Kinase Inhibitor Treatment of Patients with Advanced Cancer Results in High Tumor Drug Concentrations and in Specific Alterations of the Tumor Phosphoproteome

 , ,
, ,  , , ,
, , ,
Abstract
:1. Introduction
2. Results
2.1. Patient Accrual and Characteristics
2.2. Accumulation of Protein Kinase Inhibitors in Tumor Tissue
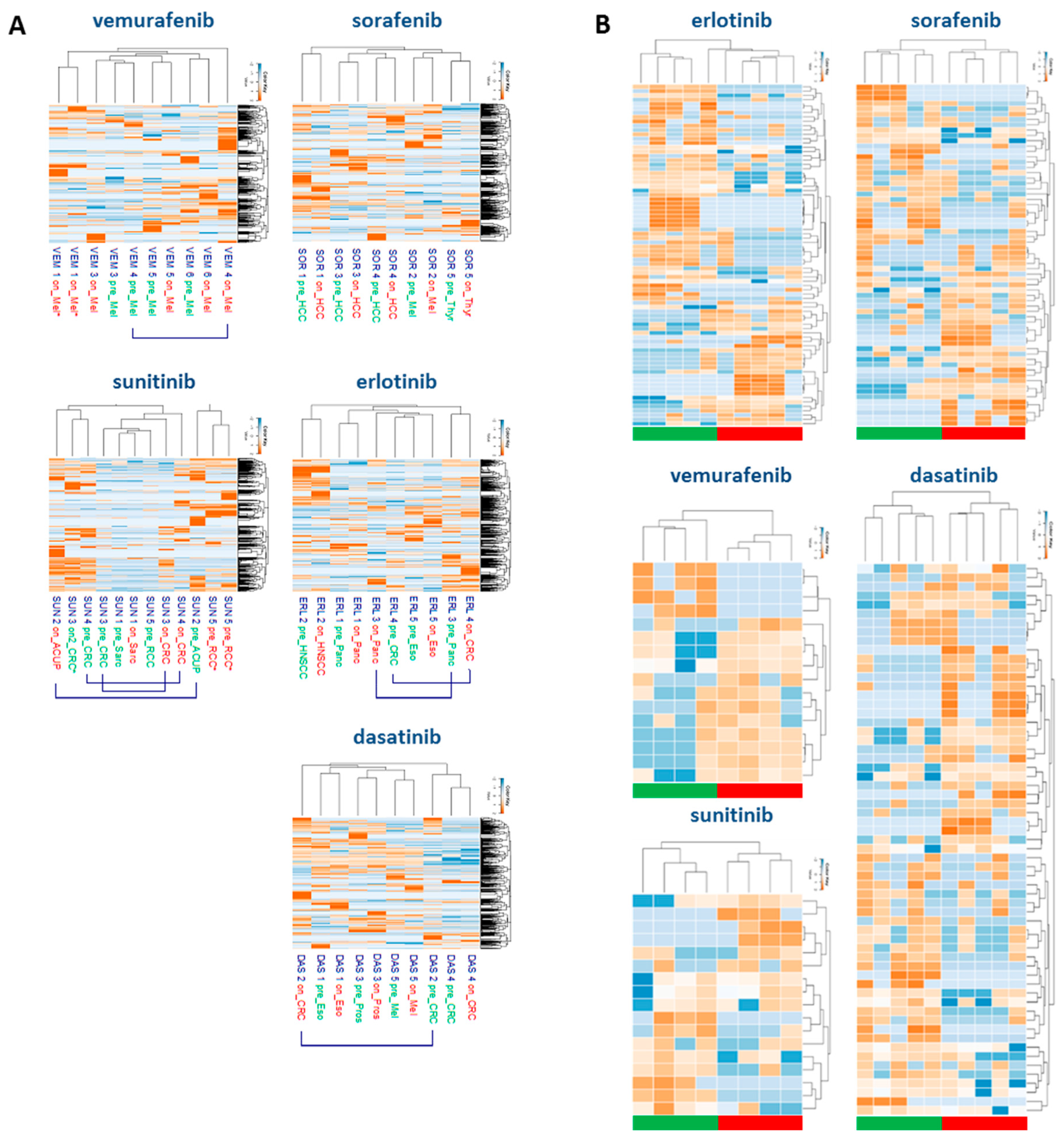
2.3. Tyrosine-Phosphoproteomic Profiling of Pre- and On-Treatment Tumor Biopsies
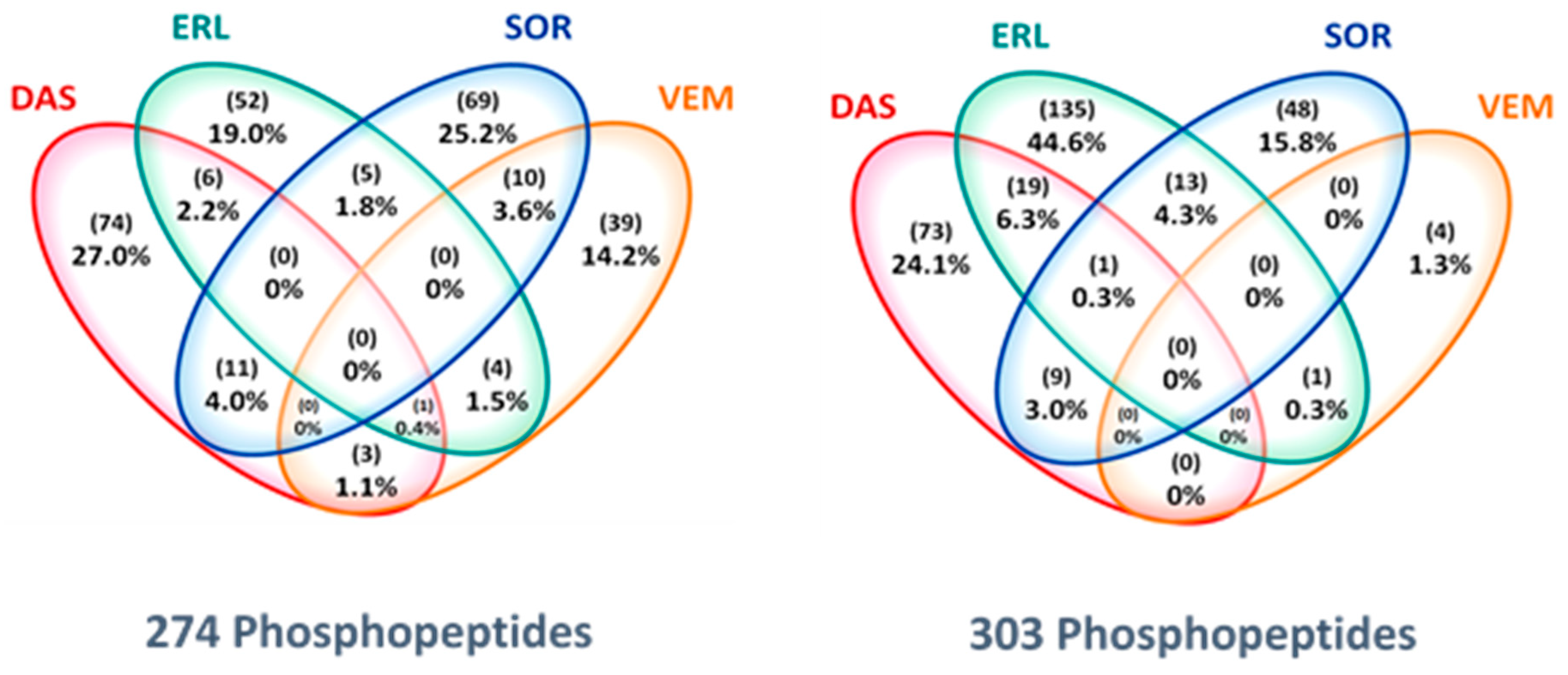
2.4. Drug-Specific Alterations of Peptide Phosphorylation upon Treatment
2.5. Protein Networks of Up- and Downregulated Phosphopeptides
2.6. Correlation between Tumor Concentration and Inhibition of Peptide Phosphorylation
3. Discussion
4. Methods
4.1. Study Design
4.2. Patient Selection and Characteristics
4.3. Tumor biopsies
4.4. Drug concentration Measurements
4.5. Biopsy Preparation for Phosphoproteomics
4.6. Phosphotyrosine Phosphoproteomics Profiling and Data Analysis
5. Conclusions
Supplementary Materials
Data and Materials Availability
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schwaederle, M.; Zhao, M.; Lee, J.J.; Eggermont, A.M.; Schilsky, R.L.; Mendelsohn, J.; Lazar, V.; Kurzrock, R. Impact of Precision Medicine in Diverse Cancers: A Meta-Analysis of Phase II Clinical Trials. J. Clin. Oncol. 2015, 33, 3817–3825. [Google Scholar] [CrossRef] [PubMed]
- Dienstmann, R.; Vermeulen, L.; Guinney, J.; Kopetz, S.; Tejpar, S.; Tabernero, J. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat. Rev. Cancer 2017, 17, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Voest, E.E.; Bernards, R. DNA-Guided Precision Medicine for Cancer: A Case of Irrational Exuberance? Cancer Discov. 2016, 6, 130–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, W.; Elemento, O. Cancer systems biology: Embracing complexity to develop better anticancer therapeutic strategies. Oncogene 2015, 34, 3215–3225. [Google Scholar] [CrossRef] [PubMed]
- Klaeger, S.; Heinzlmeir, S.; Wilhelm, M.; Polzer, H.; Vick, B.; Koenig, P.A.; Reinecke, M.; Ruprecht, B.; Petzoldt, S.; Meng, C.; et al. The target landscape of clinical kinase drugs. Science 2017, 358, 6367. [Google Scholar] [CrossRef] [Green Version]
- Minchinton, A.I.; Tannock, I.F. Drug penetration in solid tumours. Nat. Rev. Cancer 2006, 6, 583–592. [Google Scholar] [CrossRef]
- Gross, S.; Rahal, R.; Stransky, N.; Lengauer, C.; Hoeflich, K.P. Targeting cancer with kinase inhibitors. J. Clin. Investig. 2015, 125, 1780–1789. [Google Scholar] [CrossRef]
- Widmer, N.; Bardin, C.; Chatelut, E.; Paci, A.; Beijnen, J.; Leveque, D.; Veal, G.; Astier, A. Review of therapeutic drug monitoring of anticancer drugs part two--targeted therapies. Eur. J. Cancer 2014, 50, 2020–2036. [Google Scholar] [CrossRef]
- Cutillas, P.R. Role of phosphoproteomics in the development of personalized cancer therapies. Proteomics Clin. Appl. 2015, 9, 383–395. [Google Scholar] [CrossRef]
- Jimenez, C.R.; Verheul, H.M. Mass spectrometry-based proteomics: From cancer biology to protein biomarkers, drug targets, and clinical applications. Am. Soc. Clin. Oncol. Educ. Book 2014. [Google Scholar] [CrossRef]
- Rikova, K.; Guo, A.; Zeng, Q.; Possemato, A.; Yu, J.; Haack, H.; Nardone, J.; Lee, K.; Reeves, C.; Li, Y.; et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 2007, 131, 1190–1203. [Google Scholar] [CrossRef] [Green Version]
- Zagorac, I.; Fernandez-Gaitero, S.; Penning, R.; Post, H.; Bueno, M.J.; Mouron, S.; Manso, L.; Morente, M.M.; Alonso, S.; Serra, V.; et al. In vivo phosphoproteomics reveals kinase activity profiles that predict treatment outcome in triple-negative breast cancer. Nat. Commun. 2018, 9, 3501. [Google Scholar] [CrossRef] [PubMed]
- Labots, M.; van der Mijn, J.C.; Beekhof, R.; Piersma, S.R.; de Goeij-de Haas, R.R.; Pham, T.V.; Knol, J.C.; Dekker, H.; van Grieken, N.C.T.; Verheul, H.M.W.; et al. Phosphotyrosine-based-phosphoproteomics scaled-down to biopsy level for analysis of individual tumor biology and treatment selection. J. Proteomics 2017, 162, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.S.; Whittle, M.C.; Nakamura, K.; Abell, A.N.; Midland, A.A.; Zawistowski, J.S.; Johnson, N.L.; Granger, D.A.; Jordan, N.V.; Darr, D.B.; et al. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell 2012, 149, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, J.W.; Eder, J.P.; Ryan, D.; Lathia, C.; Lenz, H.J. Safety and pharmacokinetics of the dual action Raf kinase and vascular endothelial growth factor receptor inhibitor, BAY 43-9006, in patients with advanced, refractory solid tumors. Clin. Cancer Res. 2005, 11, 5472–5480. [Google Scholar] [CrossRef] [Green Version]
- Lankheet, N.A.; Knapen, L.M.; Schellens, J.H.; Beijnen, J.H.; Steeghs, N.; Huitema, A.D. Plasma concentrations of tyrosine kinase inhibitors imatinib, erlotinib, and sunitinib in routine clinical outpatient cancer care. Ther. Drug Monit. 2014, 36, 326–334. [Google Scholar] [CrossRef]
- Demetri, G.D.; Lo Russo, P.; MacPherson, I.R.; Wang, D.; Morgan, J.A.; Brunton, V.G.; Paliwal, P.; Agrawal, S.; Voi, M.; Evans, T.R. Phase I dose-escalation and pharmacokinetic study of dasatinib in patients with advanced solid tumors. Clin. Cancer Res. 2009, 15, 6232–6240. [Google Scholar] [CrossRef] [Green Version]
- Funck-Brentano, E.; Alvarez, J.C.; Longvert, C.; Abe, E.; Beauchet, A.; Funck-Brentano, C.; Saiag, P. Plasma vemurafenib concentrations in advanced BRAFV600mut melanoma patients: Impact on tumour response and tolerance. Ann. Oncol. 2015, 26, 1470–1475. [Google Scholar] [CrossRef]
- Faivre, S.; Delbaldo, C.; Vera, K.; Robert, C.; Lozahic, S.; Lassau, N.; Bello, C.; Deprimo, S.; Brega, N.; Massimini, G.; et al. Safety, pharmacokinetic, and antitumor activity of SU11248, a novel oral multitarget tyrosine kinase inhibitor, in patients with cancer. J. Clin. Oncol. 2006, 24, 25–35. [Google Scholar] [CrossRef]
- Lind, J.S.; Dingemans, A.M.; Groen, H.J.; Thunnissen, F.B.; Bekers, O.; Heideman, D.A.; Honeywell, R.J.; Giovannetti, E.; Peters, G.J.; Postmus, P.E.; et al. A multicenter phase II study of erlotinib and sorafenib in chemotherapy-naive patients with advanced non-small cell lung cancer. Clin. Cancer Res. 2010, 16, 3078–3087. [Google Scholar] [CrossRef] [Green Version]
- Knight, Z.A.; Shokat, K.M. Features of selective kinase inhibitors. Chem. Biol. 2005, 12, 621–637. [Google Scholar] [CrossRef] [Green Version]
- Van der Mijn, J.C.; Broxterman, H.J.; Knol, J.C.; Piersma, S.R.; De Haas, R.R.; Dekker, H.; Pham, T.V.; Van Beusechem, V.W.; Halmos, B.; Mier, J.W.; et al. Sunitinib activates Axl signaling in renal cell cancer. Int. J. Cancer 2016, 138, 3002–3010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, E.; Waters, B.; Spiegel, K.; Alnadaf, T.; Manley, P.W.; Buchdunger, E.; Walker, C.; Jarai, G. Inhibition of collagen-induced discoidin domain receptor 1 and 2 activation by imatinib, nilotinib and dasatinib. Eur. J. Pharmacol. 2008, 599, 44–53. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, G.M.; Seo, S.; Serebriiskii, I.G.; Lessin, S.R.; Golemis, E.A. A new central scaffold for metastasis: Parsing HEF1/Cas-L/NEDD9. Cancer Res. 2007, 67, 8975–8979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moorcraft, S.Y.; Marriott, C.; Peckitt, C.; Cunningham, D.; Chau, I.; Starling, N.; Watkins, D.; Rao, S. Patients’ willingness to participate in clinical trials and their views on aspects of cancer research: Results of a prospective patient survey. Trials 2016, 17, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seah, D.S.; Scott, S.M.; Najita, J.; Openshaw, T.; Krag, K.; Frank, E.; Sohl, J.; Stadler, Z.K.; Garrett, M.; Silverman, S.G.; et al. Attitudes of patients with metastatic breast cancer toward research biopsies. Ann. Oncol. 2013, 24, 1853–1859. [Google Scholar] [CrossRef] [PubMed]
- Gotink, K.J.; Broxterman, H.J.; Labots, M.; de Haas, R.R.; Dekker, H.; Honeywell, R.J.; Rudek, M.A.; Beerepoot, L.V.; Musters, R.J.; Jansen, G.; et al. Lysosomal sequestration of sunitinib: A novel mechanism of drug resistance. Clin. Cancer Res. 2011, 17, 7337–7346. [Google Scholar] [CrossRef] [Green Version]
- Hammers, H.J.; Verheul, H.M.; Salumbides, B.; Sharma, R.; Rudek, M.; Jaspers, J.; Shah, P.; Ellis, L.; Shen, L.; Paesante, S.; et al. Reversible epithelial to mesenchymal transition and acquired resistance to sunitinib in patients with renal cell carcinoma: Evidence from a xenograft study. Mol. Cancer Ther. 2010, 9, 1525–1535. [Google Scholar] [CrossRef] [Green Version]
- Petty, W.J.; Dragnev, K.H.; Memoli, V.A.; Ma, Y.; Desai, N.B.; Biddle, A.; Davis, T.H.; Nugent, W.C.; Memoli, N.; Hamilton, M.; et al. Epidermal growth factor receptor tyrosine kinase inhibition represses cyclin D1 in aerodigestive tract cancers. Clin. Cancer Res. 2004, 10, 7547–7554. [Google Scholar] [CrossRef] [Green Version]
- Herviou, P.; Thivat, E.; Richard, D.; Roche, L.; Dohou, J.; Pouget, M.; Eschalier, A.; Durando, X.; Authier, N. Therapeutic drug monitoring and tyrosine kinase inhibitors. Oncol. Lett. 2016, 12, 1223–1232. [Google Scholar] [CrossRef]
- Karaman, M.W.; Herrgard, S.; Treiber, D.K.; Gallant, P.; Atteridge, C.E.; Campbell, B.T.; Chan, K.W.; Ciceri, P.; Davis, M.I.; Edeen, P.T.; et al. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2008, 26, 127–132. [Google Scholar] [CrossRef]
- Fabian, M.A.; Biggs, W.H., 3rd; Treiber, D.K.; Atteridge, C.E.; Azimioara, M.D.; Benedetti, M.G.; Carter, T.A.; Ciceri, P.; Edeen, P.T.; Floyd, M.; et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 2005, 23, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Dazert, E.; Colombi, M.; Boldanova, T.; Moes, S.; Adametz, D.; Quagliata, L.; Roth, V.; Terracciano, L.; Heim, M.H.; Jenoe, P.; et al. Quantitative proteomics and phosphoproteomics on serial tumor biopsies from a sorafenib-treated HCC patient. Proc. Natl. Acad. Sci. USA 2016, 113, 1381–1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fojo, T. Commentary: Novel therapies for cancer: Why dirty might be better. Oncologist 2008, 13, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Conradt, L.; Godl, K.; Schaab, C.; Tebbe, A.; Eser, S.; Diersch, S.; Michalski, C.W.; Kleeff, J.; Schnieke, A.; Schmid, R.M.; et al. Disclosure of erlotinib as a multikinase inhibitor in pancreatic ductal adenocarcinoma. Neoplasia 2011, 13, 1026–1034. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Lee, J.C.; Lin, L.; Olivas, V.; Au, V.; LaFramboise, T.; Abdel-Rahman, M.; Wang, X.; Levine, A.D.; Rho, J.K.; et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet. 2012, 44, 852–860. [Google Scholar] [CrossRef]
- Zhou, L.; Liu, X.D.; Sun, M.; Zhang, X.; German, P.; Bai, S.; Ding, Z.; Tannir, N.; Wood, C.G.; Matin, S.F.; et al. Targeting MET and AXL overcomes resistance to sunitinib therapy in renal cell carcinoma. Oncogene 2016, 35, 2687–2697. [Google Scholar] [CrossRef]
- Yoshida, T.; Zhang, G.; Smith, M.A.; Lopez, A.S.; Bai, Y.; Li, J.; Fang, B.; Koomen, J.; Rawal, B.; Fisher, K.J.; et al. Tyrosine phosphoproteomics identifies both codrivers and cotargeting strategies for T790M-related EGFR-TKI resistance in non-small cell lung cancer. Clin. Cancer Res. 2014, 20, 4059–4074. [Google Scholar] [CrossRef] [Green Version]
- Astsaturov, I.; Ratushny, V.; Sukhanova, A.; Einarson, M.B.; Bagnyukova, T.; Zhou, Y.; Devarajan, K.; Silverman, J.S.; Tikhmyanova, N.; Skobeleva, N.; et al. Synthetic lethal screen of an EGFR-centered network to improve targeted therapies. Sci. Signal. 2010, 3, ra67. [Google Scholar] [CrossRef] [Green Version]
- Casado, P.; Rodriguez-Prados, J.C.; Cosulich, S.C.; Guichard, S.; Vanhaesebroeck, B.; Joel, S.; Cutillas, P.R. Kinase-substrate enrichment analysis provides insights into the heterogeneity of signaling pathway activation in leukemia cells. Sci. Signal. 2013, 6, rs6. [Google Scholar] [CrossRef]
- Drake, J.M.; Paull, E.O.; Graham, N.A.; Lee, J.K.; Smith, B.A.; Titz, B.; Stoyanova, T.; Faltermeier, C.M.; Uzunangelov, V.; Carlin, D.E.; et al. Phosphoproteome Integration Reveals Patient-Specific Networks in Prostate Cancer. Cell 2016, 166, 1041–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkes, E.H.; Casado, P.; Rajeeve, V.; Cutillas, P.R. Kinase activity ranking using phosphoproteomics data (KARP) quantifies the contribution of protein kinases to the regulation of cell viability. Mol. Cell Proteomics 2017, 16, 1694–1704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beekhof, R.; van Alphen, C.; Henneman, A.A.; Knol, J.C.; Pham, T.V.; Rolfs, F.; Labots, M.; Henneberry, E.; Le Large, T.Y.; de Haas, R.R.; et al. INKA, an integrative data analysis pipeline for phosphoproteomic inference of active kinases. Mol. Syst. Biol. 2019, 15, e8250. [Google Scholar] [CrossRef] [PubMed]
- Di Gion, P.; Kanefendt, F.; Lindauer, A.; Scheffler, M.; Doroshyenko, O.; Fuhr, U.; Wolf, J.; Jaehde, U. Clinical pharmacokinetics of tyrosine kinase inhibitors: focus on pyrimidines, pyridines and pyrroles. Clin. Pharmacokinet. 2011, 50, 551–603. [Google Scholar] [CrossRef]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Honeywell, R.; Yarzadah, K.; Giovannetti, E.; Losekoot, N.; Smit, E.F.; Walraven, M.; Lind, J.S.; Tibaldi, C.; Verheul, H.M.; Peters, G.J. Simple and selective method for the determination of various tyrosine kinase inhibitors used in the clinical setting by liquid chromatography tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2010, 878, 1059–1068. [Google Scholar] [CrossRef]
- Holdhoff, M.; Supko, J.G.; Gallia, G.L.; Hann, C.L.; Bonekamp, D.; Ye, X.; Cao, B.; Olivi, A.; Grossman, S.A. Intratumoral concentrations of imatinib after oral administration in patients with glioblastoma multiforme. J. Neurooncol. 2010, 97, 241–245. [Google Scholar] [CrossRef] [Green Version]
- Piersma, S.R.; Knol, J.C.; de Reus, I.; Labots, M.; Sampadi, B.K.; Pham, T.V.; Ishihama, Y.; Verheul, H.M.; Jimenez, C.R. Feasibility of label-free phosphoproteomics and application to base-line signaling of colorectal cancer cell lines. J. Proteomics 2015, 127, 247–258. [Google Scholar] [CrossRef]
- Van der Mijn, J.C.; Labots, M.; Piersma, S.R.; Pham, T.V.; Knol, J.C.; Broxterman, H.J.; Verheul, H.M.; Jimenez, C.R. Evaluation of different phospho-tyrosine antibodies for label-free phosphoproteomics. J. Proteomics 2015, 127, 259–263. [Google Scholar] [CrossRef]
- Rush, J.; Moritz, A.; Lee, K.A.; Guo, A.; Goss, V.L.; Spek, E.J.; Zhang, H.; Zha, X.M.; Polakiewicz, R.D.; Comb, M.J. Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat. Biotechnol. 2005, 23, 94–101. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Marx, H.; Lemeer, S.; Schliep, J.E.; Matheron, L.; Mohammed, S.; Cox, J.; Mann, M.; Heck, A.J.; Kuster, B. A large synthetic peptide and phosphopeptide reference library for mass spectrometry-based proteomics. Nat. Biotechnol. 2013, 31, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Vizcaino, J.A.; Deutsch, E.W.; Wang, R.; Csordas, A.; Reisinger, F.; Rios, D.; Dianes, J.A.; Sun, Z.; Farrah, T.; Bandeira, N.; et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 2014, 32, 223–226. [Google Scholar] [CrossRef]
- Horn, H.; Schoof, E.M.; Kim, J.; Robin, X.; Miller, M.L.; Diella, F.; Palma, A.; Cesareni, G.; Jensen, L.J.; Linding, R. KinomeXplorer: An integrated platform for kinome biology studies. Nat. Methods 2014, 11, 603–604. [Google Scholar] [CrossRef] [PubMed]
- Linding, R.; Jensen, L.J.; Ostheimer, G.J.; van Vugt, M.A.; Jorgensen, C.; Miron, I.M.; Diella, F.; Colwill, K.; Taylor, L.; Elder, K.; et al. Systematic discovery of in vivo phosphorylation networks. Cell 2007, 129, 1415–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Patient ID | Gender, Age | Tumor Type | Biopsied Site | Skin Biopsy | Post-Study Treatment (Best Response) |
|---|---|---|---|---|---|
| SOR 1 | M, 69 | Hepatocellular | Liver | + | Sorafenib (SD) |
| SOR 2 | M, 65 | Melanoma | Subcutaneous | + | Dacarbazine (PD) |
| SOR 3 | M, 71 | Hepatocellular | Liver | + | Sorafenib (PD) |
| SOR 4 | M, 62 | Hepatocellular | Liver | + | Sorafenib (PD) |
| SOR 5 | F, 74 | Thyroid, papillary | Muscle | + | Sorafenib (SD) |
| ERL 1 | M, 53 | Pancreatic | Liver | + | FOLFIRINOX (NE) |
| ERL 2 | M, 57 | HNSCC | Subcutaneous | + | Cisplatin/5FU/cetuximab (MR) |
| ERL 3 | M, 71 | Pancreatic | Liver | + | FOLFIRINOX (SD) |
| ERL 4 | F, 48 | Rectal | Liver | + | CAPOX-B (PR) |
| ERL 5 | M, 68 | Esophageal | Liver | + | EOX (PR) |
| DAS 1 | M, 58 | Esophageal | Esophagus | + | Gemcitabine/cisplatin (SD) |
| DAS 2 | M, 69 | Colorectal | Liver | - | Cetuximab (NE, clinical PD) |
| DAS 3 | M, 62 | Prostate | Lymph node | + | Abirateron (SD) |
| DAS 4 | M, 72 | Colorectal | Chest wall | + | Irinotecan (PD) |
| DAS 5 | M, 67 | Melanoma | Subcutaneous | + | Vemurafenib (PR) |
| VEM 1 | F, 51 | Melanoma | Subcutaneous | + | Vemurafenib (SD) |
| VEM 2 | M, 77 | Melanoma | Lymph node | + | Vemurafenib (CR) |
| VEM 3 | M, 48 | Melanoma | Cutaneous | + | Vemurafenib (NE, clinical PD) |
| VEM 4 | M, 70 | Melanoma | Subcutaneous | + | Dabrafenib/trametinib (PR) |
| VEM 5 | M, 61 | Melanoma | Subcutaneous | + | Vemurafenib (PR) |
| VEM 6 | M, 82 | Melanoma | Lymph node | + | Dabrafenib (PR) |
| SUN 1 | F, 20 | Clear cell sarcoma | Lymph node | - | Doxorubicin (PD) |
| SUN 2 | M, 65 | ACUP | Subcutaneous | + | Gemcitabin/cisplatin (NE) |
| SUN 3 | M, 59 | Colorectal | Subcutaneous | - | CAPOX (NE) |
| SUN 4 | M, 62 | Colorectal | Lung | + | CAPOX-B (PR) |
| SUN 5 | M, 69 | Renal cell | Lymph node | + | Sunitinib (SD) |
| EVE 1 | M, 67 | Renal cell | Subcutaneous | - | Everolimus (SD) |
| EVE 2 | M, 57 | Renal cell | Adrenal gland | + | Everolimus/cyclophosphamide (SD) |
| EVE 3 | M, 74 | Renal cell | Subcutaneous | + | Sunitinib/dalteparin (PR) |
| EVE 4 | M, 75 | Pancreatic NET | Liver | + | Everolimus (SD) |
| EVE 5 | F, 51 | NET | Subcutaneous | + | Sandostatin (SD) |
| PKI Cohort | Tumor µM (Range) | Skin µM (Range) | Plasma µM (Range) | Serum µM (Range) |
|---|---|---|---|---|
| Sorafenib | 10.0 (3.7–22.0) | 6.3 (1.4–28.4) | 4.8 (3.7–12.1) | 6.9 (4.8–17.4) |
| Erlotinib | 4.2 (0.9–10.8) | 2.8 (2.1–6.7) | 1.2 (0.9–4.0) | 1.1 (0.9–4.4) |
| Dasatinib | 2.0 (0.2–64.0) | 0.4 (0.2–18.5)N = 4 | 0.012 (0.005–0.041) | 0.009 (0.017–0.037)N = 3 |
| Sunitinib | 9.0 (2.3–50.0) | 4.3 (0.5–9.7)N = 3 | 0.1 (0.1–0.2) | 0.1 (0.1–0.2) |
| Vemurafenib | 1326 (331–2347)N = 6 | 879 (120–2557)N = 8 | 98 (65–210)N = 7 | 108 (47–242)N = 8 |
| Everolimus | 3.5 (3.4–3.6)N = 2 | NE | NE | NE |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Labots, M.; Pham, T.V.; Honeywell, R.J.; Knol, J.C.; Beekhof, R.; de Goeij-de Haas, R.; Dekker, H.; Neerincx, M.; Piersma, S.R.; van der Mijn, J.C.; et al. Kinase Inhibitor Treatment of Patients with Advanced Cancer Results in High Tumor Drug Concentrations and in Specific Alterations of the Tumor Phosphoproteome. Cancers 2020, 12, 330. https://doi.org/10.3390/cancers12020330
Labots M, Pham TV, Honeywell RJ, Knol JC, Beekhof R, de Goeij-de Haas R, Dekker H, Neerincx M, Piersma SR, van der Mijn JC, et al. Kinase Inhibitor Treatment of Patients with Advanced Cancer Results in High Tumor Drug Concentrations and in Specific Alterations of the Tumor Phosphoproteome. Cancers. 2020; 12(2):330. https://doi.org/10.3390/cancers12020330
Chicago/Turabian StyleLabots, Mariette, Thang V. Pham, Richard J. Honeywell, Jaco C. Knol, Robin Beekhof, Richard de Goeij-de Haas, Henk Dekker, Maarten Neerincx, Sander R. Piersma, Johannes C. van der Mijn, and et al. 2020. "Kinase Inhibitor Treatment of Patients with Advanced Cancer Results in High Tumor Drug Concentrations and in Specific Alterations of the Tumor Phosphoproteome" Cancers 12, no. 2: 330. https://doi.org/10.3390/cancers12020330