Design and Characterization of an “All-in-One” Lentiviral Vector System Combining Constitutive Anti-GD2 CAR Expression and Inducible Cytokines

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
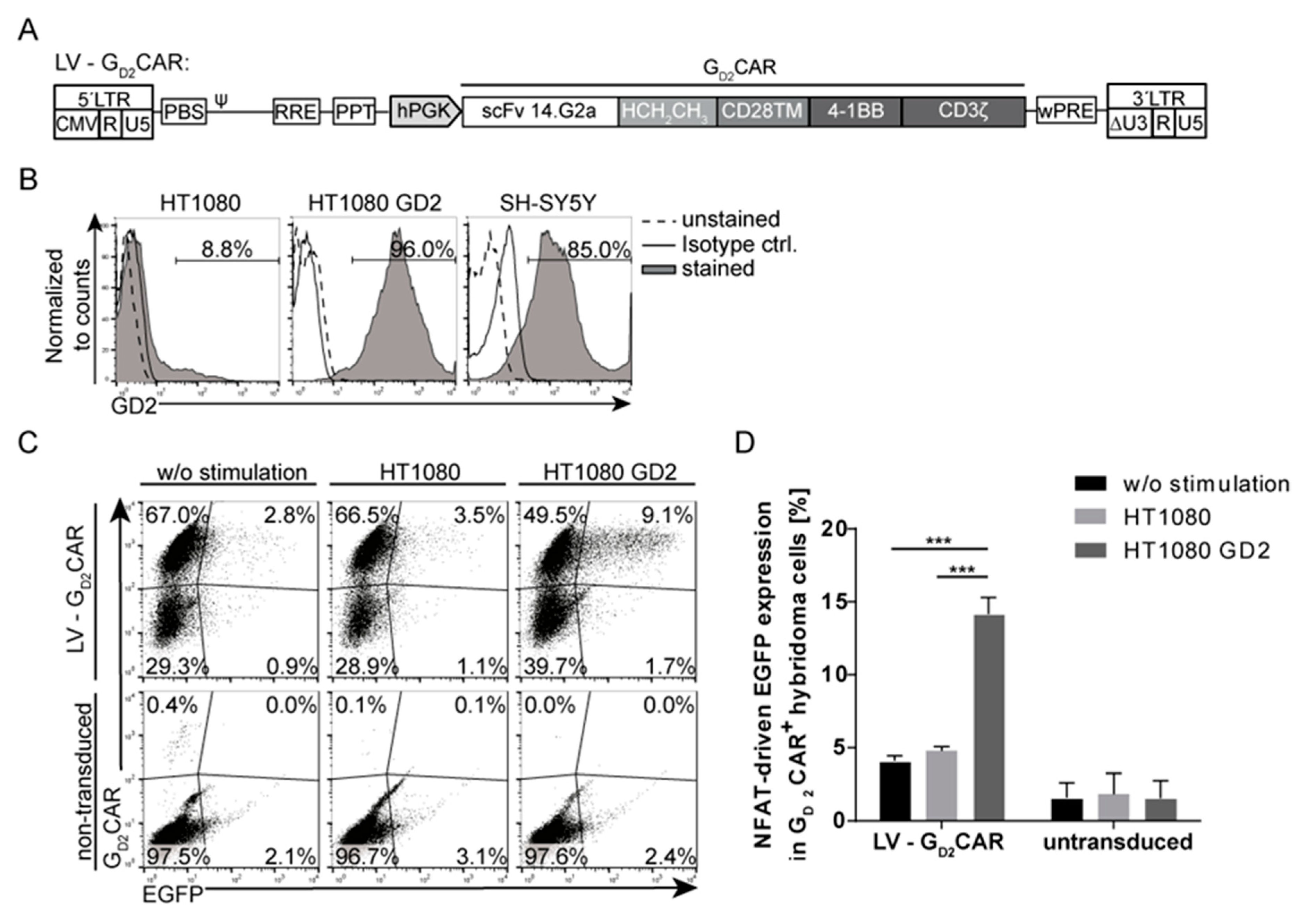
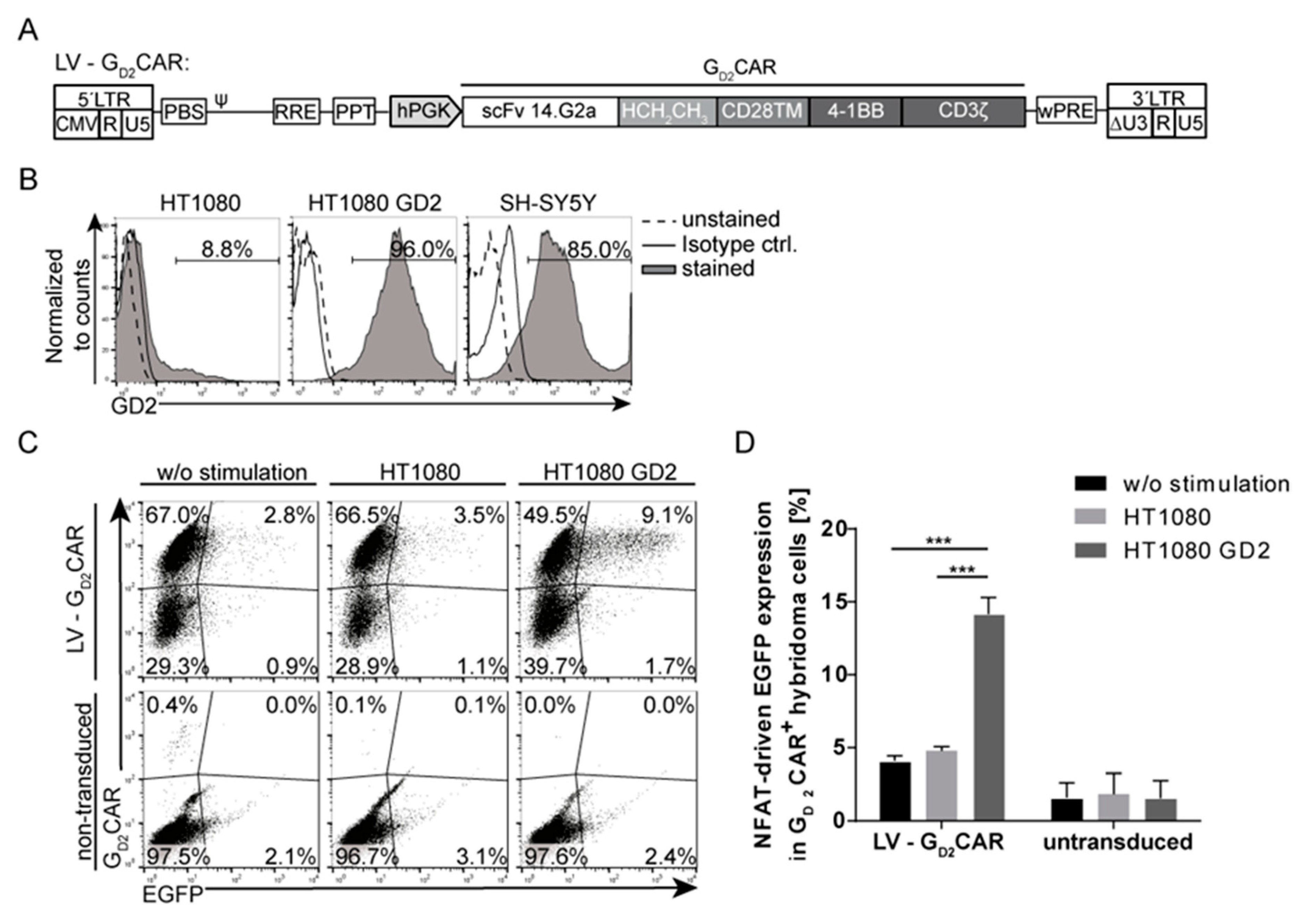
2.1. Specificity and Function of the GD2CAR
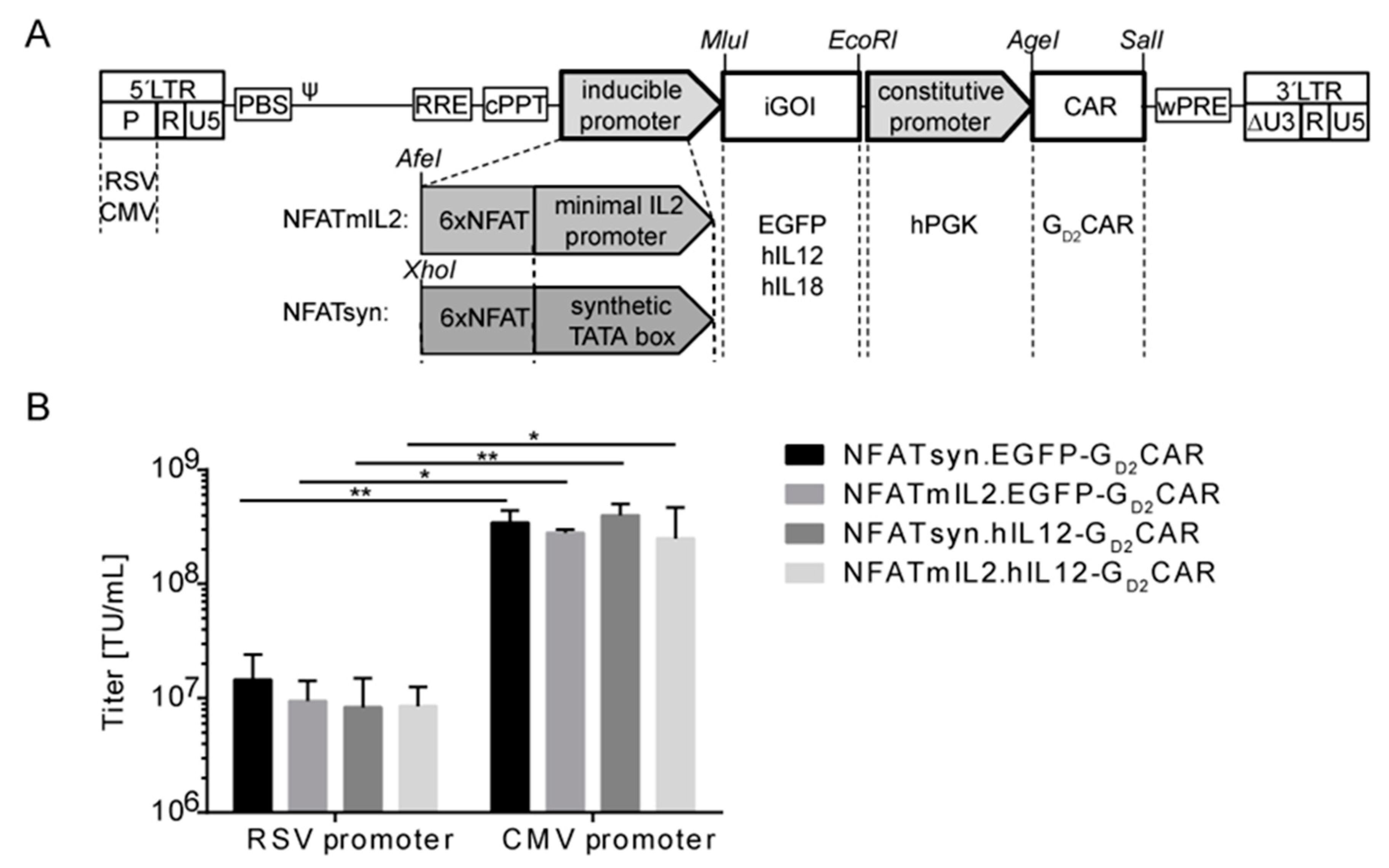
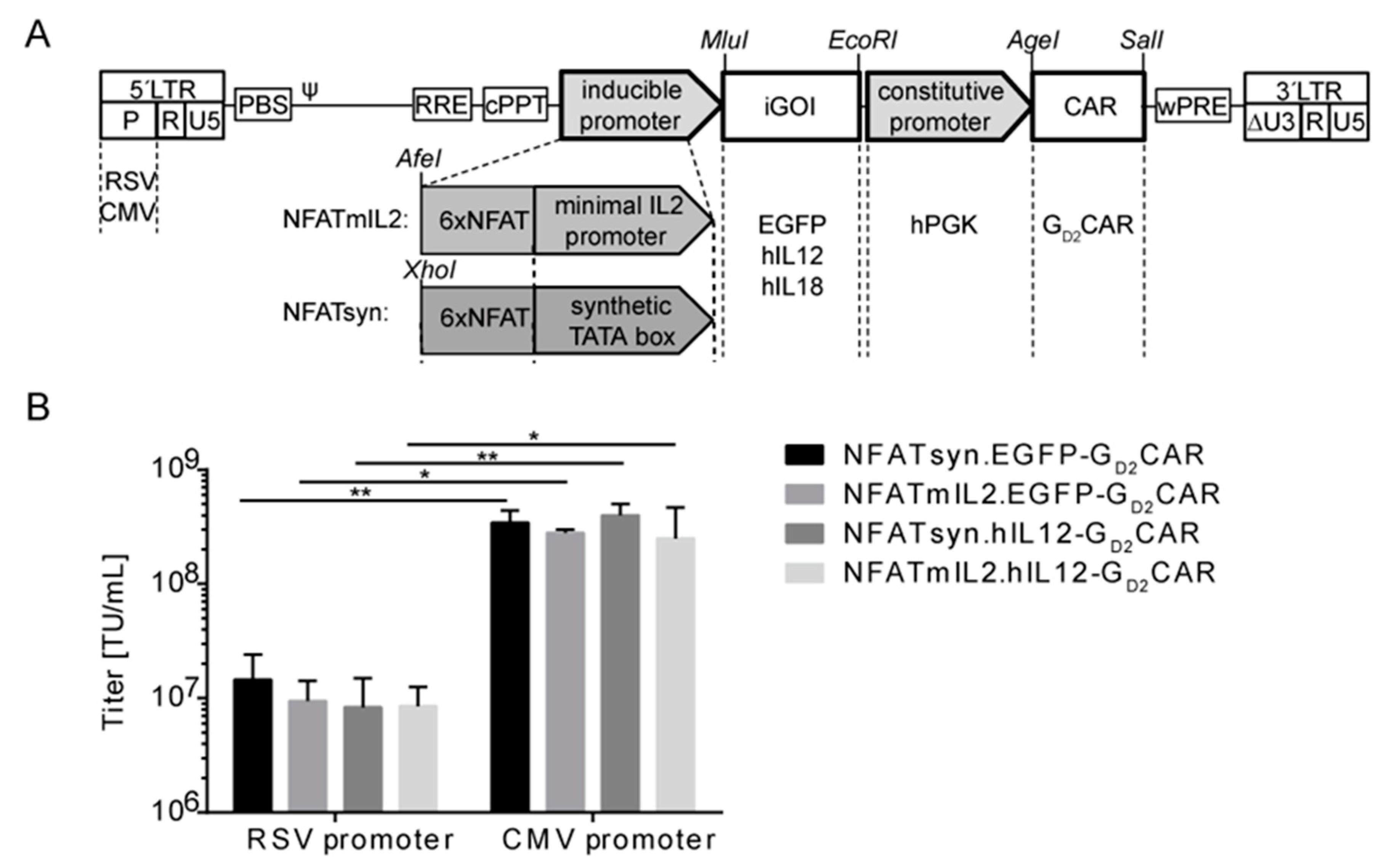
2.2. Generation of “All-in-One“ Vector Constructs for Targeted Therapy
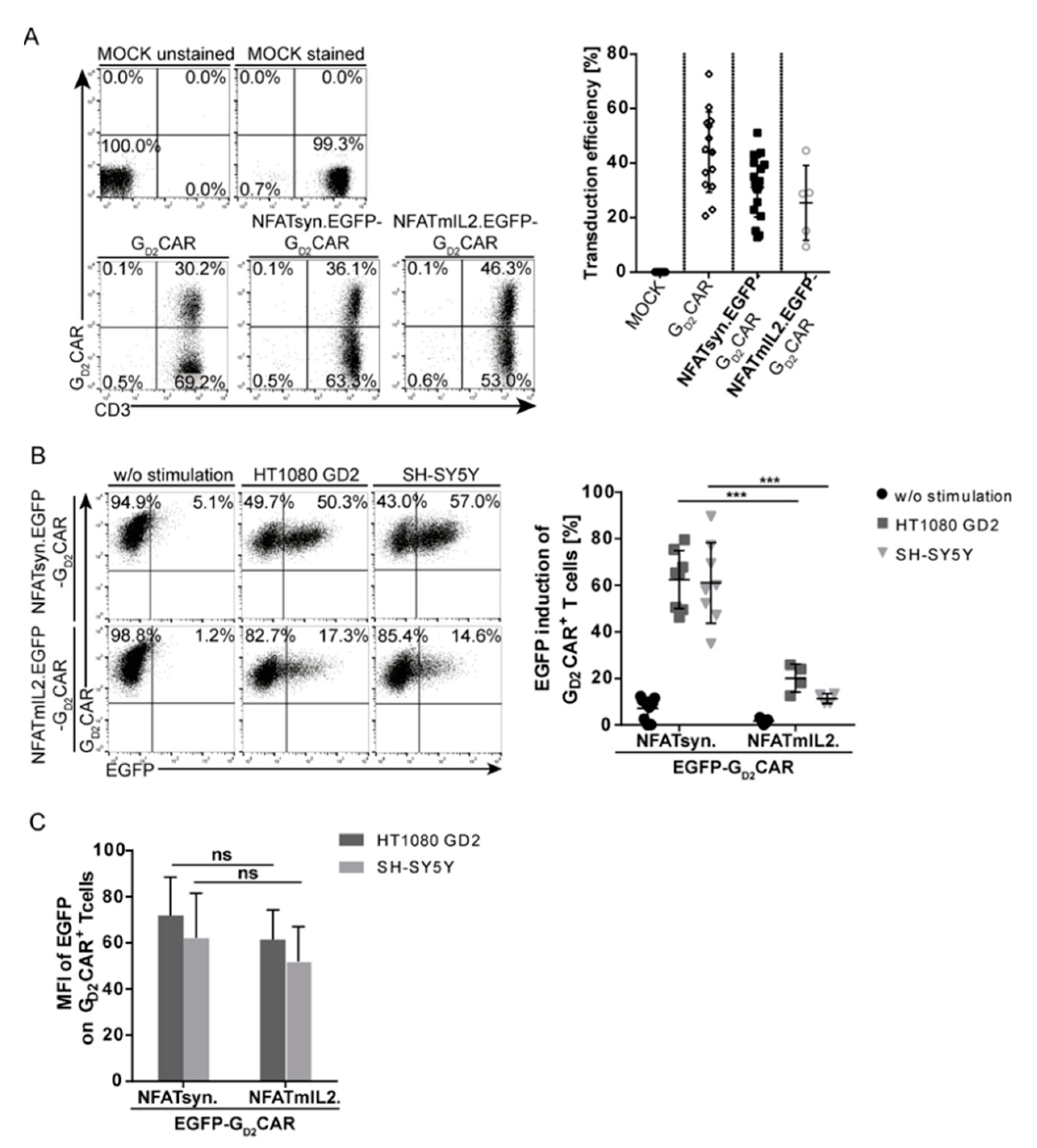
2.3. Promoter Choice of the Inducible Construct Determines the Extent of Activation
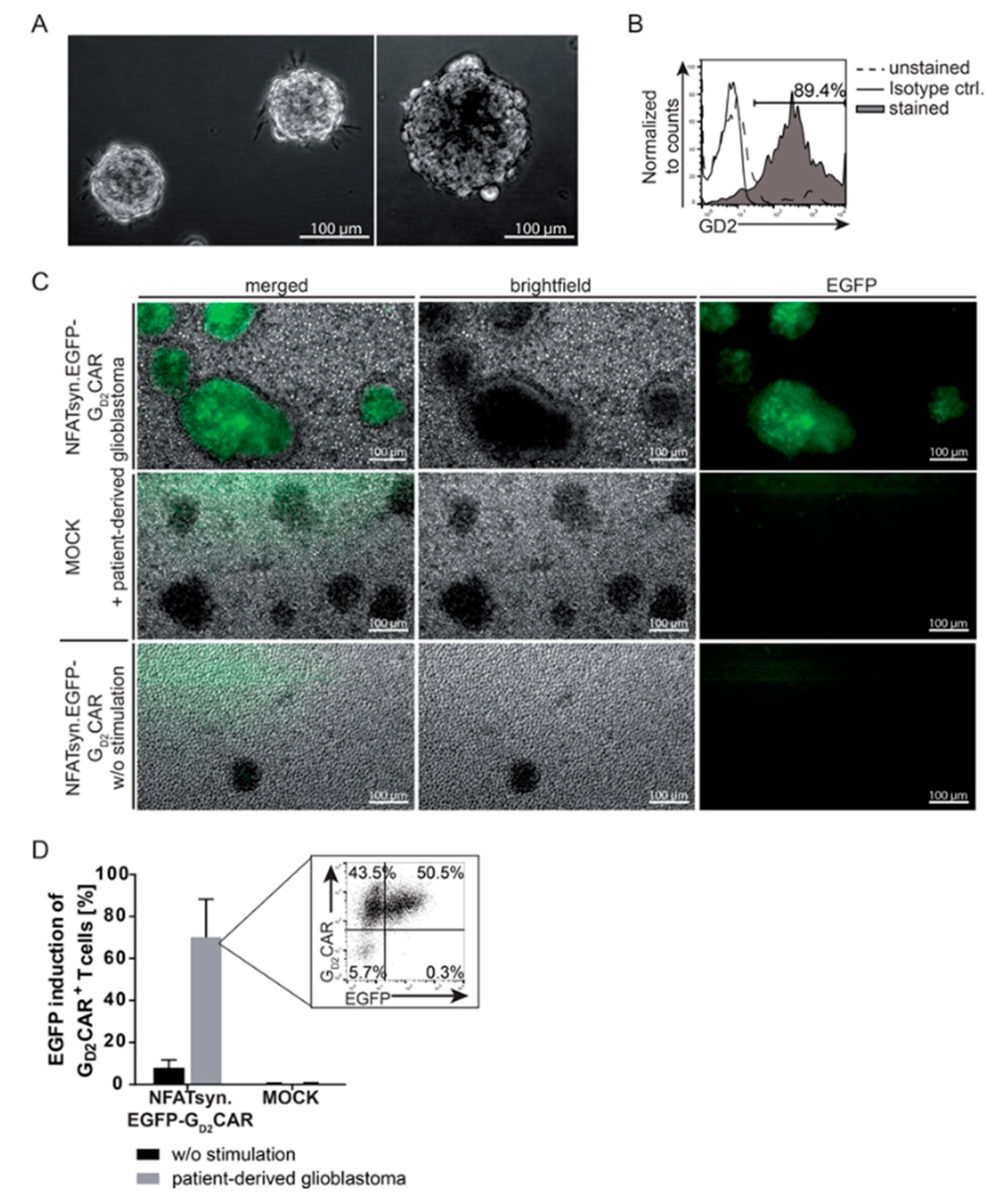
2.4. Induction of the NFATsyn Promoter after GD2CAR-Specific Activation with Patient-Derived Primary Glioblastoma Cells
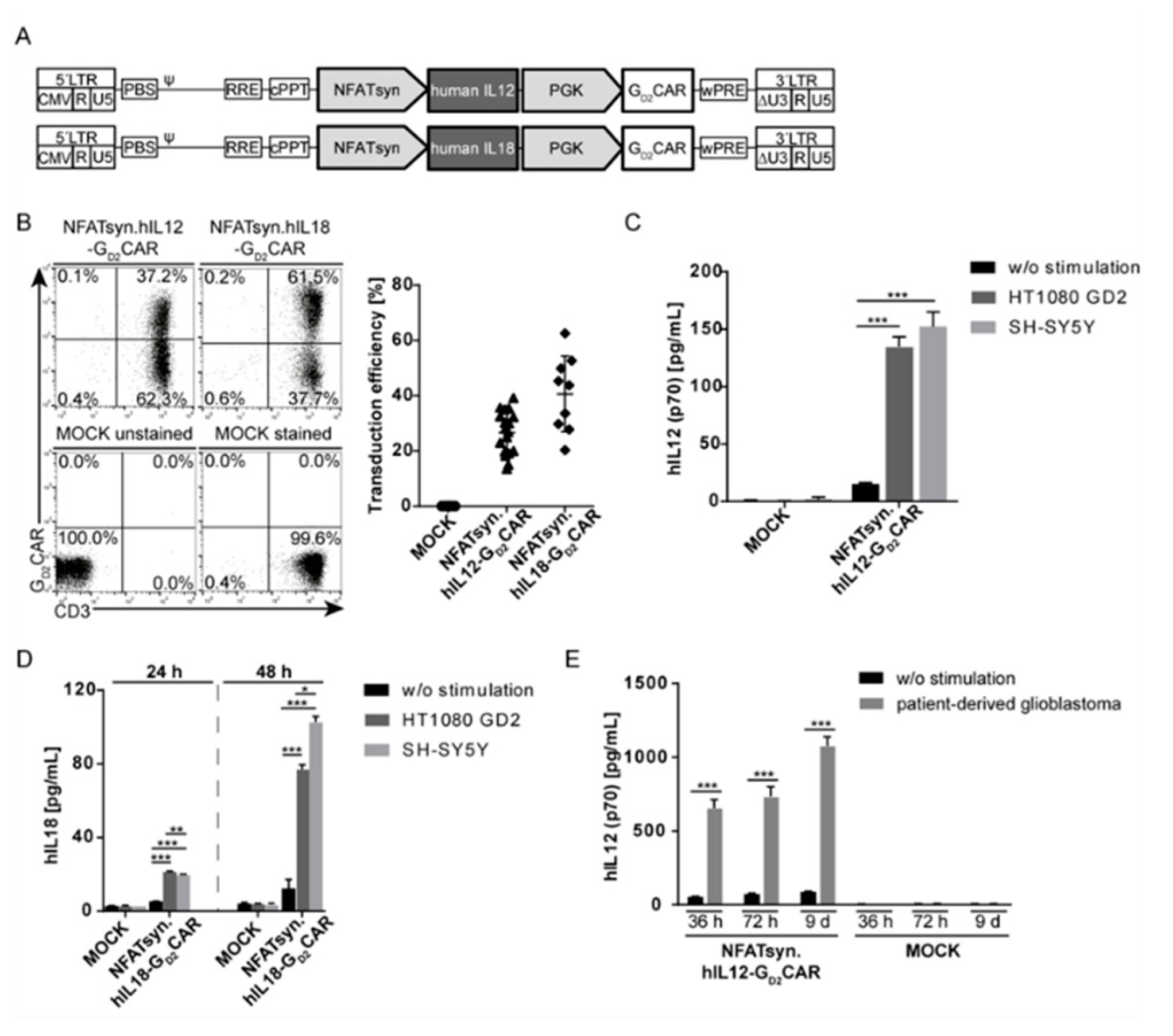
2.5. The Modular Design Facilitates Exchange of the Inducible Transgene and Allows Incorporation of the Promising Anticancer Cytokines IL12 and IL18
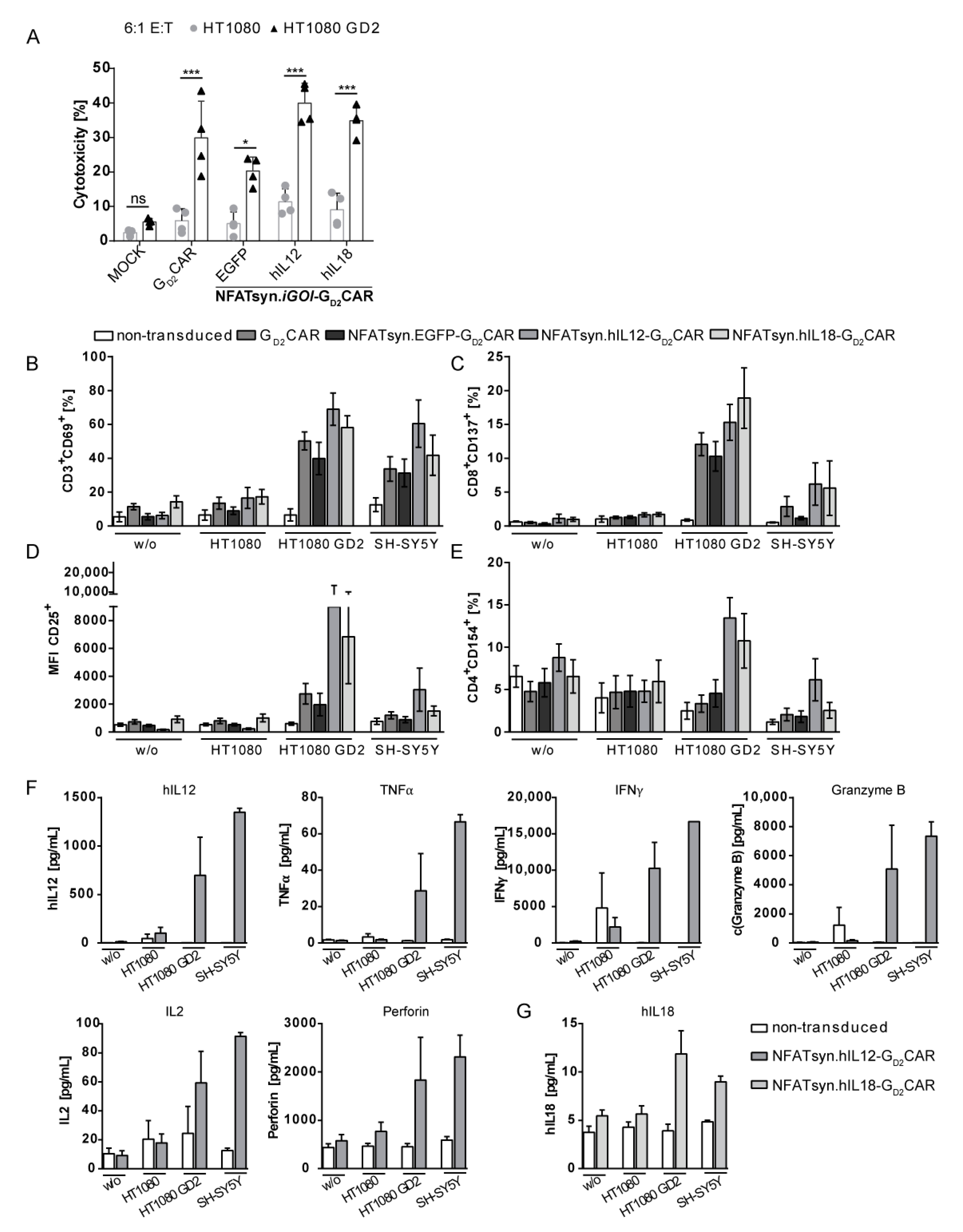
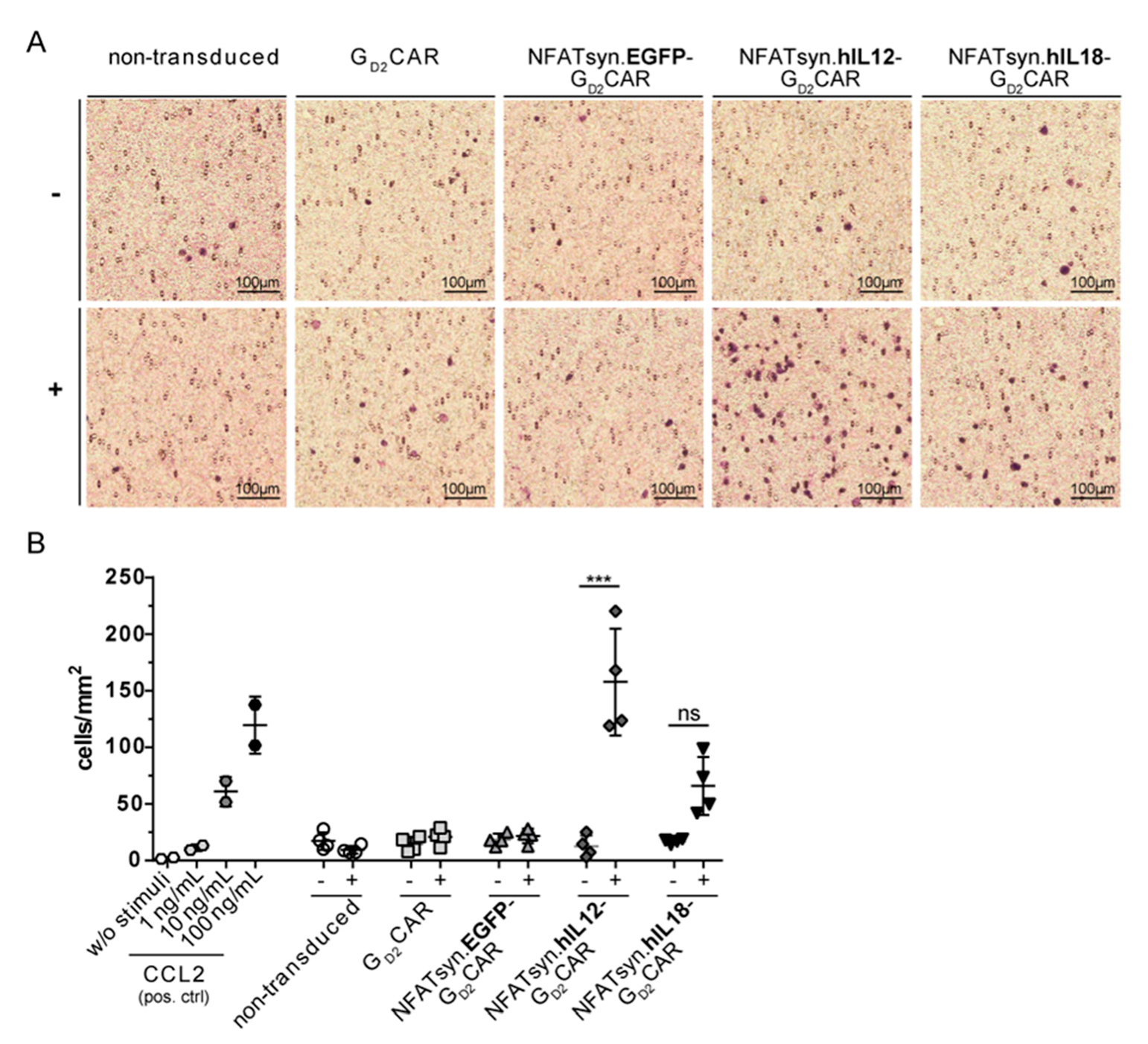
2.6. “All-in-One” Vector Construct-Transduced Primary T Cells Exhibit a Superior Response with Regard to Cytotoxicity, Activation, and Immune Cell Recruitment
3. Discussion
4. Materials and Methods
4.1. Human Material
4.2. Cell Lines
4.3. Cloning of “All-in-One” Vector Constructs, Production, and Titration of Viral Supernatants
4.4. Transduction of Cell Lines and Primary T Cells
4.5. Hybridoma Co-Culture Assay
4.6. Flow Cytometric Analysis
4.7. Cytotoxicity Assay
4.8. Cytokine Analysis by ELISA and LEGENDplex
4.9. Cell Migration Assay
4.10. Microscopy
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dotti, G.; Gottschalk, S.; Savoldo, B.; Brenner, M.K. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol. Rev. 2014, 257, 107–126. [Google Scholar] [CrossRef]
- Maus, M.V.; June, C.H. Making Better Chimeric Antigen Receptors for Adoptive T-cell Therapy. Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res. 2016, 22, 1875–1884. [Google Scholar] [CrossRef] [Green Version]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. New Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Axicabtagene ciloleucel (Yescarta) for B-cell lymphoma. Med. Lett. Drugs Ther. 2018, 60, e122–e123.
- Bach, P.B.; Giralt, S.A.; Saltz, L.B. FDA Approval of Tisagenlecleucel: Promise and Complexities of a $475000 Cancer Drug. Jama 2017, 318, 1861–1862. [Google Scholar] [CrossRef] [PubMed]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 2013, 5, 177ra38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brudno, J.N.; Somerville, R.P.; Shi, V.; Rose, J.J.; Halverson, D.C.; Fowler, D.H.; Gea-Banacloche, J.C.; Pavletic, S.Z.; Hickstein, D.D.; Lu, T.L.; et al. Allogeneic T Cells That Express an Anti-CD19 Chimeric Antigen Receptor Induce Remissions of B-Cell Malignancies That Progress After Allogeneic Hematopoietic Stem-Cell Transplantation Without Causing Graft-Versus-Host Disease. J. Clin. Oncol.: Off. J. Am. Soc. Clin. Oncol. 2016, 34, 1112–1121. [Google Scholar] [CrossRef] [Green Version]
- Kochenderfer, J.N.; Somerville, R.P.T.; Lu, T.; Yang, J.C.; Sherry, R.M.; Feldman, S.A.; McIntyre, L.; Bot, A.; Rossi, J.; Lam, N.; et al. Long-Duration Complete Remissions of Diffuse Large B Cell Lymphoma after Anti-CD19 Chimeric Antigen Receptor T Cell Therapy. Mol. Ther.: J. Am. Soc. Gene Ther. 2017, 25, 2245–2253. [Google Scholar] [CrossRef] [Green Version]
- Abken, H. Driving CARs on the Highway to Solid Cancer: Some Considerations on the Adoptive Therapy with CAR T Cells. Hum. Gene Ther. 2017, 28, 1047–1060. [Google Scholar] [CrossRef]
- Mata, M.; Gottschalk, S. Engineering for Success: Approaches to Improve Chimeric Antigen Receptor T Cell Therapy for Solid Tumors. Drugs 2019, 79, 401–415. [Google Scholar] [CrossRef]
- Morgan, M.A.; Schambach, A. Engineering CAR-T Cells for Improved Function Against Solid Tumors. Front. Immunol. 2018, 9, e2493. [Google Scholar] [CrossRef] [Green Version]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther.: J. Am. Soc. Gene Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Chinnasamy, D.; Yu, Z.; Kerkar, S.P.; Zhang, L.; Morgan, R.A.; Restifo, N.P.; Rosenberg, S.A. Local delivery of interleukin-12 using T cells targeting VEGF receptor-2 eradicates multiple vascularized tumors in mice. Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res. 2012, 18, 1672–1683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koneru, M.; Purdon, T.J.; Spriggs, D.; Koneru, S.; Brentjens, R.J. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology 2015, 4, e994446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Morgan, R.A.; Beane, J.D.; Zheng, Z.; Dudley, M.E.; Kassim, S.H.; Nahvi, A.V.; Ngo, L.T.; Sherry, R.M.; Phan, G.Q.; et al. Tumor-infiltrating lymphocytes genetically engineered with an inducible gene encoding interleukin-12 for the immunotherapy of metastatic melanoma. Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res. 2015, 21, 2278–2288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacy, M.Q.; Jacobus, S.; Blood, E.A.; Kay, N.E.; Rajkumar, S.V.; Greipp, P.R. Phase II study of interleukin-12 for treatment of plateau phase multiple myeloma (E1A96): A trial of the Eastern Cooperative Oncology Group. Leuk. Res. 2009, 33, 1485–1489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chmielewski, M.; Abken, H. CAR T cells transform to trucks: Chimeric antigen receptor-redirected T cells engineered to deliver inducible IL-12 modulate the tumour stroma to combat cancer. Cancer Immunol. Immunother. 2012, 61, 1269–1277. [Google Scholar] [CrossRef]
- Chmielewski, M.; Abken, H. CAR T Cells Releasing IL-18 Convert to T-Bet(high) FoxO1(low) Effectors that Exhibit Augmented Activity against Advanced Solid Tumors. Cell Rep. 2017, 21, 3205–3219. [Google Scholar] [CrossRef] [Green Version]
- Chmielewski, M.; Kopecky, C.; Hombach, A.A.; Abken, H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011, 71, 5697–5706. [Google Scholar] [CrossRef] [Green Version]
- Kunert, A.; Chmielewski, M.; Wijers, R.; Berrevoets, C.; Abken, H.; Debets, R. Intra-tumoral production of IL18, but not IL12, by TCR-engineered T cells is non-toxic and counteracts immune evasion of solid tumors. Oncoimmunology 2017, 7, e1378842. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Di, S.; Shi, B.; Zhang, H.; Wang, Y.; Wu, X.; Luo, H.; Wang, H.; Li, Z.; Jiang, H. Armored Inducible Expression of IL-12 Enhances Antitumor Activity of Glypican-3–Targeted Chimeric Antigen Receptor–Engineered T Cells in Hepatocellular Carcinoma. J. Immunol. 2019, 203, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Unutmaz, D.; Carroll, R.G.; Carpenito, C.; Shan, X.; Danet-Desnoyers, G.; Liu, R.; Jiang, S.; Albelda, S.M.; Golovina, T.; Coukos, G.; et al. Distinct Effects of IL-18 on the Engraftment and Function of Human Effector CD8+ T Cells and Regulatory T Cells. PLoS ONE 2008, 3, e3289. [Google Scholar] [CrossRef]
- Hu, B.; Ren, J.; Luo, Y.; Keith, B.; Young, R.M.; Scholler, J.; Zhao, Y.; June, C.H. Augmentation of Antitumor Immunity by Human and Mouse CAR T Cells Secreting IL-18. Cell Rep. 2017, 20, 3025–3033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avanzi, M.P.; Yeku, O.; Li, X.; Wijewarnasuriya, D.P.; van Leeuwen, D.G.; Cheung, K.; Park, H.; Purdon, T.J.; Daniyan, A.F.; Spitzer, M.H.; et al. Engineered Tumor-Targeted T Cells Mediate Enhanced Anti-Tumor Efficacy Both Directly and through Activation of the Endogenous Immune System. Cell Rep. 2018, 23, 2130–2141. [Google Scholar] [CrossRef] [PubMed]
- Kailayangiri, S.; Altvater, B.; Lesch, S.; Balbach, S.; Gottlich, C.; Kuhnemundt, J.; Mikesch, J.H.; Schelhaas, S.; Jamitzky, S.; Meltzer, J.; et al. EZH2 Inhibition in Ewing Sarcoma Upregulates GD2 Expression for Targeting with Gene-Modified T Cells. Mol. Ther.: J. Am. Soc. Gene Ther. 2019, 27, 933–946. [Google Scholar] [CrossRef] [PubMed]
- Golinelli, G.; Grisendi, G.; Prapa, M.; Bestagno, M.; Spano, C.; Rossignoli, F.; Bambi, F.; Sardi, I.; Cellini, M.; Horwitz, E.M.; et al. Targeting GD2-positive glioblastoma by chimeric antigen receptor empowered mesenchymal progenitors. Cancer Gene Ther. 2018. [Google Scholar] [CrossRef] [Green Version]
- Johnsen, J.I.; Dyberg, C.; Wickstrom, M. Neuroblastoma-A Neural Crest Derived Embryonal Malignancy. Front. Mol. Neurosci. 2019, 12, e9. [Google Scholar] [CrossRef]
- Kailayangiri, S.; Altvater, B.; Meltzer, J.; Pscherer, S.; Luecke, A.; Dierkes, C.; Titze, U.; Leuchte, K.; Landmeier, S.; Hotfilder, M.; et al. The ganglioside antigen GD2 is surface-expressed in Ewing sarcoma and allows for MHC-independent immune targeting. Br. J. Cancer 2012, 106, 1123–1133. [Google Scholar] [CrossRef]
- Heczey, A.; Louis, C.U.; Savoldo, B.; Dakhova, O.; Durett, A.; Grilley, B.; Liu, H.; Wu, M.F.; Mei, Z.; Gee, A.; et al. CAR T Cells Administered in Combination with Lymphodepletion and PD-1 Inhibition to Patients with Neuroblastoma. Mol. Ther.: J. Am. Soc. Gene Ther. 2017, 25, 2214–2224. [Google Scholar] [CrossRef] [Green Version]
- Louis, C.U.; Savoldo, B.; Dotti, G.; Pule, M.A.; Yvon, E.; Myers, G.D.; Rossig, C.; Russell, H.V.; Diouf, O.; Liu, E.; et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011, 118, 6050–6056. [Google Scholar] [CrossRef]
- Pule, M.A.; Savoldo, B.; Myers, G.D.; Rossig, C.; Russell, H.V.; Dotti, G.; Huls, M.H.; Liu, E.; Gee, A.P.; Mei, Z.; et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: Persistence and antitumor activity in individuals with neuroblastoma. Nature Med. 2008, 14, 1264–1270. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. New Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladenstein, R.; Pötschger, U.; Valteau-Couanet, D.; Luksch, R.; Castel, V.; Yaniv, I.; Laureys, G.; Brock, P.; Michon, J.M.; Owens, C.; et al. Interleukin 2 with anti-GD2 antibody ch14.18/CHO (dinutuximab beta) in patients with high-risk neuroblastoma (HR-NBL1/SIOPEN): A multicentre, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1617–1629. [Google Scholar] [CrossRef]
- Schambach, A.; Zychlinski, D.; Ehrnstroem, B.; Baum, C. Biosafety features of lentiviral vectors. Hum. Gene Ther. 2013, 24, 132–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noyan, F.; Zimmermann, K.; Hardtke-Wolenski, M.; Knoefel, A.; Schulde, E.; Geffers, R.; Hust, M.; Huehn, J.; Galla, M.; Morgan, M.; et al. Prevention of Allograft Rejection by Use of Regulatory T Cells With an MHC-Specific Chimeric Antigen Receptor. Am. J. Transplant.: Off. J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2017, 17, 917–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooijberg, E.; Bakker, A.Q.; Ruizendaal, J.J.; Spits, H. NFAT-controlled expression of GFP permits visualization and isolation of antigen-stimulated primary human T cells. Blood 2000, 96, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Merlet, E.; Lipskaia, L.; Marchand, A.; Hadri, L.; Mougenot, N.; Atassi, F.; Liang, L.; Hatem, S.N.; Hajjar, R.J.; Lompre, A.M. A calcium-sensitive promoter construct for gene therapy. Gene Ther. 2013, 20, 248–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeku, O.O.; Brentjens, R.J. Armored CAR T-cells: Utilizing cytokines and pro-inflammatory ligands to enhance CAR T-cell anti-tumour efficacy. Biochem. Soc. Trans. 2016, 44, 412–418. [Google Scholar] [CrossRef]
- David, R.M.; Doherty, A.T. Viral Vectors: The Road to Reducing Genotoxicity. Toxicol. Sci.: Off. J. Soc. Toxicol. 2017, 155, 315–325. [Google Scholar] [CrossRef] [Green Version]
- Biasco, L.; Rothe, M.; Schott, J.W.; Schambach, A. Integrating Vectors for Gene Therapy and Clonal Tracking of Engineered Hematopoiesis. Hematol. /Oncol. Clin. North Am. 2017, 31, 737–752. [Google Scholar] [CrossRef]
- Goswami, R.; Subramanian, G.; Silayeva, L.; Newkirk, I.; Doctor, D.; Chawla, K.; Chattopadhyay, S.; Chandra, D.; Chilukuri, N.; Betapudi, V. Gene Therapy Leaves a Vicious Cycle. Front. Oncol. 2019, 9, e297. [Google Scholar] [CrossRef] [PubMed]
- Kulemzin, S.V.; Matvienko, D.A.; Sabirov, A.H.; Sokratyan, A.M.; Chernikova, D.S.; Belovezhets, T.N.; Chikaev, A.N.; Taranin, A.V.; Gorchakov, A.A. Design and analysis of stably integrated reporters for inducible transgene expression in human T cells and CAR NK-cell lines. BMC Med Genom. 2019, 12, e44. [Google Scholar] [CrossRef] [Green Version]
- Jutz, S.; Leitner, J.; Schmetterer, K.; Doel-Perez, I.; Majdic, O.; Grabmeier-Pfistershammer, K.; Paster, W.; Huppa, J.B.; Steinberger, P. Assessment of costimulation and coinhibition in a triple parameter T cell reporter line: Simultaneous measurement of NF-kappaB, NFAT and AP-1. J. Immunol. Methods 2016, 430, 10–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rydzek, J.; Nerreter, T.; Peng, H.; Jutz, S.; Leitner, J.; Steinberger, P.; Einsele, H.; Rader, C.; Hudecek, M. Chimeric Antigen Receptor Library Screening Using a Novel NF-kappaB/NFAT Reporter Cell Platform. Mol. Ther.: J. Am. Soc. of Gene Ther. 2019, 27, 287–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy-assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Kerkar, S.P.; Yu, Z.; Zheng, Z.; Yang, S.; Restifo, N.P.; Rosenberg, S.A.; Morgan, R.A. Improving adoptive T cell therapy by targeting and controlling IL-12 expression to the tumor environment. Mol. Ther.: J. Am. Soc. Gene Ther. 2011, 19, 751–759. [Google Scholar] [CrossRef]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; van Leeuwen, D.G. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol. 2018, 36, 847–856. [Google Scholar] [CrossRef]
- Tray, N.; Weber, J.S.; Adams, S. Predictive Biomarkers for Checkpoint Immunotherapy: Current Status and Challenges for Clinical Application. Cancer Immunol. Res. 2018, 6, 1122–1128. [Google Scholar] [CrossRef] [Green Version]
- Yi, L.; Weifan, Y.; Huan, Y. Chimeric antigen receptor-engineered regulatory T lymphocytes: Promise for immunotherapy of autoimmune disease. Cytotherapy 2019, 21, 925–934. [Google Scholar] [CrossRef]
- Hasselbach, L.A.; Irtenkauf, S.M.; Lemke, N.W.; Nelson, K.K.; Berezovsky, A.D.; Carlton, E.T.; Transou, A.D.; Mikkelsen, T.; deCarvalho, A.C. Optimization of high grade glioma cell culture from surgical specimens for use in clinically relevant animal models and 3D immunochemistry. J. Vis. Exp.: JoVE 2014, 83, e51088. [Google Scholar] [CrossRef] [Green Version]
- Klapdor, R.; Wang, S.; Hacker, U.; Buning, H.; Morgan, M.; Dork, T.; Hillemanns, P.; Schambach, A. Improved Killing of Ovarian Cancer Stem Cells by Combining a Novel Chimeric Antigen Receptor-Based Immunotherapy and Chemotherapy. Hum. Gene Ther. 2017, 28, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Schambach, A.; Bohne, J.; Chandra, S.; Will, E.; Margison, G.P.; Williams, D.A.; Baum, C. Equal potency of gammaretroviral and lentiviral SIN vectors for expression of O6-methylguanine-DNA methyltransferase in hematopoietic cells. Mol. Ther.: J. Am. Soc. Gene Ther. 2006, 13, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Vanin, E.F.; Whitt, M.A.; Fornerod, M.; Zwart, R.; Schneiderman, R.D.; Grosveld, G.; Nienhuis, A.W. Inducible, High-Level production of Infectious Murine Leukemia Retroviral Vector Particles Pseudotyped with Vesicular Stomatitis Virus G Envelope Protein. Hum. Gene Ther. 1995, 6, 1203–1213. [Google Scholar] [CrossRef] [PubMed]
- Fehse, B.; Kustikova, O.S.; Bubenheim, M.; Baum, C. Pois(s)on--it’s a question of dose. Gene Ther. 2004, 11, 879–881. [Google Scholar] [CrossRef] [PubMed]
- Suerth, J.D.; Morgan, M.A.; Kloess, S.; Heckl, D.; Neudorfl, C.; Falk, C.S.; Koehl, U.; Schambach, A. Efficient generation of gene-modified human natural killer cells via alpharetroviral vectors. J. Mol. Med. 2016, 94, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Lode, H.N.; Schmidt, M.; Seidel, D.; Huebener, N.; Brackrock, D.; Bleeke, M.; Reker, D.; Brandt, S.; Mueller, H.P.; Helm, C.; et al. Vaccination with anti-idiotype antibody ganglidiomab mediates a GD(2)-specific anti-neuroblastoma immune response. Cancer Immunol. Immunother. 2013, 62, 999–1010. [Google Scholar] [CrossRef]
- Siebert, N.; Seidel, D.; Eger, C.; Brackrock, D.; Reker, D.; Schmidt, M.; Lode, H.N. Validated detection of anti-GD2 antibody ch14.18/CHO in serum of neuroblastoma patients using anti-idiotype antibody ganglidiomab. J. Immunol. Methods 2013, 398-399, 51–59. [Google Scholar] [CrossRef]
- Aichinger, M.; Wu, C.; Nedjic, J.; Klein, L. Macroautophagy substrates are loaded onto MHC class II of medullary thymic epithelial cells for central tolerance. J. Exp. Med. 2013, 210, 287–300. [Google Scholar] [CrossRef] [Green Version]
- Ziegler-Heitbrock, H.W.; Thiel, E.; Fütterer, A.; Herzog, V.; Wirtz, A.; Riethmüller, G. Establishment of a human cell line (Mono Mac 6) with characteristics of mature monocytes. Int. J. Cancer 1988, 15, 456–461. [Google Scholar] [CrossRef]
- Humphries, J.; Gossage, J.A.; Modarai, B.; Burnand, K.G.; Sisson, T.H.; Murdoch, C.; Smith, A. Monocyte urokinase-type plasminogen activator up-regulation reduces thrombus size in a model of venous thrombosis. J. Vasc. Surg. 2009, 50, 1127–1134. [Google Scholar] [CrossRef] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zimmermann, K.; Kuehle, J.; Dragon, A.C.; Galla, M.; Kloth, C.; Rudek, L.S.; Sandalcioglu, I.E.; Neyazi, B.; Moritz, T.; Meyer, J.; et al. Design and Characterization of an “All-in-One” Lentiviral Vector System Combining Constitutive Anti-GD2 CAR Expression and Inducible Cytokines. Cancers 2020, 12, 375. https://doi.org/10.3390/cancers12020375
Zimmermann K, Kuehle J, Dragon AC, Galla M, Kloth C, Rudek LS, Sandalcioglu IE, Neyazi B, Moritz T, Meyer J, et al. Design and Characterization of an “All-in-One” Lentiviral Vector System Combining Constitutive Anti-GD2 CAR Expression and Inducible Cytokines. Cancers. 2020; 12(2):375. https://doi.org/10.3390/cancers12020375
Chicago/Turabian StyleZimmermann, Katharina, Johannes Kuehle, Anna Christina Dragon, Melanie Galla, Christina Kloth, Loreen Sophie Rudek, I. Erol Sandalcioglu, Belal Neyazi, Thomas Moritz, Johann Meyer, and et al. 2020. "Design and Characterization of an “All-in-One” Lentiviral Vector System Combining Constitutive Anti-GD2 CAR Expression and Inducible Cytokines" Cancers 12, no. 2: 375. https://doi.org/10.3390/cancers12020375
APA StyleZimmermann, K., Kuehle, J., Dragon, A. C., Galla, M., Kloth, C., Rudek, L. S., Sandalcioglu, I. E., Neyazi, B., Moritz, T., Meyer, J., Rossig, C., Altvater, B., Eiz-Vesper, B., Morgan, M. A., Abken, H., & Schambach, A. (2020). Design and Characterization of an “All-in-One” Lentiviral Vector System Combining Constitutive Anti-GD2 CAR Expression and Inducible Cytokines. Cancers, 12(2), 375. https://doi.org/10.3390/cancers12020375