specific T-cells enhances antitumor activity to suboptimal levels of weakly-immunogenic tumor-antigens. Cancer Res. 2010, 70, 7442–7454. [Google Scholar] [CrossRef]
- Seki, E.; Tsutsui, H.; Tsuji, N.M.; Hayashi, N.; Adachi, K.; Nakano, H.; Futatsugi-Yumikura, S.; Takeuchi, O.; Hoshino, K.; Akira, S. Critical roles of myeloid differentiation factor 88-dependent proinflammatory cytokine release in early phase clearance of Listeria monocytogenes in mice. J. Immunol. 2002, 169, 3863–3868. [Google Scholar] [CrossRef]
- Seki, E.; Brenner, D.A. Toll-like receptors and adaptor molecules in liver disease: update. Hepatology 2008, 48, 322–335. [Google Scholar] [CrossRef]
- Zanin-Zhorov, A.; Nussbaum, G.; Franitza, S.; Cohen, I.R.; Lider, O. T cells respond to heat shock protein 60 via TLR2: activation of adhesion and inhibition of chemokine receptors. FASEB J. 2003, 17, 1567–1569. [Google Scholar] [CrossRef]
- Mueller, K.L.; Daniels, M.A.; Felthauser, A.; Kao, C.; Jameson, S.C.; Shimizu, Y. Cutting edge: LFA-1 integrin-dependent T cell adhesion is regulated by both ag specificity and sensitivity. J. Immunol. 2004, 173, 2222–2226. [Google Scholar] [CrossRef]
- Kobayashi, N.; Takata, H.; Yokota, S.; Takiguchi, M. Down-regulation of CXCR4 expression on human CD8+ T cells during peripheral differentiation. Eur. J. Immunol. 2004, 34, 3370–3378. [Google Scholar] [CrossRef]
- Liu, H.; Komai-Koma, M.; Xu, D.; Liew, F.Y. Toll-like receptor 2 signaling modulates the functions of CD4+ CD25+ regulatory T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 7048–7053. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.H.; Taylor, D.K.; Turka, L.A. The contribution of direct TLR signaling to T cell responses. Immunol. Res. 2009, 45, 25–36. [Google Scholar] [CrossRef]
- Salem, M.L. Triggering of toll-like receptor signaling pathways in T cells contributes to the anti-tumor efficacy of T cell responses. Immunol. Lett. 2011, 137, 9–14. [Google Scholar] [CrossRef]
- Sutmuller, R.P.; den Brok, M.H.; Kramer, M.; Bennink, E.J.; Toonen, L.W.; Kullberg, B.-J.; Joosten, L.A.; Akira, S.; Netea, M.G.; Adema, G.J. Toll-like receptor 2 controls expansion and function of regulatory T cells. J. Clin. Invest. 2006, 116, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A. Progress in human tumour immunology and immunotherapy. Nature 2001, 411, 380. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Restifo, N.P. Cancer immunotherapy: moving beyond current vaccines. Nat. Med. 2004, 10, 909. [Google Scholar] [CrossRef]
- Mercier, B.C.; Cottalorda, A.; Coupet, C.-A.; Marvel, J.; Bonnefoy-Bérard, N. TLR2 engagement on CD8 T cells enables generation of functional memory cells in response to a suboptimal TCR signal. J. Immunol. 2009, 182, 1860–1867. [Google Scholar] [CrossRef] [PubMed]
- Kaech, S.M.; Tan, J.T.; Wherry, E.J.; Konieczny, B.T.; Surh, C.D.; Ahmed, R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat. Immunol. 2003, 4, 1191. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.-M.; Yao, Y.-M.; Liang, H.-P.; Xu, C.-T.; Dong, N.; Yu, Y.; Sheng, Z.-Y. High mobility group box-1 protein regulate immunosuppression of regulatory T cells through toll-like receptor 4. Cytokine 2011, 54, 296–304. [Google Scholar] [CrossRef]
- Day, E.B.; Zeng, W.; Doherty, P.C.; Jackson, D.C.; Kedzierska, K.; Turner, S.J. The context of epitope presentation can influence functional quality of recalled influenza A virus-specific memory CD8+ T cells. J. Immunol. 2007, 179, 2187–2194. [Google Scholar] [CrossRef] [PubMed]
- Deetz, C.O.; Hebbeler, A.M.; Propp, N.A.; Cairo, C.; Tikhonov, I.; Pauza, C.D. Gamma interferon secretion by human Vγ2Vδ2 T cells after stimulation with antibody against the T-cell receptor plus the Toll-Like receptor 2 agonist Pam3Cys. Infect. Immun. 2006, 74, 4505–4511. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lancioni, C.L.; Li, Q.; Thomas, J.J.; Ding, X.; Thiel, B.; Drage, M.G.; Pecora, N.D.; Ziady, A.G.; Shank, S.; Harding, C.V. Mycobacterium tuberculosis lipoproteins directly regulate human memory CD4+ T cell activation via Toll-like receptors 1 and 2. Infect. Immun. 2011, 79, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Gelman, A.E.; Zhang, J.; Choi, Y.; Turka, L.A. Toll-like receptor ligands directly promote activated CD4+ T cell survival. J. Immunol. 2004, 172, 6065–6073. [Google Scholar] [CrossRef]
- Tabiasco, J.; Devêvre, E.; Rufer, N.; Salaun, B.; Cerottini, J.-C.; Speiser, D.; Romero, P. Human effector CD8+ T lymphocytes express TLR3 as a functional coreceptor. J. Immunol. 2006, 177, 8708–8713. [Google Scholar] [CrossRef]
- Salem, M.L.; Diaz-Montero, C.M.; El-Naggar, S.A.; Chen, Y.; Moussa, O.; Cole, D.J. The TLR3 agonist poly (I: C) targets CD8+ T cells and augments their antigen-specific responses upon their adoptive transfer into naive recipient mice. Vaccine 2009, 27, 549–557. [Google Scholar] [CrossRef]
- Wesch, D.; Beetz, S.; Oberg, H.-H.; Marget, M.; Krengel, K.; Kabelitz, D. Direct costimulatory effect of TLR3 ligand poly (I: C) on human γδ T lymphocytes. J. Immunol. 2006, 176, 1348–1354. [Google Scholar] [CrossRef]
- Shojaei, H.; Oberg, H.-H.; Juricke, M.; Marischen, L.; Kunz, M.; Mundhenke, C.; Gieseler, F.; Kabelitz, D.; Wesch, D. Toll-like receptors 3 and 7 agonists enhance tumor cell lysis by human γδ T cells. Cancer Res. 2009, 0008-5472, CAN-0009-1602. [Google Scholar]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef]
- Doyle, S.E.; O’Connell, R.; Vaidya, S.A.; Chow, E.K.; Yee, K.; Cheng, G. Toll-like receptor 3 mediates a more potent antiviral response than Toll-like receptor 4. J. Immunol. 2003, 170, 3565–3571. [Google Scholar] [CrossRef]
- Davey, G.M.; Wojtasiak, M.; Proietto, A.I.; Carbone, F.R.; Heath, W.R.; Bedoui, S. Cutting edge: priming of CD8 T cell immunity to herpes simplex virus type 1 requires cognate TLR3 expression in vivo. J. Immunol. 2010, 184, 2243–2246. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.; Zammit, D.J.; Lefrancois, L.; Vella, A.T. Effects of LPS-mediated bystander activation in the innate immune system. J. Leukoc. Biol. 2006, 80, 1251–1261. [Google Scholar] [CrossRef] [PubMed]
- Vogel, S.; Hilfiker, M.; Caulfield, M. Endotoxin-induced T lymphocyte proliferation. J. Immunol. 1983, 130, 1774–1779. [Google Scholar]
- Komai-Koma, M.; Gilchrist, D.S.; Xu, D. Direct recognition of LPS by human but not murine CD8+ T cells via TLR4 complex. Eur. J. Immunol. 2009, 39, 1564–1572. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, J.M.; Martinez, G.J.; Chung, Y.; Dong, C. Toll-like receptor 4 signaling in T cells promotes autoimmune inflammation. Proc. Natl. Acad. Sci. USA 2012, 109, 13064–13069. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, J.M.; Pappu, B.P.; Peng, J.; Martinez, G.J.; Zhang, Y.; Chung, Y.; Ma, L.; Yang, X.O.; Nurieva, R.I.; Tian, Q. Toll-like receptor 2 signaling in CD4+ T lymphocytes promotes T helper 17 responses and regulates the pathogenesis of autoimmune disease. Immunity 2010, 32, 692–702. [Google Scholar] [CrossRef] [PubMed]
- González-Navajas, J.M.; Fine, S.; Law, J.; Datta, S.K.; Nguyen, K.P.; Yu, M.; Corr, M.; Katakura, K.; Eckman, L.; Lee, J. TLR4 signaling in effector CD4+ T cells regulates TCR activation and experimental colitis in mice. J. Clin. Invest. 2010, 120, 570–581. [Google Scholar] [CrossRef]
- Caramalho, I.; Lopes-Carvalho, T.; Ostler, D.; Zelenay, S.; Haury, M.; Demengeot, J. Regulatory T cells selectively express toll-like receptors and are activated by lipopolysaccharide. J. Exp. Med. 2003, 197, 403–411. [Google Scholar] [CrossRef]
- Caron, G.; Duluc, D.; Frémaux, I.; Jeannin, P.; David, C.; Gascan, H.; Delneste, Y. Direct stimulation of human T cells via TLR5 and TLR7/8: flagellin and R-848 up-regulate proliferation and IFN-γ production by memory CD4+ T cells. J. Immunol. 2005, 175, 1551–1557. [Google Scholar] [CrossRef]
- Crellin, N.K.; Garcia, R.V.; Hadisfar, O.; Allan, S.E.; Steiner, T.S.; Levings, M.K. Human CD4+ T cells express TLR5 and its ligand flagellin enhances the suppressive capacity and expression of FOXP3 in CD4+ CD25+ T regulatory cells. J. Immunol. 2005, 175, 8051–8059. [Google Scholar] [CrossRef]
- Peng, G.; Guo, Z.; Kiniwa, Y.; shin Voo, K.; Peng, W.; Fu, T.; Wang, D.Y.; Li, Y.; Wang, H.Y.; Wang, R.-F. Toll-like receptor 8-mediated reversal of CD4+ regulatory T cell function. Science 2005, 309, 1380–1384. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Wang, H.Y.; Peng, W.; Kiniwa, Y.; Seo, K.H.; Wang, R.-F. Tumor-infiltrating γδ T cells suppress T and dendritic cell function via mechanisms controlled by a unique toll-like receptor signaling pathway. Immunity 2007, 27, 334–348. [Google Scholar] [CrossRef] [PubMed]
- Marsland, B.J.; Nembrini, C.; Grün, K.; Reissmann, R.; Kurrer, M.; Leipner, C.; Kopf, M. TLR ligands act directly upon T cells to restore proliferation in the absence of protein kinase C-θ signaling and promote autoimmune myocarditis. J. Immunol. 2007, 178, 3466–3473. [Google Scholar] [CrossRef] [PubMed]
- Chiffoleau, E.; Heslan, J.-M.; Heslan, M.; Louvet, C.; Condamine, T.; Cuturi, M.-C. TLR9 ligand enhances proliferation of rat CD4+ T cell and modulates suppressive activity mediated by CD4+ CD25+ T cell. Int. Immunol. 2007, 19, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Bendigs, S.; Salzer, U.; Lipford, G.B.; Wagner, H.; Heeg, K. CpG-oligodeoxynucleotides co-stimulate primary T cells in the absence of antigen-presenting cells. Eur. J. Immunol. 1999, 29, 1209–1218. [Google Scholar] [CrossRef]
- Zheng, L.; Asprodites, N.; Keene, A.H.; Rodriguez, P.; Brown, K.D.; Davila, E. TLR9 engagement on CD4 T lymphocytes represses γ-radiation–induced apoptosis through activation of checkpoint kinase response elements. Blood 2008, 111, 2704–2713. [Google Scholar] [CrossRef]
- Curtin, J.F.; Liu, N.; Candolfi, M.; Xiong, W.; Assi, H.; Yagiz, K.; Edwards, M.R.; Michelsen, K.S.; Kroeger, K.M.; Liu, C. HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PLoS Med. 2009, 6, e1000010. [Google Scholar] [CrossRef]
- Ribeiro, R.A.; Wanderley, C.W.; Wong, D.V.; Mota, J.M.S.; Leite, C.A.; Souza, M.H.; Cunha, F.Q.; Lima-Junior, R.C. Irinotecan-and 5-fluorouracil-induced intestinal mucositis: insights into pathogenesis and therapeutic perspectives. Cancer Chemother. Pharmacol. 2016, 78, 881–893. [Google Scholar] [CrossRef]
- Kuo, W.-T.; Lee, T.-C.; Yu, L.C.-H. Eritoran suppresses colon cancer by altering a functional balance in Toll-like receptors that bind lipopolysaccharide. Cancer Res. 2016, 76, 4684–4695. [Google Scholar] [CrossRef]
- Prakash, H.; Nadella, V.; Singh, S.; Schmitz-Winnenthal, H. CD14/TLR4 priming potentially recalibrates and exerts anti-tumor efficacy in tumor associated macrophages in a mouse model of pancreatic carcinoma. Sci. Rep. 2016, 6, 31490. [Google Scholar] [CrossRef]
- Llitjos, J.F.; Auffray, C.; Alby-Laurent, F.; Rousseau, C.; Merdji, H.; Bonilla, N.; Toubiana, J.; Belaïdouni, N.; Mira, J.P.; Lucas, B. Sepsis-induced expansion of granulocytic myeloid-derived suppressor cells promotes tumour growth through Toll-like receptor 4. J. Pathol. 2016, 239, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Fukata, M.; Chen, A.; Vamadevan, A.S.; Cohen, J.; Breglio, K.; Krishnareddy, S.; Hsu, D.; Xu, R.; Harpaz, N.; Dannenberg, A.J. Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterology 2007, 133, 1869–1869. e1814. [Google Scholar] [CrossRef] [PubMed]
- Furrie, E.; Macfarlane, S.; Thomson, G.; Macfarlane, G.T.; Microbiology, T.T.; Gut Biology Group; Bank, T. Toll-like receptors-2,-3 and-4 expression patterns on human colon and their regulation by mucosal-associated bacteria. Immunology 2005, 115, 565–574. [Google Scholar] [CrossRef]
- Zhou, M.; McFarland-Mancini, M.M.; Funk, H.M.; Husseinzadeh, N.; Mounajjed, T.; Drew, A.F. Toll-like receptor expression in normal ovary and ovarian tumors. Cancer Immunol. Immunother. 2009, 58, 1375–1385. [Google Scholar] [CrossRef] [PubMed]
- Kelly, M.G.; Alvero, A.B.; Chen, R.; Silasi, D.-A.; Abrahams, V.M.; Chan, S.; Visintin, I.; Rutherford, T.; Mor, G. TLR-4 signaling promotes tumor growth and paclitaxel chemoresistance in ovarian cancer. Cancer Res. 2006, 66, 3859–3868. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-J.; Wu, M.-S.; Lin, J.-T.; Chen, C.-C. Helicobacter pylori-induced invasion and angiogenesis of gastric cells is mediated by cyclooxygenase-2 induction through TLR2/TLR9 and promoter regulation. J. Immunol. 2005, 175, 8242–8252. [Google Scholar] [CrossRef] [PubMed]
- Niedzielska, I.; Orawczyk, T.; Ziaja, K.; Tkacz, M.; Starzewski, J.; Ciopala, M.; Kalacinski, J.; Niedzielski, Z.; Mazurek, U.; Ziaja, D. Toll-like receptors and the tendency of normal mucous membrane to transform to polyp or colorectal cancer. Int. J. Oral Maxillofac. Surg. 2009, 38, 574. [Google Scholar] [CrossRef]
- Naugler, W.E.; Sakurai, T.; Kim, S.; Maeda, S.; Kim, K.; Elsharkawy, A.M.; Karin, M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science 2007, 317, 121–124. [Google Scholar] [CrossRef]
- Harmey, J.H.; Bucana, C.D.; Lu, W.; Byrne, A.M.; McDonnell, S.; Lynch, C.; Bouchier-Hayes, D.; Dong, Z. Lipopolysaccharide-induced metastatic growth is associated with increased angiogenesis, vascular permeability and tumor cell invasion. Int. J. Cancer 2002, 101, 415–422. [Google Scholar] [CrossRef]
- Yang, H.; Zhou, H.; Feng, P.; Zhou, X.; Wen, H.; Xie, X.; Shen, H.; Zhu, X. Reduced expression of Toll-like receptor 4 inhibits human breast cancer cells proliferation and inflammatory cytokines secretion. J. Exp. Clin. Cancer Res. 2010, 29, 92. [Google Scholar] [CrossRef]
- Bhattacharya, D.; Yusuf, N. Expression of toll-like receptors on breast tumors: taking a toll on tumor microenvironment. Int. J. Breast Cancer 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Zhao, J.; Li, H.; He, K.-L.; Chen, Y.; Mayer, L.; Unkeless, J.C.; Xiong, H. Toll-like receptors on tumor cells facilitate evasion of immune surveillance. Cancer Res. 2005, 65, 5009–5014. [Google Scholar] [CrossRef] [PubMed]
- Song, E.-J.; Kang, M.-J.; Kim, Y.-S.; Kim, S.-M.; Lee, S.-E.; Kim, C.-H.; Kim, D.-J.; Park, J.-H. Flagellin promotes the proliferation of gastric cancer cells via the Toll-like receptor 5. Int. J. Mol. Med. 2011, 28, 115–119. [Google Scholar]
- Husseinzadeh, N.; Davenport, S.M. Role of toll-like receptors in cervical, endometrial and ovarian cancers: a review. Gynecol. Oncol. 2014, 135, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Cherfils-Vicini, J.; Platonova, S.; Gillard, M.; Laurans, L.; Validire, P.; Caliandro, R.; Magdeleinat, P.; Mami-Chouaib, F.; Dieu-Nosjean, M.-C.; Fridman, W.-H. Triggering of TLR7 and TLR8 expressed by human lung cancer cells induces cell survival and chemoresistance. J. Clin. Invest. 2010, 120, 1285–1297. [Google Scholar] [CrossRef] [PubMed]
- Belmont, L.; Rabbe, N.; Antoine, M.; Cathelin, D.; Guignabert, C.; Kurie, J.; Cadranel, J.; Wislez, M. Expression of TLR9 in tumor-infiltrating mononuclear cells enhances angiogenesis and is associated with a worse survival in lung cancer. Int. J. Cancer 2014, 134, 765–777. [Google Scholar] [CrossRef]
- Luo, Y.; Jiang, Q.-W.; Wu, J.-Y.; Qiu, J.-G.; Zhang, W.-J.; Mei, X.-L.; Shi, Z.; Di, J.-M. Regulation of migration and invasion by Toll-like receptor-9 signaling network in prostate cancer. Oncotarget 2015, 6, 22564. [Google Scholar] [CrossRef]
- Bevers, R.; Kurth, K.; Schamhart, D. Role of urothelial cells in BCG immunotherapy for superficial bladder cancer. Br. J. Cancer 2004, 91, 607. [Google Scholar] [CrossRef]
- Paone, A.; Starace, D.; Galli, R.; Padula, F.; De Cesaris, P.; Filippini, A.; Ziparo, E.; Riccioli, A. Toll-like receptor 3 triggers apoptosis of human prostate cancer cells through a PKC-α-dependent mechanism. Carcinogenesis 2008, 29, 1334–1342. [Google Scholar] [CrossRef]
- Taura, M.; Fukuda, R.; Suico, M.A.; Eguma, A.; Koga, T.; Shuto, T.; Sato, T.; Morino-Koga, S.; Kai, H. TLR3 induction by anticancer drugs potentiates poly I: C-induced tumor cell apoptosis. Cancer Sci. 2010, 101, 1610–1617. [Google Scholar] [CrossRef]
- Salaun, B.; Lebecque, S.; Matikainen, S.; Rimoldi, D.; Romero, P. Toll-like receptor 3 expressed by melanoma cells as a target for therapy? Clin. Cancer Res. 2007, 13, 4565–4574. [Google Scholar] [CrossRef] [PubMed]
- Bauer, A.K.; Dixon, D.; DeGraff, L.M.; Cho, H.-Y.; Walker, C.R.; Malkinson, A.M.; Kleeberger, S.R. Toll-like receptor 4 in butylated hydroxytoluene–induced mouse pulmonary inflammation and tumorigenesis. J. Natl. Cancer Inst. 2005, 97, 1778–1781. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Sanchez, A.; Shi, Z.; Zhang, T.; Liu, M.; Zhang, D. Activation of Toll-like receptor 5 on breast cancer cells by flagellin suppresses cell proliferation and tumor growth. Cancer Res. 2011, 71, 2466–2475. [Google Scholar] [CrossRef] [PubMed]
- Burdelya, L.G.; Gleiberman, A.S.; Toshkov, I.; Aygun-Sunar, S.; Bapardekar, M.; Manderscheid-Kern, P.; Bellnier, D.; Krivokrysenko, V.I.; Feinstein, E.; Gudkov, A.V. Toll-like receptor 5 agonist protects mice from dermatitis and oral mucositis caused by local radiation: implications for head-and-neck cancer radiotherapy. Int. J. Radiat. Oncol. Biol. Physics 2012, 83, 228–234. [Google Scholar] [CrossRef]
- Li, X.; Liu, D.; Liu, X.; Jiang, W.; Zhou, W.; Yan, W.; Cen, Y.; Li, B.; Cao, G.; Ding, G. CpG ODN107 potentiates radiosensitivity of human glioma cells via TLR9-mediated NF-κB activation and NO production. Tumor Biol. 2012, 33, 1607–1618. [Google Scholar] [CrossRef]
- Damiano, V.; Rosa, R.; Formisano, L.; Nappi, L.; Gelardi, T.; Marciano, R.; Cozzolino, I.; Troncone, G.; Agrawal, S.; Veneziani, B. Toll-like receptor 9 agonist IMO cooperates with everolimus in renal cell carcinoma by interfering with tumour growth and angiogenesis. Br. J. Cancer 2013, 108, 1616. [Google Scholar] [CrossRef]
- Guha, M. Anticancer TLR agonists on the ropes. Nat. Rev. Drug Discov. 2012, 11, 503–505. [Google Scholar] [CrossRef]
- Chefetz, I.; Alvero, A.; Holmberg, J.; Lebowitz, N.; Craveiro, V.; Yang-Hartwich, Y.; Yin, G.; Squillace, L.; Gurrea Soteras, M.; Aldo, P. TLR2 enhances ovarian cancer stem cell self-renewal and promotes tumor repair and recurrence. Cell Cycle 2013, 12, 511–521. [Google Scholar] [CrossRef]
- Kim, S.; Takahashi, H.; Lin, W.-W.; Descargues, P.; Grivennikov, S.; Kim, Y.; Luo, J.-L.; Karin, M. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 2009, 457, 102. [Google Scholar] [CrossRef]
- Li, D.; Wang, X.; Wu, J.-L.; Quan, W.-Q.; Ma, L.; Yang, F.; Wu, K.-Y.; Wan, H.-Y. Tumor-produced versican V1 enhances hCAP18/LL-37 expression in macrophages through activation of TLR2 and vitamin D3 signaling to promote ovarian cancer progression in vitro. PLoS One 2013, 8, e56616. [Google Scholar] [CrossRef]
- Xie, W.; Wang, Y.; Huang, Y.; Yang, H.; Wang, J.; Hu, Z. Toll-like receptor 2 mediates invasion via activating NF-κB in MDA-MB-231 breast cancer cells. Biochem. Biophys. Res. Commun. 2009, 379, 1027–1032. [Google Scholar] [CrossRef]
- Boraska Jelavić, T.; Barišić, M.; Drmic Hofman, I.; Boraska, V.; Vrdoljak, E.; Peruzović, M.; Hozo, I.; Puljiz, Ž.; Terzić, J. Microsatelite GT polymorphism in the toll-like receptor 2 is associated with colorectal cancer. Clin. Genet. 2006, 70, 156–160. [Google Scholar] [CrossRef]
- Theodoropoulos, G.E.; Saridakis, V.; Karantanos, T.; Michalopoulos, N.V.; Zagouri, F.; Kontogianni, P.; Lymperi, M.; Gazouli, M.; Zografos, G.C. Toll-like receptors gene polymorphisms may confer increased susceptibility to breast cancer development. Breast 2012, 21, 534–538. [Google Scholar] [CrossRef] [PubMed]
- Tomomitsu, T.; Tomiyasu, A.; Fangyu, W.; Tomoyuki, S.; Masakatsu, N.; Mikijyu, S.; Ichiro, H.; Hiroshi, N. Toll-like receptor 2 –196 to 174del polymorphism influences the susceptibility of Japanese people to gastric cancer. Cancer Sci. 2007, 98, 1790–1794. [Google Scholar] [CrossRef]
- Hans-Dieter, N.; Martin, C.; Cordula, B.; Katharina, A.; Tobias, M.; Thomas, B.; Benjamin, K.; Christian, K.; Margarete, O.; Falko, S.; et al. The toll-like receptor 2 (TLR2) -196 to -174 del/ins polymorphism affects viral loads and susceptibility to hepatocellular carcinoma in chronic hepatitis C. Int. J. Cancer 2012, 130, 1470–1475. [Google Scholar] [CrossRef]
- Xie, W.; Huang, Y.; Xie, W.; Guo, A.; Wu, W. Bacteria Peptidoglycan Promoted Breast Cancer Cell Invasiveness and Adhesiveness by Targeting Toll-Like Receptor 2 in the Cancer Cells. PLoS One 2010, 5, e10850. [Google Scholar] [CrossRef]
- He, W.; Liu, Q.; Wang, L.; Chen, W.; Li, N.; Cao, X. TLR4 signaling promotes immune escape of human lung cancer cells by inducing immunosuppressive cytokines and apoptosis resistance. Mol. Immunol. 2007, 44, 2850–2859. [Google Scholar] [CrossRef]
- Szajnik, M.; Szczepanski, M.J.; Czystowska, M.; Elishaev, E.; Mandapathil, M.; Nowak-Markwitz, E.; Spaczynski, M.; Whiteside, T.L. TLR4 signaling induced by lipopolysaccharide or paclitaxel regulates tumor survival and chemoresistance in ovarian cancer. Oncogene 2009, 28, 4353. [Google Scholar] [CrossRef]
- Hsu, R.Y.; Chan, C.H.; Spicer, J.D.; Rousseau, M.C.; Giannias, B.; Rousseau, S.; Ferri, L.E. LPS-induced TLR4 signaling in human colorectal cancer cells increases β1 integrin-mediated cell adhesion and liver metastasis. Cancer Res. 2011, 71, 1989–1998. [Google Scholar] [CrossRef]
- Yuan, X.; Zhou, Y.; Wang, W.; Li, J.; Xie, G.; Zhao, Y.; Xu, D.; Shen, L. Activation of TLR4 signaling promotes gastric cancer progression by inducing mitochondrial ROS production. Cell Death Dis. 2013, 4, e794. [Google Scholar] [CrossRef]
- Liu, X.; Pei, C.; Yan, S.; Liu, G.; Liu, G.; Chen, W.; Cui, Y.; Liu, Y. NADPH oxidase 1-dependent ROS is crucial for TLR4 signaling to promote tumor metastasis of non-small cell lung cancer. Tumor Biol. 2015, 36, 1493–1502. [Google Scholar] [CrossRef] [PubMed]
- Ikebe, M.; Kitaura, Y.; Nakamura, M.; Tanaka, H.; Yamasaki, A.; Nagai, S.; Wada, J.; Yanai, K.; Koga, K.; Sato, N. Lipopolysaccharide (LPS) increases the invasive ability of pancreatic cancer cells through the TLR4/MyD88 signaling pathway. J. Surg. Oncol. 2009, 100, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Sfondrini, L.; Rossini, A.; Besusso, D.; Merlo, A.; Tagliabue, E.; Mènard, S.; Balsari, A. Antitumor activity of the TLR-5 ligand flagellin in mouse models of cancer. J. Immunol. 2006, 176, 6624–6630. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Crozet, L.; Damotte, D.; Iribarren, K.; Schramm, C.; Alifano, M.; Lupo, A.; Cherfils-Vicini, J.; Goc, J.; Katsahian, S. TLR7 promotes tumor progression, chemotherapy resistance, and poor clinical outcomes in non–small cell lung cancer. Cancer Res. 2014, 74, 5008–5018. [Google Scholar] [CrossRef]
- Grimmig, T.; Matthes, N.; Hoeland, K.; Tripathi, S.; Chandraker, A.; Grimm, M.; Moench, R.; Moll, E.-M.; Friess, H.; Tsaur, I. TLR7 and TLR8 expression increases tumor cell proliferation and promotes chemoresistance in human pancreatic cancer. Int. J. Oncol. 2015, 47, 857–866. [Google Scholar] [CrossRef]
- Ren, T.; Wen, Z.-K.; Liu, Z.-M.; Liang, Y.-J.; Guo, Z.-L.; Xu, L. Functional expression of TLR9 is associated to the metastatic potential of human lung cancer cell. Cancer Biol. Ther. 2007, 6, 1704–1709. [Google Scholar] [CrossRef]
- Ren, T.; Xu, L.; Jiao, S.; Wang, Y.; Cai, Y.; Liang, Y.; Zhou, Y.; Zhou, H.; Wen, Z. TLR9 signaling promotes tumor progression of human lung cancer cell in vivo. Pathol. Oncol. Res. 2009, 15, 623–630. [Google Scholar] [CrossRef]
- Moreira, D.; Zhang, Q.; Hossain, D.M.S.; Nechaev, S.; Li, H.; Kowolik, C.M.; D’Apuzzo, M.; Forman, S.; Jones, J.; Pal, S.K. TLR9 signaling through NF-κB/RELA and STAT3 promotes tumor-propagating potential of prostate cancer cells. Oncotarget 2015, 6, 17302. [Google Scholar] [CrossRef]
- Ilvesaro, J.M.; Merrell, M.A.; Swain, T.M.; Davidson, J.; Zayzafoon, M.; Harris, K.W.; Selander, K.S. Toll like receptor-9 agonists stimulate prostate cancer invasion in vitro. Prostate 2007, 67, 774–781. [Google Scholar] [CrossRef]
- Xin, C.; Zhang, H.; Liu, Z. miR-154 suppresses colorectal cancer cell growth and motility by targeting TLR2. Mol. Cell. Biochem. 2014, 387, 271–277. [Google Scholar] [CrossRef]
- Salaun, B.; Coste, I.; Rissoan, M.-C.; Lebecque, S.J.; Renno, T. TLR3 can directly trigger apoptosis in human cancer cells. J. Immunol. 2006, 176, 4894–4901. [Google Scholar] [CrossRef] [PubMed]
- Harashima, N.; Inao, T.; Imamura, R.; Okano, S.; Suda, T.; Harada, M. Roles of the PI3K/Akt pathway and autophagy in TLR3 signaling-induced apoptosis and growth arrest of human prostate cancer cells. Cancer Immunol. Immunother. 2012, 61, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Ang, B.; Xu, X.; Huang, X.; Wu, Y.; Sun, Y.; Wang, W.; Li, N.; Cao, X.; Wan, T. TLR4 is essential for dendritic cell activation and anti-tumor T-cell response enhancement by DAMPs released from chemically stressed cancer cells. Cell. Mol. Immunol. 2014, 11, 150. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Kim, Y.J.; Kim, J.S.; Ryu, H.S.; Lee, H.K.; Kang, J.S.; Kim, H.M.; Hong, J.T.; Kim, Y.; Han, S.-B. Adjuvant effect of a natural TLR4 ligand on dendritic cell-based cancer immunotherapy. Cancer Lett. 2011, 313, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.T.; Hong, S.H.; Sin, J.-I.; Vu, H.V.D.; Jeong, K.; Cho, K.O.; Uematsu, S.; Akira, S.; Lee, S.E.; Rhee, J.H. Flagellin enhances tumor-specific CD8+ T cell immune responses through TLR5 stimulation in a therapeutic cancer vaccine model. Vaccine 2013, 31, 3879–3887. [Google Scholar] [CrossRef]
- Shi, M.; Yao, Y.; Han, F.; Li, Y.; Li, Y. MAP1S controls breast cancer cell TLR5 signaling pathway and promotes TLR5 signaling-based tumor suppression. PloS One 2014, 9, e86839. [Google Scholar] [CrossRef]
- Zhou, H.; Chen, J.-h.; Hu, J.; Luo, Y.-z.; Li, F.; Xiao, L.; Zhong, M.-z. High expression of Toll-like receptor 5 correlates with better prognosis in non-small-cell lung cancer: an anti-tumor effect of TLR5 signaling in non-small cell lung cancer. J. Cancer Res. Clin. Oncol. 2014, 140, 633–643. [Google Scholar] [CrossRef]
- Spinetti, T.; Spagnuolo, L.; Mottas, I.; Secondini, C.; Treinies, M.; Rüegg, C.; Hotz, C.; Bourquin, C. TLR7-based cancer immunotherapy decreases intratumoral myeloid-derived suppressor cells and blocks their immunosuppressive function. Oncoimmunology 2016, 5, e1230578. [Google Scholar] [CrossRef]
- Ito, H.; Ando, T.; Ogiso, H.; Arioka, Y.; Seishima, M. Inhibition of induced nitric oxide synthase enhances the anti-tumor effects on cancer immunotherapy using TLR7 agonist in mice. Cancer Immunol. Immunother. 2015, 64, 429–436. [Google Scholar] [CrossRef]
- Brignole, C.; Marimpietri, D.; Di Paolo, D.; Perri, P.; Morandi, F.; Pastorino, F.; Zorzoli, A.; Pagnan, G.; Loi, M.; Caffa, I. Therapeutic targeting of TLR9 inhibits cell growth and induces apoptosis in neuroblastoma. Cancer Res. 2010, 70, 9816–9826. [Google Scholar] [CrossRef]
- Pashenkov, M.; Goëss, G.; Wagner, C.; Hörmann, M.; Jandl, T.; Moser, A.; Britten, C.M.; Smolle, J.; Koller, S.; Mauch, C. Phase II trial of a Toll-like receptor 9–activating oligonucleotide in patients with metastatic melanoma. J. Clin. Oncol. 2006, 24, 5716–5724. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vacchelli, E.; Eggermont, A.; Fridman, W.H.; Galon, J.; Sautès-Fridman, C.; Tartour, E.; Zitvogel, L.; Kroemer, G. Trial Watch: Experimental Toll-like receptor agonists for cancer therapy. Oncoimmunology 2012, 1, 699–739. [Google Scholar] [CrossRef] [PubMed]
- Bekierkunst, A.; Levij, I.; Yarkoni, E.; Vilkas, E.; Lederer, E. Suppression of urethan-induced lung adenomas in mice treated with trehalose-6, 6-dimycolate (cord factor) and living bacillus Calmette Guérin. Science 1971, 174, 1240–1242. [Google Scholar] [CrossRef] [PubMed]
- Sjögren, H.O.; Ankerst, J. Effect of BCG and allogeneic tumour cells on adenovirus type 12 tumorigenesis in mice. Nature 1969, 221, 863–864. [Google Scholar] [CrossRef]
- Zbar, B.; Bernstein, I.; Tanaka, T.; Rapp, H.J. Tumor immunity produced by the intradermal inoculation of living tumor cells and living Mycobacterium bovis (strain BCG). Science 1970, 170, 1217–1218. [Google Scholar] [CrossRef]
- Zbar, B.; Tanaka, T. Immunotherapy of cancer: regression of tumors after intralesional injection of living Mycobacterium bovis. Science 1971, 271–273. [Google Scholar] [CrossRef]
- Morales, A.; Eidinger, D.; Bruce, A. Intracavitary Bacillus Calmette-Guerin in the treatment of superficial bladder tumors. J. Urol. 1976, 116, 180–182. [Google Scholar] [CrossRef]
- Hall, M.C.; Chang, S.S.; Dalbagni, G.; Pruthi, R.S.; Seigne, J.D.; Skinner, E.C.; Wolf, J.S.; Schellhammer, P.F. Guideline for the management of nonmuscle invasive bladder cancer (stages Ta, T1, and Tis): 2007 update. J. Urol. 2007, 178, 2314–2330. [Google Scholar] [CrossRef]
- Babjuk, M.; Oosterlinck, W.; Sylvester, R.; Kaasinen, E.; Böhle, A.; Palou-Redorta, J.; Rouprêt, M. EAU guidelines on non–muscle-invasive urothelial carcinoma of the bladder, the 2011 update. Eur. Urol. 2011, 59, 997–1008. [Google Scholar] [CrossRef]
- Kroemer, G.; Zitvogel, L.; Galluzzi, L. Victories and Deceptions in Tumor Immunology: Stimuvax®; Taylor & Francis: Milton, UK, 2013. [Google Scholar]
- Chin, A.I.; Miyahira, A.K.; Covarrubias, A.; Teague, J.; Guo, B.; Dempsey, P.W.; Cheng, G. Toll-like receptor 3–mediated suppression of TRAMP prostate cancer shows the critical role of type I interferons in tumor immune surveillance. Cancer Res. 2010, 70, 2595–2603. [Google Scholar] [CrossRef]
- Forte, G.; Rega, A.; Morello, S.; Luciano, A.; Arra, C.; Pinto, A.; Sorrentino, R. Polyinosinic-polycytidylic acid limits tumor outgrowth in a mouse model of metastatic lung cancer. J. Immunol. 2012, 188, 5357–5364. [Google Scholar] [CrossRef] [PubMed]
- Garaude, J.; Kent, A.; van Rooijen, N.; Blander, J.M. Simultaneous targeting of toll-and nod-like receptors induces effective tumor-specific immune responses. Sci. Transl. Med. 2012, 4, ra116–ra120. [Google Scholar] [CrossRef] [PubMed]
- Burdelya, L.G.; Krivokrysenko, V.I.; Tallant, T.C.; Strom, E.; Gleiberman, A.S.; Gupta, D.; Kurnasov, O.V.; Fort, F.L.; Osterman, A.L.; DiDonato, J.A. An agonist of toll-like receptor 5 has radioprotective activity in mouse and primate models. Science 2008, 320, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.; Kandimalla, E.R.; Yu, D.; Bhagat, L.; Li, Y.; Wang, D.; Zhu, F.; Tang, J.X.; Putta, M.R.; Cong, Y. Stabilized immune modulatory RNA compounds as agonists of Toll-like receptors 7 and 8. Proc. Natl. Acad. Sci. USA 2007, 104, 13750–13755. [Google Scholar] [CrossRef]
- Geisse, J.; Caro, I.; Lindholm, J.; Golitz, L.; Stampone, P.; Owens, M. Imiquimod 5% cream for the treatment of superficial basal cell carcinoma: results from two phase III, randomized, vehicle-controlled studies. J. Am. Acad. Dermatol. 2004, 50, 722–733. [Google Scholar] [CrossRef]
- Love, W.E.; Bernhard, J.D.; Bordeaux, J.S. Topical imiquimod or fluorouracil therapy for basal and squamous cell carcinoma: a systematic review. Arch. Dermatol. 2009, 145, 1431–1438. [Google Scholar] [CrossRef]
- Rajpar, S.; Marsden, J. Imiquimod in the treatment of lentigo maligna. Br. J. Dermatol. 2006, 155, 653–656. [Google Scholar] [CrossRef]
- Krieg, A.M. Development of TLR9 agonists for cancer therapy. J. Clin. Invest. 2007, 117, 1184–1194. [Google Scholar] [CrossRef]
- Baines, J.; Celis, E. Immune-mediated tumor regression induced by CpG-containing oligodeoxynucleotides. Clin. Cancer Res. 2003, 9, 2693–2700. [Google Scholar]
- Carpentier, A.F.; Chen, L.; Maltonti, F.; Delattre, J.-Y. Oligodeoxynucleotides containing CpG motifs can induce rejection of a neuroblastoma in mice. Cancer Res. 1999, 59, 5429–5432. [Google Scholar]
- Heckelsmiller, K.; Rall, K.; Beck, S.; Schlamp, A.; Seiderer, J.; Jahrsdörfer, B.; Krug, A.; Rothenfusser, S.; Endres, S.; Hartmann, G. Peritumoral CpG DNA elicits a coordinated response of CD8 T cells and innate effectors to cure established tumors in a murine colon carcinoma model. J. Immunol. 2002, 169, 3892–3899. [Google Scholar] [CrossRef]
- Goutagny, N.; Estornes, Y.; Hasan, U.; Lebecque, S.; Caux, C. Targeting pattern recognition receptors in cancer immunotherapy. Target. Oncol. 2012, 7, 29–54. [Google Scholar] [CrossRef]
- Kolida, S.; Gibson, G.R. Synbiotics in health and disease. Annu. Rev. Food Sci. Technol. 2011, 2, 373–393. [Google Scholar] [CrossRef] [PubMed]
- Le Leu, R.K.; Hu, Y.; Brown, I.L.; Woodman, R.J.; Young, G.P. Synbiotic intervention of Bifidobacterium lactis and resistant starch protects against colorectal cancer development in rats. Carcinogenesis 2009, 31, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Rowland, I.; Rumney, C.; Coutts, J.; Lievense, L. Effect of Bifidobacterium longum and inulin on gut bacterial metabolism and carcinogen-induced aberrant crypt foci in rats. Carcinogenesis 1998, 19, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-L.; Yu, L.-X.; Yang, W.; Tang, L.; Lin, Y.; Wu, H.; Zhai, B.; Tan, Y.-X.; Shan, L.; Liu, Q. Profound impact of gut homeostasis on chemically-induced pro-tumorigenic inflammation and hepatocarcinogenesis in rats. J. Hepatol. 2012, 57, 803–812. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Wang, K.; Mucida, D.; Stewart, C.A.; Schnabl, B.; Jauch, D.; Taniguchi, K.; Yu, G.-Y.; Österreicher, C.H.; Hung, K.E. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 2012, 491, 254. [Google Scholar] [CrossRef]
- Couturier-Maillard, A.; Secher, T.; Rehman, A.; Normand, S.; De Arcangelis, A.; Haesler, R.; Huot, L.; Grandjean, T.; Bressenot, A.; Delanoye-Crespin, A. NOD2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J. Clin. Invest. 2013, 123, 700–711. [Google Scholar] [CrossRef]
- Dapito, D.H.; Mencin, A.; Gwak, G.-Y.; Pradere, J.-P.; Jang, M.-K.; Mederacke, I.; Caviglia, J.M.; Khiabanian, H.; Adeyemi, A.; Bataller, R. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell 2012, 21, 504–516. [Google Scholar] [CrossRef]
- Yu, L.X.; Yan, H.X.; Liu, Q.; Yang, W.; Wu, H.P.; Dong, W.; Tang, L.; Lin, Y.; He, Y.Q.; Zou, S.S. Endotoxin accumulation prevents carcinogen-induced apoptosis and promotes liver tumorigenesis in rodents. Hepatology 2010, 52, 1322–1333. [Google Scholar] [CrossRef]
- Bass, N.M.; Mullen, K.D.; Sanyal, A.; Poordad, F.; Neff, G.; Leevy, C.B.; Sigal, S.; Sheikh, M.Y.; Beavers, K.; Frederick, T. Rifaximin treatment in hepatic encephalopathy. N. Engl. J. Med. 2010, 362, 1071–1081. [Google Scholar] [CrossRef] [PubMed]
- Fort, M.M.; Mozaffarian, A.; Stöver, A.G.; da Silva Correia, J.; Johnson, D.A.; Crane, R.T.; Ulevitch, R.J.; Persing, D.H.; Bielefeldt-Ohmann, H.; Probst, P. A synthetic TLR4 antagonist has anti-inflammatory effects in two murine models of inflammatory bowel disease. J. Immunol. 2005, 174, 6416–6423. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, D.P.; Lynn, M. Antagonism of in vivo and ex vivo response to endotoxin by E5564, a synthetic lipid A analogue. J. Endotoxin Res. 2002, 8, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Sha, T.; Sunamoto, M.; Kitazaki, T.; Sato, J.; Ii, M.; Iizawa, Y. Therapeutic effects of TAK-242, a novel selective Toll-like receptor 4 signal transduction inhibitor, in mouse endotoxin shock model. Eur. J. Pharmacol. 2007, 571, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Arslan, F.; Houtgraaf, J.H.; Keogh, B.; Kazemi, K.; de Jong, R.; McCormack, W.J.; O’Neill, L.A.; McGuirk, P.; Timmers, L.; Smeets, M.B. Treatment with OPN-305, a humanized anti–Toll-like receptor-2 antibody, reduces myocardial ischemia/reperfusion injury in pigs. Circ. Cardiovasc. Interv. 2012, 5, 279–287. [Google Scholar] [CrossRef]
- Palucka, K.; Banchereau, J. Cancer immunotherapy via dendritic cells. Nat. Rev. Cancer 2012, 12, 265. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| TLRs | Agonist/Ligand | Mechanism | Cancer Type | Enhanced Cancer Characteristics | References |
|---|---|---|---|---|---|
| TLR2 | Peptidoglycan (PGN) | Synergistic effect of wound-associated injury and PGN | Epithelial ovarian cancer | Self-renewal, repair, and recurrence | [229] |
| Versican | Inflammatory microenvironment | Lewis lung carcinoma | Metastasis | [230] | |
| hCAP18/LL-37 overexpression | Ovarian tumor | Growth and invasion | [231] | ||
| pg-LPS | Increased NF-κB signaling; IL-6, TGF-β, VEGF, and MMP9 secretion | MDA-MB-231 breast cancer cells | Invasion | [232] | |
| Arg753Gln and (GT)n microsatellite polymorphisms | TLR2 overexpression and increased NF-κB signaling | Colorectal cancer | Growth, progression, and invasion | [233] | |
| −196 to −174del | Decreased transcription of TLR2 gene | Breast cancer, gastric cancer, hepatocellular carcinoma | Tumor progression due to weaker immune response | [234,235,236] | |
| Bacterial PGN | Augmentation of NF-κB, STAT3, and Smad3 activities | Breast cancer | Invasion and adhesion | [237] | |
| TLR4 | LPS | Increased secretion of TGF-β, VEGF, and IL-8 | Lung cancer, ovarian cancer | Immune evasion and apoptosis resistance | [238,239] |
| Activation of PI3K–AKT signaling and promotion of β1 integrin function | Colorectal cancer | Increased adhesiveness and metastasis | [240] | ||
| Increased mitochondrial ROS production | Gastric cancer, non–small cell lung cancer | Increased cell proliferation | [241,242] | ||
| Increased NF-κB signaling | Pancreatic cancer | Increased invasion and progression | [243] | ||
| TLR5 | Flagellin | Decreased IFNγ:IL-4 ratio and increased number of CD4+CD25+ Treg cells | Tumor mouse model | Tumor growth | [244] |
| Enhanced activity of NF-κB, IL-8, and ERK | Gastric cancer | Cell proliferation | [214] | ||
| TLR7/8 | ssRNA | Activated NF-κB, upregulation of Bcl-2 | Lung cancer | Survival and chemoresistance | [216] |
| Loxoribine | Enhanced signaling | NSCLC | Progression and chemoresistance | [245] | |
| Resiquimod (R848) | Elevated NF-κB and COX2 expression | Pancreatic cancer | Proliferation and chemoresistance | [246] | |
| TLR9 | CpG ODN | Elevated expression of IL-1α, IL-8, CXCR4, ICAM1, and MMP2 | Human lung cancer | Metastasis | [247,248] |
| Greater response of NF-κB/RELA and STAT3 pathways | Prostate cancer | Cell proliferation | [249,250] |
| TLRs | Agonist/Ligand | Mechanism | Cancer Type | Inhibited Cancer Characteristics | References |
|---|---|---|---|---|---|
| TLR2 | MicroRNA-154 | TLR2 downregulation at post-transcription level | Colorectal cancer | Tumor growth, migration, and invasion | [251] |
| Krestin | Stimulation of CD8+ T cells and NK cells | Breast cancer | Growth | [154] | |
| TLR3 | Synthetic dsRNA | Elevated signaling | Breast cancer | Tumor survival | [252] |
| Poly(I:C) | PI3K/AKT pathway and autophagy | Prostate cancer | Growth and survival | [253] | |
| TLR4 | DAMPs | Antitumor T cells response with activation of DCs | Colorectal cancer | Cell proliferation | [254] |
| Angelan | Enhanced DC maturation | Melanoma | Tumor growth | [255] | |
| TLR5 | Flagellin | Increased IFNγ:IL-4 ratio and decreased number of CD4+CD25+ Treg cells | Tumor mouse model | Tumor growth | [244] |
| CD8+ CTL immune responses | Tumor model | Growth and survival | [256] | ||
| Increased MAP1S expression | Breast cancer | Tumor cell growth and migration | [257] | ||
| Increased signaling | NSCLC | Cell proliferation, migration, and invasion | [258] | ||
| Activated signaling | Breast cancer | Cell growth and proliferation | [224] | ||
| TLR7/8 | Imiquimod | Establishment of proimmunogenic microenvironment | Breast cancer | Metastasis | [216] |
| Resiquimod (R848) | Maturation and differentiation of MDSCs | Tumor model | Growth | [259] | |
| Imiquimod | Inhibition of nitric oxide synthase | Tumor model | Growth | [260] | |
| TLR9 | CpG ODN | Enhanced signaling | Neuroblastoma | Growth and survival | [261] |
| PF-3512676 | Enhanced signaling | Melanoma | Metastasis | [150,262] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Javaid, N.; Choi, S. Toll-like Receptors from the Perspective of Cancer Treatment. Cancers 2020, 12, 297. https://doi.org/10.3390/cancers12020297
Javaid N, Choi S. Toll-like Receptors from the Perspective of Cancer Treatment. Cancers. 2020; 12(2):297. https://doi.org/10.3390/cancers12020297
Chicago/Turabian StyleJavaid, Nasir, and Sangdun Choi. 2020. "Toll-like Receptors from the Perspective of Cancer Treatment" Cancers 12, no. 2: 297. https://doi.org/10.3390/cancers12020297
APA StyleJavaid, N., & Choi, S. (2020). Toll-like Receptors from the Perspective of Cancer Treatment. Cancers, 12(2), 297. https://doi.org/10.3390/cancers12020297