Mutation Analysis of Colorectal and Gastric Carcinomas Originating from Adenomas: Insights into Genomic Evolution Associated with Malignant Progression


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
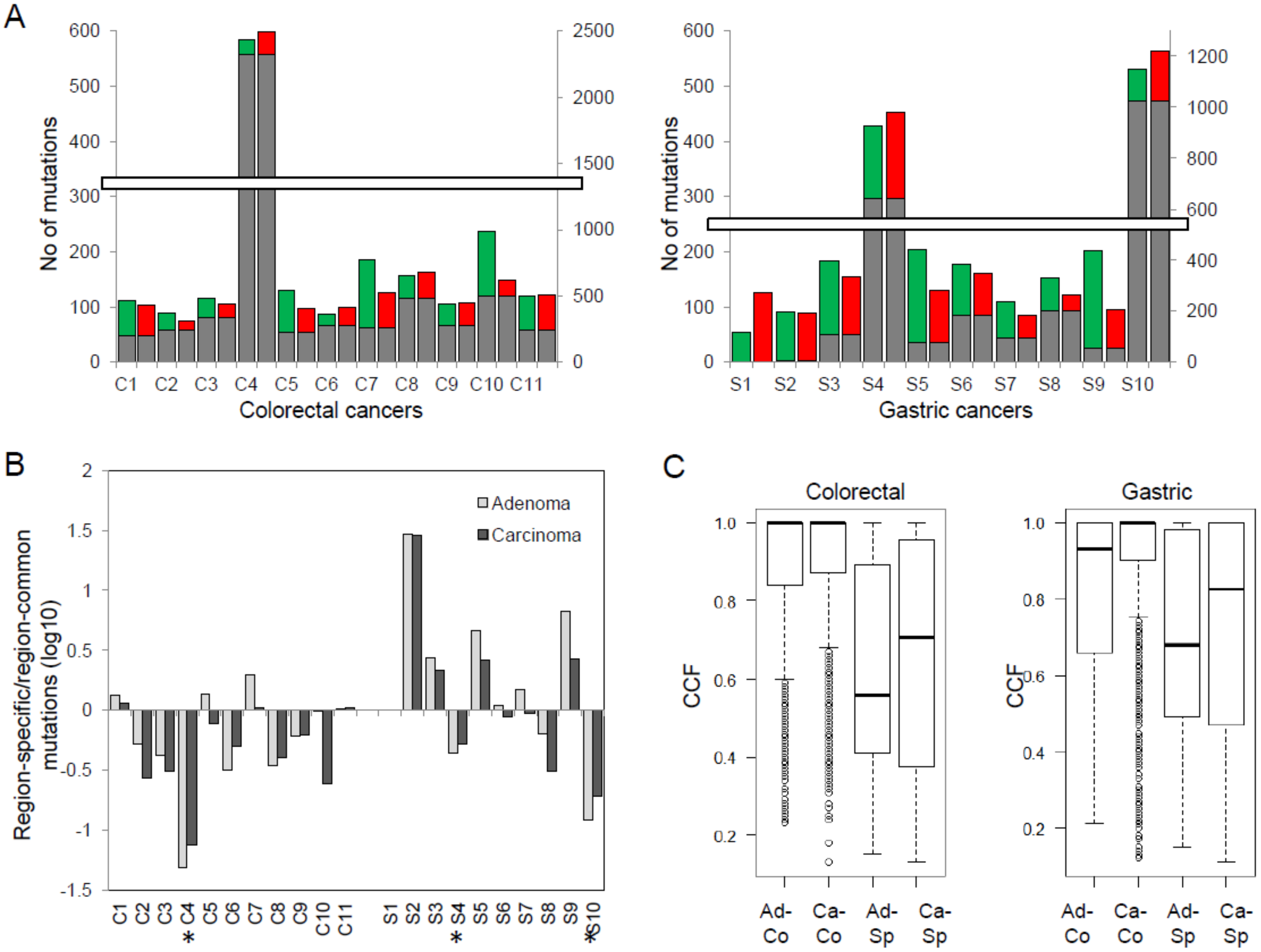
2. Results
2.1. Whole-Exome Sequencing and Somatic Variants
2.2. Genes Enriched to Regional Mutation Classes
2.3. Mutation Clusters between Adenoma–Carcinoma Pairs
2.4. Copy Number Profiles
2.5. Microsatellite Instability and Hypermutation
3. Discussion
4. Materials and Methods
4.1. Patient and Tumor Specimens
4.2. DNA Sequencing
4.3. Somatic Mutations
4.4. Copy Number Alterations
4.5. Microsatellite Instability
4.6. Clustering of Tumor Cell Populations Using PyClone
4.7. Driver Mutation Identification
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fedewa, S.A.; Ahnen, D.J.; Meester, R.G.S.; Barzi, A.; Jemal, A. Colorectal cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 177–193. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fearon, E.R. Molecular genetics of colorectal cancer. Annu. Rev. Pathol. 2011, 6, 479–507. [Google Scholar] [CrossRef]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Correa, P. Human gastric carcinogenesis: A multistep and multifactorial process—First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res. 1992, 52, 6735–6740. [Google Scholar] [PubMed]
- Brini, C.; Bogliolo, G.; Pontone, S.; Ierfone, N.; Chessa, A.; Pontone, P. Adenomas with severe dysplasia and early carcinoma of the colon-rectum: Our experience of 27 patients. Giornale Chirurgia 2001, 22, 26–28. [Google Scholar]
- Meyerson, M.; Gabriel, S.; Getz, G. Advances in understanding cancer genomes through second-generation sequencing. Nat. Rev. Genet. 2010, 11, 685–696. [Google Scholar] [CrossRef]
- Yachida, S.; Jones, S.; Bozic, I.; Antal, T.; Leary, R.; Fu, B.; Kamiyama, M.; Hruban, R.H.; Eshleman, J.R.; Nowak, M.A.; et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010, 467, 1114–1117. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.M.; An, C.H.; Rhee, J.K.; Jung, S.H.; Lee, S.H.; Baek, I.P.; Kim, M.S.; Chung, Y.J. Clonal origins and parallel evolution of regionally synchronous colorectal adenoma and carcinoma. Oncotarget 2015, 6, 27725–27735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landau, D.A.; Carter, S.L.; Stojanov, P.; McKenna, A.; Stevenson, K.; Lawrence, M.S.; Sougnez, C.; Stewart, C.; Sivachenko, A.; Wang, L.; et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell 2013, 152, 714–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Wolff, R.K.; Hoffman, M.D.; Wolff, E.C.; Herrick, J.S.; Sakoda, L.C.; Samowitz, W.S.; Slattery, M.L. Mutation analysis of adenomas and carcinomas of the colon: Early and late drivers. Genes Chromosomes Cancer 2018, 57, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Irahara, N.; Baba, Y.; Nosho, K.; Shima, K.; Yan, L.; Dias-Santagata, D.; Iafrate, A.J.; Fuchs, C.S.; Haigis, K.M.; Ogino, S. NRAS mutations are rare in colorectal cancer. Diagn. Mol. Pathol. 2010, 19, 157–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.H.; Raju, G.S.; Huff, C.; Ye, Y.; Gu, J.; Chen, J.S.; Hildebrandt, M.A.T.; Liang, H.; Menter, D.G.; Morris, J.; et al. The somatic mutation landscape of premalignant colorectal adenoma. Gut 2017. [Google Scholar] [CrossRef] [Green Version]
- Bloushtain-Qimron, N.; Yao, J.; Snyder, E.L.; Shipitsin, M.; Campbell, L.L.; Mani, S.A.; Hu, M.; Chen, H.; Ustyansky, V.; Antosiewicz, J.E.; et al. Cell type-specific DNA methylation patterns in the human breast. Proc. Natl. Acad. Sci. USA 2008, 105, 14076–14081. [Google Scholar] [CrossRef] [Green Version]
- Han, B.; Bhowmick, N.; Qu, Y.; Chung, S.; Giuliano, A.E.; Cui, X. FOXC1: An emerging marker and therapeutic target for cancer. Oncogene 2017, 36, 3957–3963. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.H.; Kim, S.Y.; An, C.H.; Lee, S.H.; Jung, E.S.; Park, H.C.; Kim, M.S.; Chung, Y.J.; Lee, S.H. Clonal Structures of Regionally Synchronous Gastric Adenomas and Carcinomas. Clin. Cancer Res. 2018, 24, 4715–4725. [Google Scholar] [CrossRef] [Green Version]
- Wright, S. The Roles of Mutation, Inbreeding, Crossbreeding, and Selection in Evolution. Proc. Natl. Acad. Sci. USA 1932, 1, 356–366. [Google Scholar]
- Cross, W.; Kovac, M.; Mustonen, V.; Temko, D.; Davis, H.; Baker, A.M.; Biswas, S.; Arnold, R.; Chegwidden, L.; Gatenbee, C.; et al. The evolutionary landscape of colorectal tumorigenesis. Nat. Ecol. Evol. 2018, 2, 1661–1672. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.M.; Laird, P.W.; Park, P.J. The landscape of microsatellite instability in colorectal and endometrial cancer genomes. Cell 2013, 155, 858–868. [Google Scholar] [CrossRef] [Green Version]
- Stachler, M.D.; Taylor-Weiner, A.; Peng, S.; McKenna, A.; Agoston, A.T.; Odze, R.D.; Davison, J.M.; Nason, K.S.; Loda, M.; Leshchiner, I.; et al. Paired exome analysis of Barrett’s esophagus and adenocarcinoma. Nat. Genet. 2015, 47, 1047–1055. [Google Scholar] [CrossRef] [Green Version]
- Shibata, D. Mutation and epigenetic molecular clocks in cancer. Carcinogenesis 2011, 32, 123–128. [Google Scholar] [CrossRef] [Green Version]
- Sottoriva, A.; Kang, H.; Ma, Z.; Graham, T.A.; Salomon, M.P.; Zhao, J.; Marjoram, P.; Siegmund, K.; Press, M.F.; Shibata, D.; et al. A Big Bang model of human colorectal tumor growth. Nat. Genet. 2015, 47, 209–216. [Google Scholar] [CrossRef]
- Walter, M.J.; Shen, D.; Ding, L.; Shao, J.; Koboldt, D.C.; Chen, K.; Larson, D.E.; McLellan, M.D.; Dooling, D.; Abbott, R.; et al. Clonal architecture of secondary acute myeloid leukemia. N. Engl. J. Med. 2012, 366, 1090–1098. [Google Scholar] [CrossRef] [Green Version]
- Yap, T.A.; Gerlinger, M.; Futreal, P.A.; Pusztai, L.; Swanton, C. Intratumor heterogeneity: Seeing the wood for the trees. Sci. Transl. Med. 2012, 4, 127ps110. [Google Scholar] [CrossRef] [Green Version]
- Hudler, P. Challenges of deciphering gastric cancer heterogeneity. World J. Gastroenterol. 2015, 21, 10510–10527. [Google Scholar] [CrossRef]
- Huang, J.; Papadopoulos, N.; McKinley, A.J.; Farrington, S.M.; Curtis, L.J.; Wyllie, A.H.; Zheng, S.; Willson, J.K.; Markowitz, S.D.; Morin, P.; et al. APC mutations in colorectal tumors with mismatch repair deficiency. Proc. Natl. Acad. Sci. USA 1996, 93, 9049–9054. [Google Scholar] [CrossRef] [Green Version]
- Shibata, D.; Navidi, W.; Salovaara, R.; Li, Z.H.; Aaltonen, L.A. Somatic microsatellite mutations as molecular tumor clocks. Nat. Med. 1996, 2, 676–681. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [Green Version]
- Olshen, A.B.; Venkatraman, E.S.; Lucito, R.; Wigler, M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 2004, 5, 557–572. [Google Scholar] [CrossRef]
- Carter, S.L.; Cibulskis, K.; Helman, E.; McKenna, A.; Shen, H.; Zack, T.; Laird, P.W.; Onofrio, R.C.; Winckler, W.; Weir, B.A.; et al. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 2012, 30, 413–421. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Roth, A.; Khattra, J.; Yap, D.; Wan, A.; Laks, E.; Biele, J.; Ha, G.; Aparicio, S.; Bouchard-Cote, A.; Shah, S.P. PyClone: Statistical inference of clonal population structure in cancer. Nat. Methods 2014, 11, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Perez, A.; Perez-Llamas, C.; Deu-Pons, J.; Tamborero, D.; Schroeder, M.P.; Jene-Sanz, A.; Santos, A.; Lopez-Bigas, N. IntOGen-mutations identifies cancer drivers across tumor types. Nat. Methods 2013, 10, 1081–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamborero, D.; Rubio-Perez, C.; Deu-Pons, J.; Schroeder, M.P.; Vivancos, A.; Rovira, A.; Tusquets, I.; Albanell, J.; Rodon, J.; Tabernero, J.; et al. Cancer Genome Interpreter annotates the biological and clinical relevance of tumor alterations. Genome Med. 2018, 10, 25. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Perez, C.; Tamborero, D.; Schroeder, M.P.; Antolin, A.A.; Deu-Pons, J.; Perez-Llamas, C.; Mestres, J.; Gonzalez-Perez, A.; Lopez-Bigas, N. In silico prescription of anticancer drugs to cohorts of 28 tumor types reveals targeting opportunities. Cancer Cell 2015, 27, 382–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.H.; Yoo, J.; Song, Y.S.; Lim, C.-H.; Kim, T.-M. Mutation Analysis of Colorectal and Gastric Carcinomas Originating from Adenomas: Insights into Genomic Evolution Associated with Malignant Progression. Cancers 2020, 12, 325. https://doi.org/10.3390/cancers12020325
Lee SH, Yoo J, Song YS, Lim C-H, Kim T-M. Mutation Analysis of Colorectal and Gastric Carcinomas Originating from Adenomas: Insights into Genomic Evolution Associated with Malignant Progression. Cancers. 2020; 12(2):325. https://doi.org/10.3390/cancers12020325
Chicago/Turabian StyleLee, Sung Hak, Jinseon Yoo, Young Soo Song, Chul-Hyun Lim, and Tae-Min Kim. 2020. "Mutation Analysis of Colorectal and Gastric Carcinomas Originating from Adenomas: Insights into Genomic Evolution Associated with Malignant Progression" Cancers 12, no. 2: 325. https://doi.org/10.3390/cancers12020325