Combined Inhibition of Epigenetic Readers and Transcription Initiation Targets the EWS-ETS Transcriptional Program in Ewing Sarcoma

 , , , , , ,
, , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
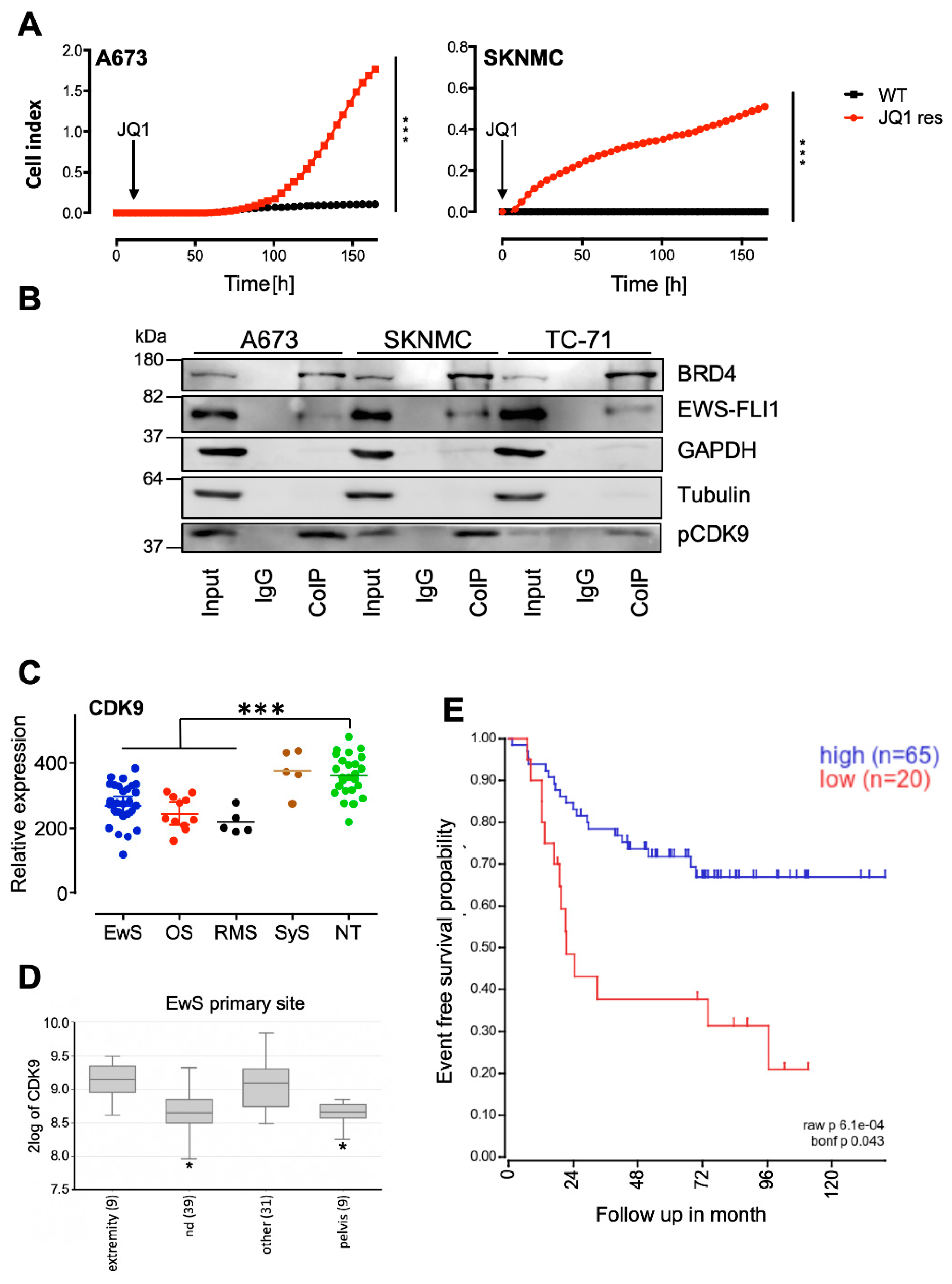
2.1. BRD4 Interacts with CDK9, and Its Inhibition Promotes the Development of Resistant Cells
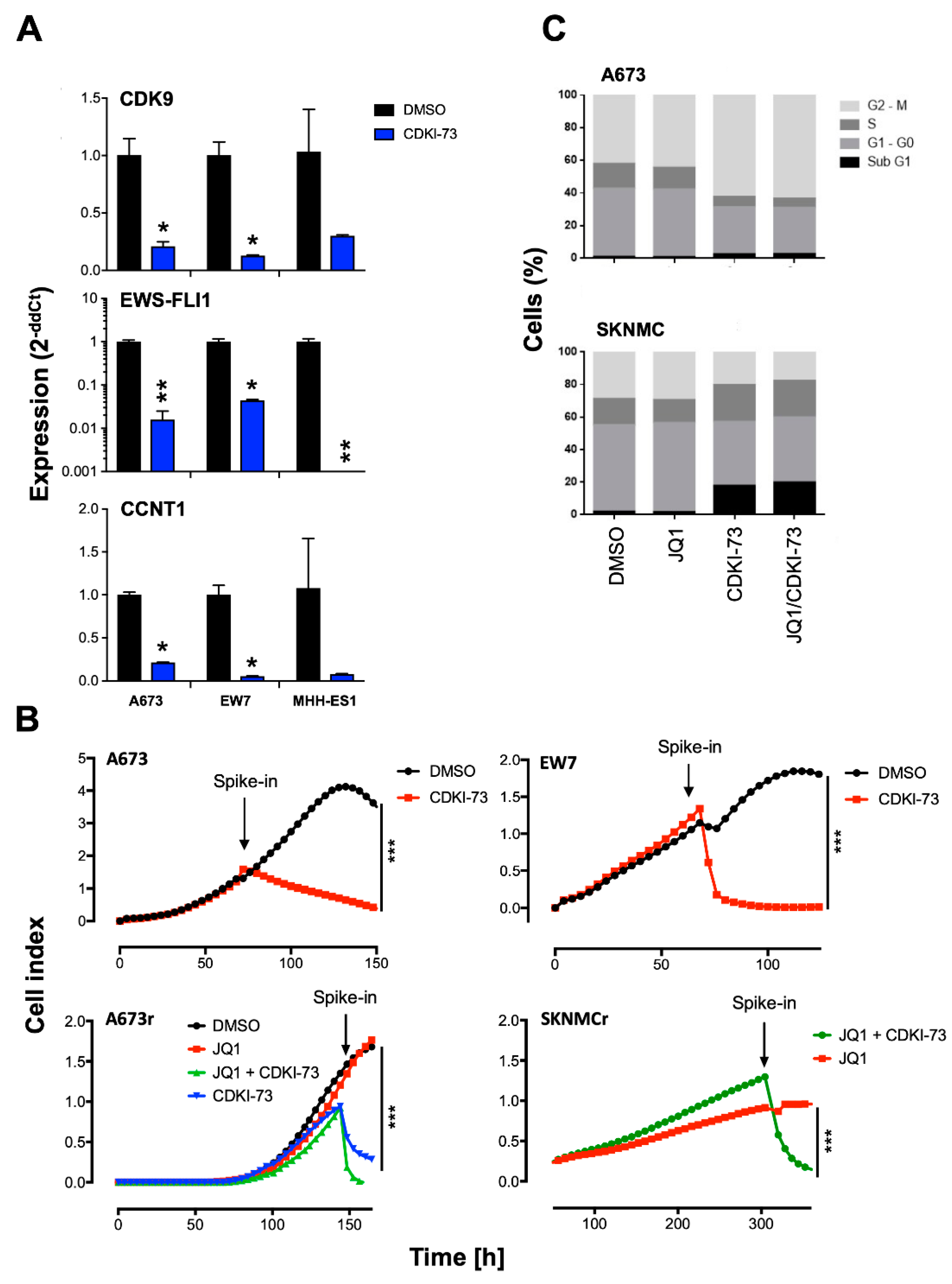
2.2. CDK9 Inhibitors Block EWS-FLI1 Expression and Proliferation of EwS
2.3. Expression Profile of EwS Cells after Treatment with CDK9 Inhibitors
2.4. Combined Targeting of CDK9 and BRD4 Inhibits Proliferation and Induces Apoptosis
2.5. Combined Targeting of CDK9 and BRD4 Inhibits Tumor Growth
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Chemical Compounds
4.3. Proliferation Assay
4.4. Co-Immunoprecipitation (Co-IP)
4.5. Western Blot Analysis
4.6. Cell Cycle Analysis
4.7. RNA Isolation
4.8. Quantitative Real Time-PCR (qRT-PCR)
4.9. Microarray Analysis
4.10. Animal Model
4.11. In Vivo Experiments
4.12. Histology
4.13. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Balamuth, N.J.; Womer, R.B. Ewing’s sarcoma. Lancet Oncol. 2010, 11, 184–192. [Google Scholar] [CrossRef]
- Delattre, O.; Zucman, J.; Plougastel, B.; Desmaze, C.; Melot, T.; Peter, M.; Kovar, H.; Joubert, I.; de Jong, P.; Rouleau, G.; et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature 1992, 359, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Lessnick, S.L.; Ladanyi, M. Molecular pathogenesis of Ewing sarcoma: new therapeutic and transcriptional targets. Annu. Rev. Pathol. 2012, 7, 145–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackintosh, C.; Madoz-Gurpide, J.; Ordonez, J.L.; Osuna, D.; Herrero-Martin, D. The molecular pathogenesis of Ewing’s sarcoma. Cancer Biol. Ther. 2010, 9, 655–667. [Google Scholar] [CrossRef]
- Brohl, A.S.; Solomon, D.A.; Chang, W.; Wang, J.; Song, Y.; Sindiri, S.; Patidar, R.; Hurd, L.; Chen, L.; Shern, J.F.; et al. The genomic landscape of the Ewing Sarcoma family of tumors reveals recurrent STAG2 mutation. PLoS Genet. 2014, 10, e1004475. [Google Scholar] [CrossRef] [Green Version]
- Crompton, B.D.; Stewart, C.; Taylor-Weiner, A.; Alexe, G.; Kurek, K.C.; Calicchio, M.L.; Kiezun, A.; Carter, S.L.; Shukla, S.A.; Mehta, S.S.; et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov. 2014, 4, 1326–1341. [Google Scholar] [CrossRef] [Green Version]
- Tirode, F.; Surdez, D.; Ma, X.; Parker, M.; Le Deley, M.C.; Bahrami, A.; Zhang, Z.; Lapouble, E.; Grossetete-Lalami, S.; Rusch, M.; et al. Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov. 2014, 4, 1342–1353. [Google Scholar] [CrossRef] [Green Version]
- Agelopoulos, K.; Richter, G.H.; Schmidt, E.; Dirksen, U.; von Heyking, K.; Moser, B.; Klein, H.U.; Kontny, U.; Dugas, M.; Poos, K.; et al. Deep sequencing in conjunction with expression and functional analyses reveals activation of FGFR1 in Ewing sarcoma. Clin. Cancer Res. 2015. [Google Scholar] [CrossRef] [Green Version]
- Grobner, S.N.; Worst, B.C.; Weischenfeldt, J.; Buchhalter, I.; Kleinheinz, K.; Rudneva, V.A.; Johann, P.D.; Balasubramanian, G.P.; Segura-Wang, M.; Brabetz, S.; et al. The landscape of genomic alterations across childhood cancers. Nature 2018, 555, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Riggi, N.; Knoechel, B.; Gillespie, S.M.; Rheinbay, E.; Boulay, G.; Suva, M.L.; Rossetti, N.E.; Boonseng, W.E.; Oksuz, O.; Cook, E.B.; et al. EWS-FLI1 utilizes divergent chromatin remodeling mechanisms to directly activate or repress enhancer elements in Ewing sarcoma. Cancer Cell 2014, 26, 668–681. [Google Scholar] [CrossRef] [Green Version]
- Tomazou, E.M.; Sheffield, N.C.; Schmidl, C.; Schuster, M.; Schonegger, A.; Datlinger, P.; Kubicek, S.; Bock, C.; Kovar, H. Epigenome mapping reveals distinct modes of gene regulation and widespread enhancer reprogramming by the oncogenic fusion protein EWS-FLI1. Cell Rep. 2015, 10, 1082–1095. [Google Scholar] [CrossRef] [Green Version]
- Dawson, M.A.; Prinjha, R.K.; Dittmann, A.; Giotopoulos, G.; Bantscheff, M.; Chan, W.I.; Robson, S.C.; Chung, C.W.; Hopf, C.; Savitski, M.M.; et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011, 478, 529–533. [Google Scholar] [CrossRef] [Green Version]
- Dey, A.; Ellenberg, J.; Farina, A.; Coleman, A.E.; Maruyama, T.; Sciortino, S.; Lippincott-Schwartz, J.; Ozato, K. A bromodomain protein, MCAP, associates with mitotic chromosomes and affects G(2)-to-M transition. Mol. Cell. Biol. 2000, 20, 6537–6549. [Google Scholar] [CrossRef]
- Wang, R.; Li, Q.; Helfer, C.M.; Jiao, J.; You, J. Bromodomain protein Brd4 associated with acetylated chromatin is important for maintenance of higher-order chromatin structure. J. Biol. Chem. 2012, 287, 10738–10752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filippakopoulos, P.; Knapp, S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 2014, 13, 337–356. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Vakoc, C.R. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol. Cell 2014, 54, 728–736. [Google Scholar] [CrossRef] [Green Version]
- Shu, S.; Polyak, K. BET Bromodomain Proteins as Cancer Therapeutic Targets. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 123–129. [Google Scholar] [CrossRef] [Green Version]
- Hensel, T.; Giorgi, C.; Schmidt, O.; Calzada-Wack, J.; Neff, F.; Buch, T.; Niggli, F.K.; Schafer, B.W.; Burdach, S.; Richter, G.H. Targeting the EWS-ETS transcriptional program by BET bromodomain inhibition in Ewing sarcoma. Oncotarget 2016, 7, 1451–1463. [Google Scholar] [CrossRef] [Green Version]
- Zhao, R.; Nakamura, T.; Fu, Y.; Lazar, Z.; Spector, D.L. Gene bookmarking accelerates the kinetics of post-mitotic transcriptional re-activation. Nat. Cell Biol. 2011, 13, 1295–1304. [Google Scholar] [CrossRef]
- Peterlin, B.M.; Price, D.H. Controlling the elongation phase of transcription with P-TEFb. Mol. Cell 2006, 23, 297–305. [Google Scholar] [CrossRef]
- Bisgrove, D.A.; Mahmoudi, T.; Henklein, P.; Verdin, E. Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription. Proc. Natl. Acad. Sci. USA 2007, 104, 13690–13695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, M.K.; Mochizuki, K.; Zhou, M.; Jeong, H.S.; Brady, J.N.; Ozato, K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol. Cell 2005, 19, 523–534. [Google Scholar] [CrossRef]
- Schroder, S.; Cho, S.; Zeng, L.; Zhang, Q.; Kaehlcke, K.; Mak, L.; Lau, J.; Bisgrove, D.; Schnolzer, M.; Verdin, E.; et al. Two-pronged binding with bromodomain-containing protein 4 liberates positive transcription elongation factor b from inactive ribonucleoprotein complexes. J. Biol. Chem. 2012, 287, 1090–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shim, E.Y.; Walker, A.K.; Shi, Y.; Blackwell, T.K. CDK-9/cyclin T (P-TEFb) is required in two postinitiation pathways for transcription in the C. elegans embryo. Genes Dev. 2002, 16, 2135–2146. [Google Scholar] [CrossRef] [Green Version]
- Fujinaga, K.; Irwin, D.; Huang, Y.; Taube, R.; Kurosu, T.; Peterlin, B.M. Dynamics of human immunodeficiency virus transcription: P-TEFb phosphorylates RD and dissociates negative effectors from the transactivation response element. Mol. Cell. Biol. 2004, 24, 787–795. [Google Scholar] [CrossRef] [Green Version]
- Marshall, N.F.; Price, D.H. Purification of P-TEFb, a transcription factor required for the transition into productive elongation. J. Biol. Chem. 1995, 270, 12335–12338. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Pandey, S.; Travers, M.; Sun, H.; Morton, G.; Madzo, J.; Chung, W.; Khowsathit, J.; Perez-Leal, O.; Barrero, C.A.; et al. Targeting CDK9 Reactivates Epigenetically Silenced Genes in Cancer. Cell 2018, 175, 1244–1258 e1226. [Google Scholar] [CrossRef] [Green Version]
- Krystof, V.; Baumli, S.; Furst, R. Perspective of cyclin-dependent kinase 9 (CDK9) as a drug target. Curr. Pharm. Des. 2012, 18, 2883–2890. [Google Scholar] [CrossRef] [Green Version]
- Sonawane, Y.A.; Taylor, M.A.; Napoleon, J.V.; Rana, S.; Contreras, J.I.; Natarajan, A. Cyclin Dependent Kinase 9 Inhibitors for Cancer Therapy. J. Med. Chem. 2016, 59, 8667–8684. [Google Scholar] [CrossRef]
- Ma, H.; Seebacher, N.A.; Hornicek, F.J.; Duan, Z. Cyclin-dependent kinase 9 (CDK9) is a novel prognostic marker and therapeutic target in osteosarcoma. EBioMedicine 2019, 39, 182–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Seebacher, N.A.; Xiao, T.; Hornicek, F.J.; Duan, Z. Targeting regulation of cyclin dependent kinase 9 as a novel therapeutic strategy in synovial sarcoma. J. Orthop. Res. 2019, 37, 510–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gollavilli, P.N.; Pawar, A.; Wilder-Romans, K.; Natesan, R.; Engelke, C.G.; Dommeti, V.L.; Krishnamurthy, P.M.; Nallasivam, A.; Apel, I.J.; Xu, T.; et al. EWS/ETS-Driven Ewing Sarcoma Requires BET Bromodomain Proteins. Cancer Res. 2018, 78, 4760–4773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurimchak, A.M.; Shelton, C.; Duncan, K.E.; Johnson, K.J.; Brown, J.; O’Brien, S.; Gabbasov, R.; Fink, L.S.; Li, Y.; Lounsbury, N.; et al. Resistance to BET Bromodomain Inhibitors Is Mediated by Kinome Reprogramming in Ovarian Cancer. Cell Rep. 2016, 16, 1273–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itzen, F.; Greifenberg, A.K.; Bosken, C.A.; Geyer, M. Brd4 activates P-TEFb for RNA polymerase II CTD phosphorylation. Nucleic Acids Res. 2014, 42, 7577–7590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsby, E.; Pratt, G.; Shao, H.; Abbas, A.Y.; Fischer, P.M.; Bradshaw, T.D.; Brennan, P.; Fegan, C.; Wang, S.; Pepper, C. A novel Cdk9 inhibitor preferentially targets tumor cells and synergizes with fludarabine. Oncotarget 2014, 5, 375–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Yik, J.H.; Chen, R.; He, N.; Jang, M.K.; Ozato, K.; Zhou, Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell 2005, 19, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Yeh, Y.Y.; Chen, R.; Hessler, J.; Mahoney, E.; Lehman, A.M.; Heerema, N.A.; Grever, M.R.; Plunkett, W.; Byrd, J.C.; Johnson, A.J. Up-regulation of CDK9 kinase activity and Mcl-1 stability contributes to the acquired resistance to cyclin-dependent kinase inhibitors in leukemia. Oncotarget 2015, 6, 2667–2679. [Google Scholar] [CrossRef] [Green Version]
- Rahaman, M.H.; Yu, Y.; Zhong, L.; Adams, J.; Lam, F.; Li, P.; Noll, B.; Milne, R.; Peng, J.; Wang, S. CDKI-73: An orally bioavailable and highly efficacious CDK9 inhibitor against acute myeloid leukemia. Invest. New Drugs 2019, 37, 625–635. [Google Scholar] [CrossRef]
- Kong, L.J.; Chang, J.T.; Bild, A.H.; Nevins, J.R. Compensation and specificity of function within the E2F family. Oncogene 2007, 26, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Dowless, M.; Lowery, C.D.; Shackleford, T.; Renschler, M.; Stephens, J.; Flack, R.; Blosser, W.; Gupta, S.; Stewart, J.; Webster, Y.; et al. Abemaciclib Is Active in Preclinical Models of Ewing Sarcoma via Multipronged Regulation of Cell Cycle, DNA Methylation, and Interferon Pathway Signaling. Clin. Cancer Res. 2018. [Google Scholar] [CrossRef] [Green Version]
- Boulay, G.; Sandoval, G.J.; Riggi, N.; Iyer, S.; Buisson, R.; Naigles, B.; Awad, M.E.; Rengarajan, S.; Volorio, A.; McBride, M.J.; et al. Cancer-Specific Retargeting of BAF Complexes by a Prion-like Domain. Cell 2017, 171, 163–178 e119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dey, A.; Chitsaz, F.; Abbasi, A.; Misteli, T.; Ozato, K. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc. Natl. Acad. Sci. USA 2003, 100, 8758–8763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacques, C.; Lamoureux, F.; Baud’huin, M.; Rodriguez Calleja, L.; Quillard, T.; Amiaud, J.; Tirode, F.; Redini, F.; Bradner, J.E.; Heymann, D.; et al. Targeting the epigenetic readers in Ewing sarcoma inhibits the oncogenic transcription factor EWS/Fli1. Oncotarget 2016, 7, 24125–24140. [Google Scholar] [CrossRef] [Green Version]
- Lam, F.; Abbas, A.Y.; Shao, H.; Teo, T.; Adams, J.; Li, P.; Bradshaw, T.D.; Fischer, P.M.; Walsby, E.; Pepper, C.; et al. Targeting RNA transcription and translation in ovarian cancer cells with pharmacological inhibitor CDKI-73. Oncotarget 2014, 5, 7691–7704. [Google Scholar] [CrossRef] [PubMed]
- Pawar, A.; Gollavilli, P.N.; Wang, S.; Asangani, I.A. Resistance to BET Inhibitor Leads to Alternative Therapeutic Vulnerabilities in Castration-Resistant Prostate Cancer. Cell Rep. 2018, 22, 2236–2245. [Google Scholar] [CrossRef] [Green Version]
- Winter, G.E.; Mayer, A.; Buckley, D.L.; Erb, M.A.; Roderick, J.E.; Vittori, S.; Reyes, J.M.; di Iulio, J.; Souza, A.; Ott, C.J.; et al. BET Bromodomain Proteins Function as Master Transcription Elongation Factors Independent of CDK9 Recruitment. Mol. Cell 2017, 67, 5–18 e19. [Google Scholar] [CrossRef] [Green Version]
- Kinsey, M.; Smith, R.; Lessnick, S.L. NR0B1 is required for the oncogenic phenotype mediated by EWS/FLI in Ewing’s sarcoma. Mol. Cancer Res. 2006, 4, 851–859. [Google Scholar] [CrossRef] [Green Version]
- Shao, H.; Shi, S.; Huang, S.; Hole, A.J.; Abbas, A.Y.; Baumli, S.; Liu, X.; Lam, F.; Foley, D.W.; Fischer, P.M.; et al. Substituted 4-(thiazol-5-yl)-2-(phenylamino)pyrimidines are highly active CDK9 inhibitors: synthesis, X-ray crystal structures, structure-activity relationship, and anticancer activities. J. Med. Chem. 2013, 56, 640–659. [Google Scholar] [CrossRef]
- Richter, G.H.; Plehm, S.; Fasan, A.; Rossler, S.; Unland, R.; Bennani-Baiti, I.M.; Hotfilder, M.; Lowel, D.; von Luettichau, I.; Mossbrugger, I.; et al. EZH2 is a mediator of EWS/FLI1 driven tumor growth and metastasis blocking endothelial and neuro-ectodermal differentiation. Proc. Natl. Acad. Sci. USA 2009, 106, 5324–5329. [Google Scholar] [CrossRef] [Green Version]
- Richter, G.H.; Fasan, A.; Hauer, K.; Grunewald, T.G.; Berns, C.; Rossler, S.; Naumann, I.; Staege, M.S.; Fulda, S.; Esposito, I.; et al. G-Protein coupled receptor 64 promotes invasiveness and metastasis in Ewing sarcomas through PGF and MMP1. J. Pathol. 2013, 230, 70–81. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Bardou, P.; Mariette, J.; Escudie, F.; Djemiel, C.; Klopp, C. jvenn: an interactive Venn diagram viewer. BMC Bioinformatics 2014, 15, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
H.S. Richter, G.; Hensel, T.; Schmidt, O.; Saratov, V.; von Heyking, K.; Becker-Dettling, F.; Prexler, C.; Yen, H.-Y.; Steiger, K.; Fulda, S.; et al. Combined Inhibition of Epigenetic Readers and Transcription Initiation Targets the EWS-ETS Transcriptional Program in Ewing Sarcoma. Cancers 2020, 12, 304. https://doi.org/10.3390/cancers12020304
H.S. Richter G, Hensel T, Schmidt O, Saratov V, von Heyking K, Becker-Dettling F, Prexler C, Yen H-Y, Steiger K, Fulda S, et al. Combined Inhibition of Epigenetic Readers and Transcription Initiation Targets the EWS-ETS Transcriptional Program in Ewing Sarcoma. Cancers. 2020; 12(2):304. https://doi.org/10.3390/cancers12020304
Chicago/Turabian StyleH.S. Richter, Günther, Tim Hensel, Oxana Schmidt, Vadim Saratov, Kristina von Heyking, Fiona Becker-Dettling, Carolin Prexler, Hsi-Yu Yen, Katja Steiger, Simone Fulda, and et al. 2020. "Combined Inhibition of Epigenetic Readers and Transcription Initiation Targets the EWS-ETS Transcriptional Program in Ewing Sarcoma" Cancers 12, no. 2: 304. https://doi.org/10.3390/cancers12020304