1. Introduction
The pharmacokinetics of a drug are typically described as four steps: Absorption, distribution, metabolism and excretion [
1]. Absorption is linked to the transfer of the drug-to-blood circulation, distribution to its diffusion in the different organs and tissues, metabolism refers to its degradation through chemical reaction and excretion through physical pathways. In the last 30 years, nanomedicine has strongly developed as a possible solution for improving the therapeutic index of already-known drugs [
2,
3]. Indeed, numerous drugs are known to be active for certain pathologies, but are restricted by access to the site that would be treated in the most specific manner possible. Drugs might be degraded and eliminated before reaching the site, or they might diffuse in different organs and have severe side effects, as in the case of anti-cancer drugs. The encapsulation of drugs inside a nanovector is intended to overcome unfavorable pharmacokinetic properties, thereby, improving the selectivity of delivery, or increasing the efficacy by simply increasing its apparent aqueous solubility. A key element, which has prompted thousands of studies, is the discovery by Maeda in the eighties of the so-called Enhanced Permeability and Retention effect (EPR) [
4,
5]. This terminology describes the ability of nanovectors to spontaneously accumulate in tumor areas, due to the existence of disjunctions between the endothelial cells of blood vessels close to tumors and the strongly limited lymphatic system of tumors, preventing the rapid clearance of the vectors from the area. Compared to the tremendous hope carried by nanomedicine, only a few drug formulations are today on the market, less than 15 in the field of oncology [
6]. The most successful drugs are Doxil
®, a liposome-encapsulated doxorubicin, and Abraxane
®, an association of albumin and paclitaxel. One of the possible explanations of this low success rate is the over-simplification of the EPR effect, which is increasingly questioned regarding its frequency [
7,
8,
9]. An important discovery was recently made by Matsumoto et al. who observed pulses in drug transfer, which showed that the EPR might not be constant in time [
10], rendering the behavior even more complex. Another recurring hypothesis is the poor understanding of the transfer of the drug from the vector to the target site. For this, the interaction between the nanovectors and the different biological barriers encountered should be better assessed [
11,
12]. This statement is true for all nanomedicine systems, and in particular for those developed for photodynamic therapy (PDT). PDT is based on a photosensitizer molecule, the irradiation of which locally produces reactive oxygen species, such as singlet oxygen, leading to cell death. PDT is clinically used in dermatology, ophthalmology or oncology [
13,
14,
15]. However, limitations include the low water solubility of the photosensitizers and their tendency to form aggregates by π-π stacking, strongly decreasing their PDT efficiency. In order to improve this, encapsulation of photosensitizers has been assessed, either in liposomes [
16] or in polymeric nanovectors [
15,
17,
18,
19,
20,
21,
22]. These studies showed an improvement of the photocytotoxicity upon encapsulation, linked to a limitation of the aggregation and altered absorption by cells compared with free photosensitizer. In this context, we present here a focus on the delivery process of Pheophorbide
a (Pheo), a known photosensitizer, from polymeric micelles based on poly (ethylene oxide-b-ε-caprolactone) (PEO-PCL) and poly (ethylene oxide-b-styrene) (PEO-PS) towards human tumor cells or model lipid vesicles. We used block-copolymer self-assemblies in previous studies [
18,
20,
21,
22] and they showed to efficiently encapsulate Pheo. Among the assessed nanocarriers, we selected micelles with approximately the same size, but with different nature of the hydrophobic block in order to discriminate the effects of these parameters in the nanocarrier-membranes interactions. Our study brings translational information between a classical biological and physical chemistry approaches.
3. Discussion
In this paper, studies of the cellular uptake of free Pheo show that its level of internalization in cells is low, and its photocytotoxicity is very limited (
Figure 2,
Figure 6 and
Figure 7). In contrast, when Pheo is loaded in PEO-PCL or PEO-PS micelles, its cellular uptake is increased drastically, as well as its efficiency as a PDT sensitizer. This confirms the high benefit of copolymer micelles in Pheo delivery in cancerous cells for PDT applications as also mentioned in many previous studies [
15,
32].
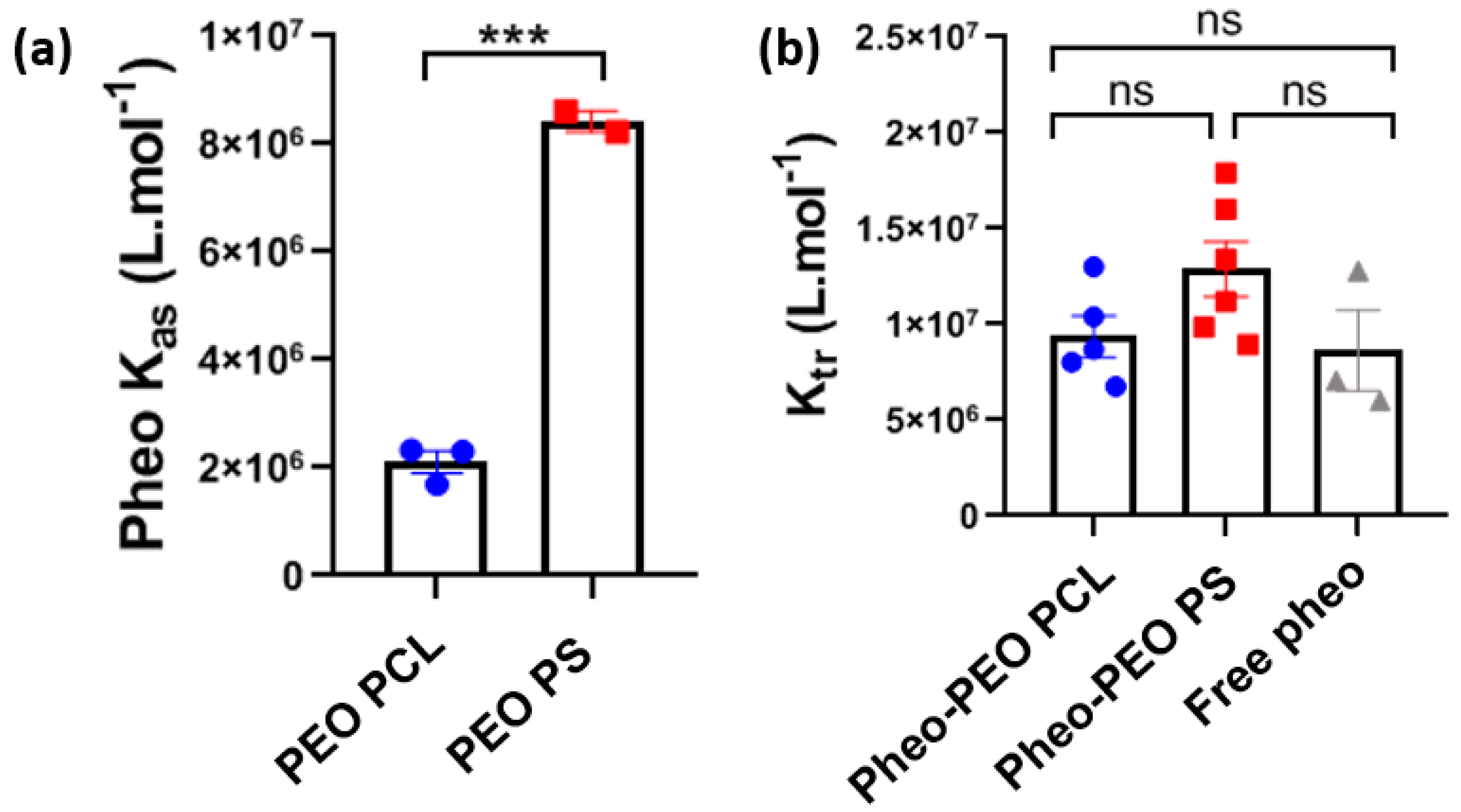
If the advantages of drug encapsulation are well established, the mechanism by which amphiphilic block copolymer micelles deliver drugs is still poorly understood. In particular little is known experimentally about the interactions with plasma membranes and the cellular processes involved in the internalization of the drug. The initial encounter between nanovectors and membrane is crucial in determining the efficiency of the drug towards its cellular target. From a general point of view, nanovectors will not only lead to increased intracellular concentration of the drug, compared to unvectorized drugs, but could modify its subcellular localization and alter its natural targets. In this study, we focused on spherical PEO-PS and PEO-PCL micelles used to encapsulate Pheo, a lipophilic photosensitizer which generates reactive oxygen species by de-excitation from its excited triplet state. Pheo fluoresces at 670 nm is known to generate primarily singlet oxygen. This compound was used both as a model drug for photodynamic therapy applications, and also as a probe thanks to its natural fluorescence, which has given insight into the relationships between cellular uptake and therapeutic efficiency. The determination of the affinity constant of Pheo for both types of block copolymer micelles reveals that Pheo has a higher affinity for PEO-PS micelles. This may be partly explained by the strength of π-π interactions between Pheo and PS which can be stronger than the hydrophobic ones between Pheo and PCL [
33]. This result suggests that Pheo will remain more easily sequestered in PEO-PS micelles than in PEO-PCL micelles. We then looked at the transfer efficiency of Pheo from micelles to DOPC LUVs, employed here as synthetic model membranes. The transfer of Pheo towards the LUVs was found to be independent of the type of micelle used, despite the differences in affinity constants. Interestingly, the presence of copolymer micelles has no effect on the thermodynamics of Pheo transfer to the LUV since the transfer constants, K
tr, are comparable in all conditions.
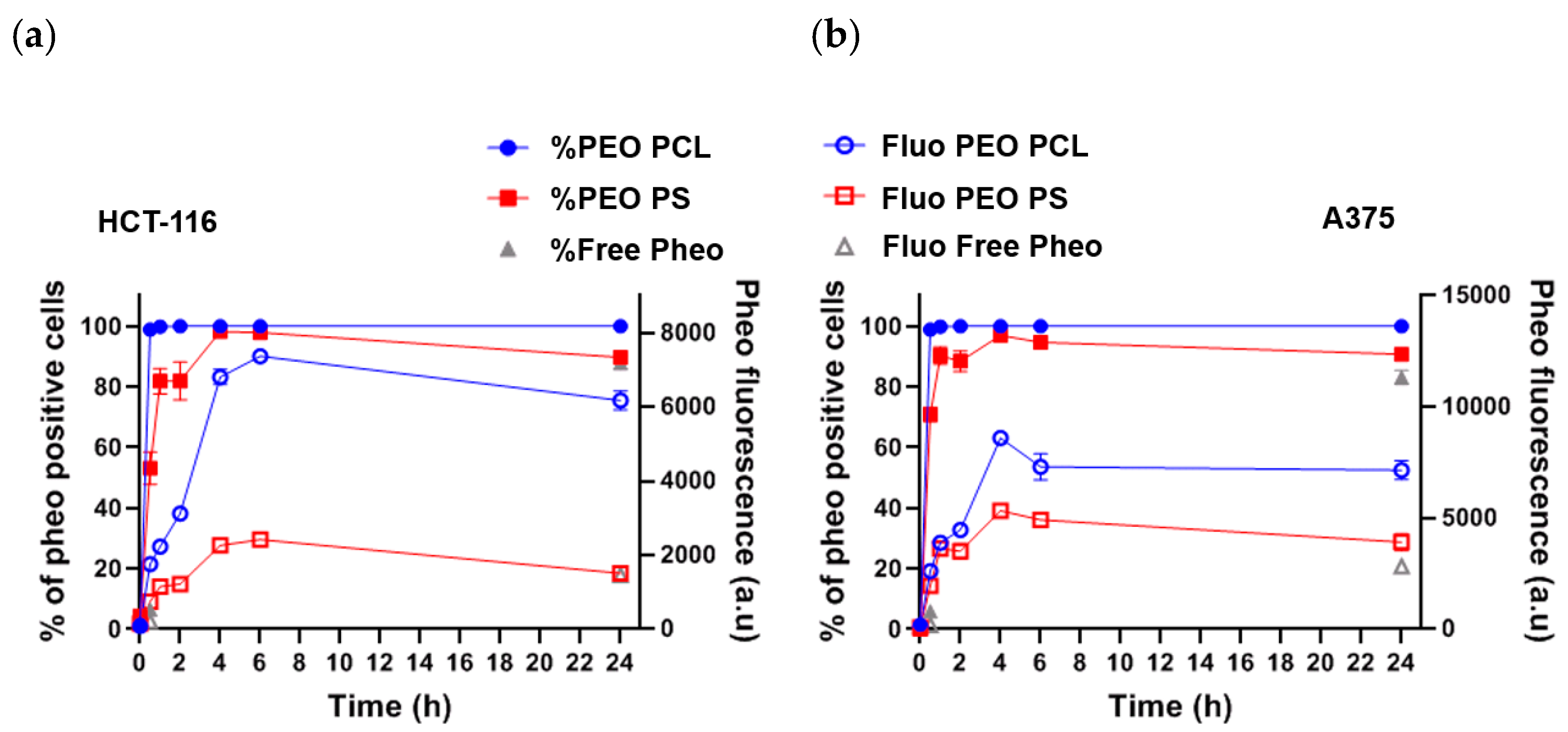
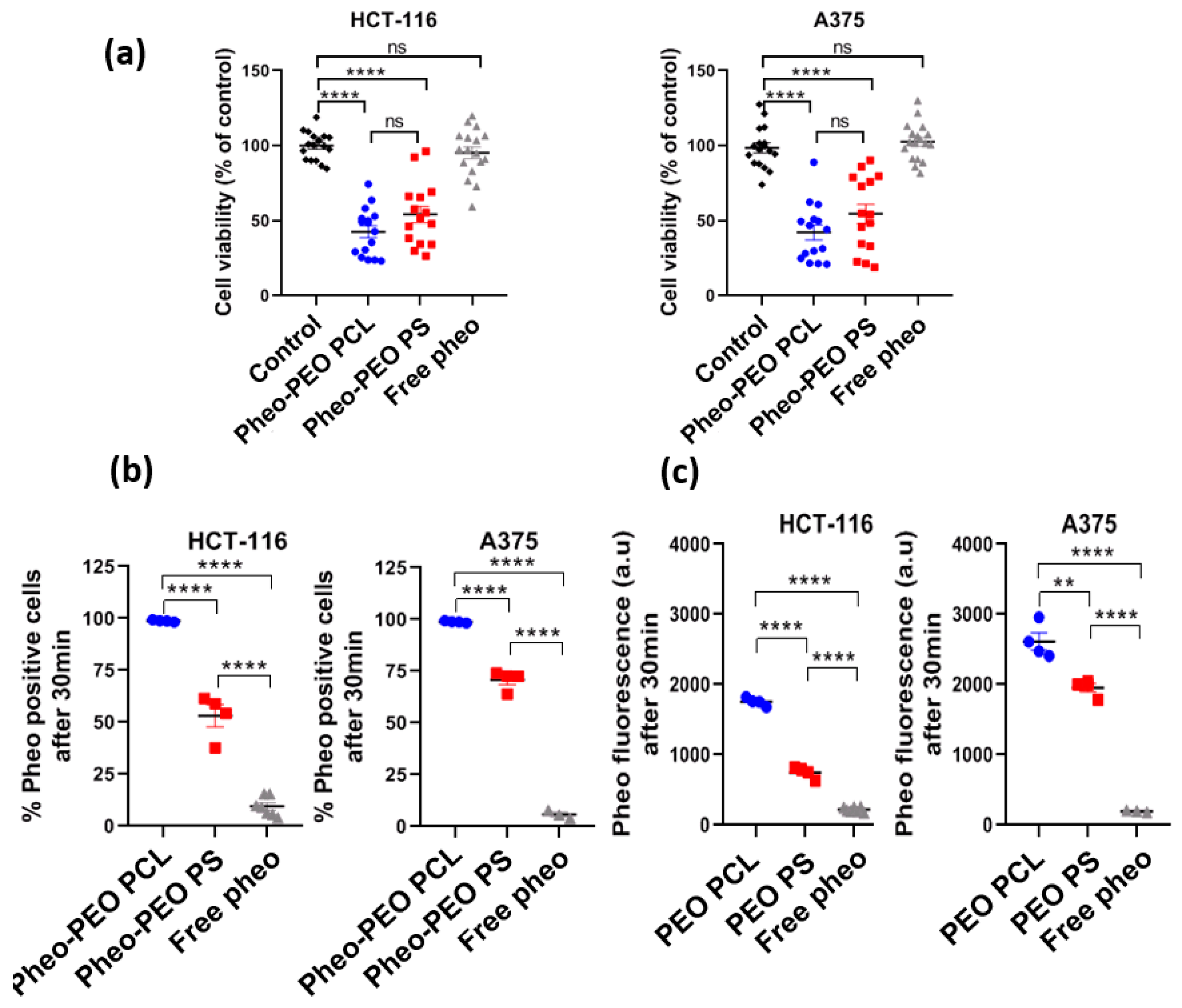
Experiments on cellular uptake of Pheo loaded PEO-PCL and Pheo loaded PEO-PS (
Figure 2) point out that whichever cancerous cell types were used, HCT-116 or A375, the Pheo incorporation is ~3 times higher after 4 h incubation when Pheo is encapsulated in PEO-PCL micelles than in PEO-PS micelles. It is important to note that with both cancerous cell types and both nanovectors, the cellular penetration of Pheo exhibits two phases, a fast one between 0 and 4 h of incubation and a slower phase leading to a plateau. This strongly suggests that the cellular penetration of encapsulated Pheo in PEO-PS or PEO-PCL involves the participation of endocytosis or another active mechanism.
100% of cells were positive for Pheo after 30 min of incubation when the carrier was PEO-PCL whereas this rate was only achieved at 4 h with PEO-PS, which indicates that Pheo loaded PEO-PCL may enter cells via different pathways compared to PEO-PS.
It has to be noted that experiments on cells were conducted with an excess of photosensitizer with respect to the available bilayer volume. In this case, Pheo detected by flow cytometry is definitely not all located at the plasma membrane, but also intracellularly, as it was already proved by previous experiments on Pheo intracellular localization [
18,
34]. As FRET experiments showed that the Pheo absorbed on the membrane was the same in the presence of the two nanocarriers, we can reasonably conclude that PEO-PCL nanocarriers lead to higher Pheo cell internalization than PEO-PS ones.
Since both types of carriers share the same PEO corona, we can exclude specific attachment of the micelles to cells as a potential explanation for this marked difference. The hydrophobic core must thus play a crucial role in the incorporation mechanism. The PS core has been shown to be more robust towards destabilization than PCL one [
18]. This could seem surprising since PCL is a semi-crystalline polymer and crystallinity has been shown to favor the stability of polymer micelles [
35]. However, in the system presented here, we have previously demonstrated that whereas empty PEO-PCL retained some crystallinity, the introduction of Pheo induced its complete disappearance [
34]. Furthermore, PS exhibits a much higher glass transition temperature (ca. 100 °C) compared to PCL (ca.−50 °C). At 37 °C, PCL is thus expected to be much softer than PS, providing higher mobility for all species. This has been very recently shown by Mosinger’s team who proved that oxygen was able to move more easily in a PCL matrix compared to a PS one [
36]. This difference in characteristics of the micellar cores leads to differences of behavior when the micelles are in the presence of target membranes.
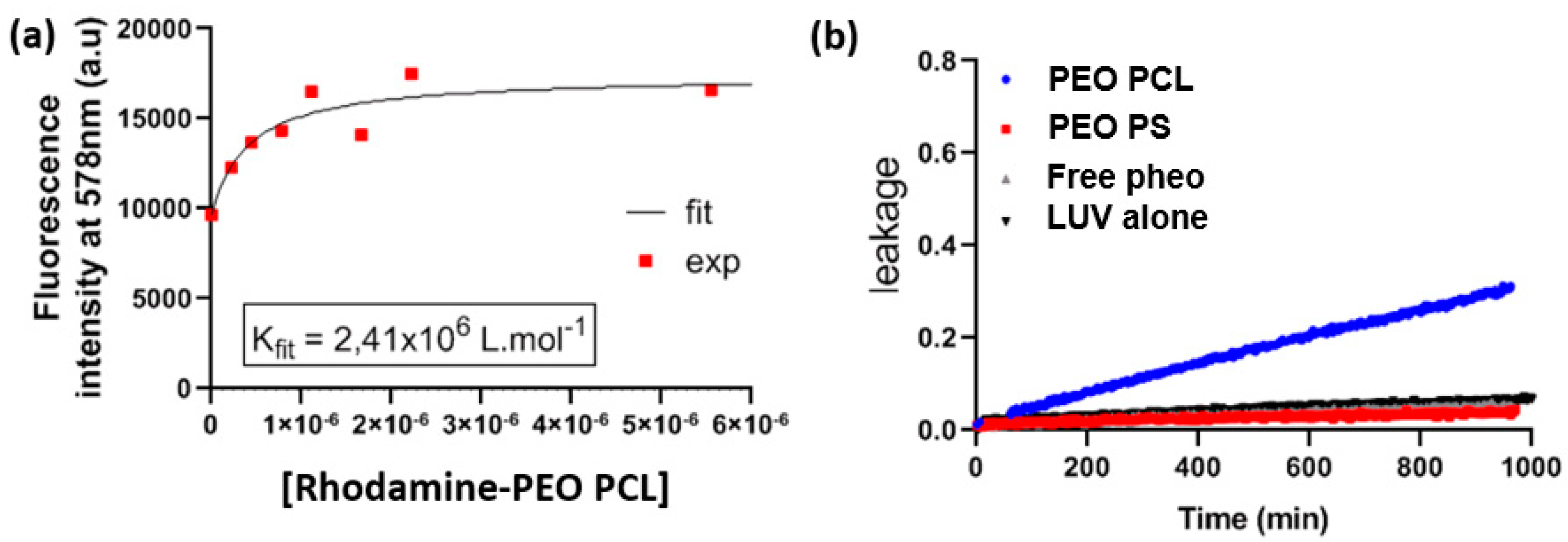
Figure 3b describes the impact of block copolymer micelles on the permeability of LUVs, indicated by the time-dependence of the leakage of a small molecule. Leakage of carboxyfluorescein from LUVs was observed only in the case of PEO-PCL micelles which suggests defects in the lipid bilayer induced by PEO-PCL. Further, we observed a steady permeabilization rather than a short-lived destabilization. FRET experiments were ideal to tackle proximity problems such as this one.
Figure 3a depicts the increase in rhodamine fluorescence due to FRET from NBD-labelled LUVs. The critical distance R
0, at which FRET efficiency is reduced to 50% was calculated to be approximately 4 nm. Previous studies have shown [
34] that the radius of the PEO-PCL micelle is ~ 9 nm according to neutron scattering measurements. These two parameters allow us to exclude the simple spatial proximity of unperturbed LUVs and micelles as a justification for the energy transfer. Rather, there has to be intermixing of copolymer into LUVs, or of labelled lipid into copolymer micelles. However, we cannot conclude on the true nature of this incorporation, and several models have been proposed [
37]. The phenomenon observed in our case is much slower than in the study by Nawaz et al. [
38], where they observed complete hemolysis in under 100 min using Pluronics as copolymers. The faster dynamics in the case of Pluronics are probably to be attributed to the higher critical micellar concentration of PEO-PPO-PEO than PEO-PCL, and the consequent higher concentration of free copolymer chains. These findings on model membranes have been confirmed by cellular studies on the uptake of rhodamine labelled PEO-PCL, and revealed the presence of polymer in the cells. For studies on both cell types, all cells were positive for rhodamine after very short interaction times thus supporting the hypothesis of polymer incorporation into cells. However, the intensity of the rhodamine fluorescence, which is representative of polymer incorporation, is steadily increasing even after 24 h of interaction. This indicates the contribution of a passive mechanism, given that saturation is not reached after such a long interaction time.
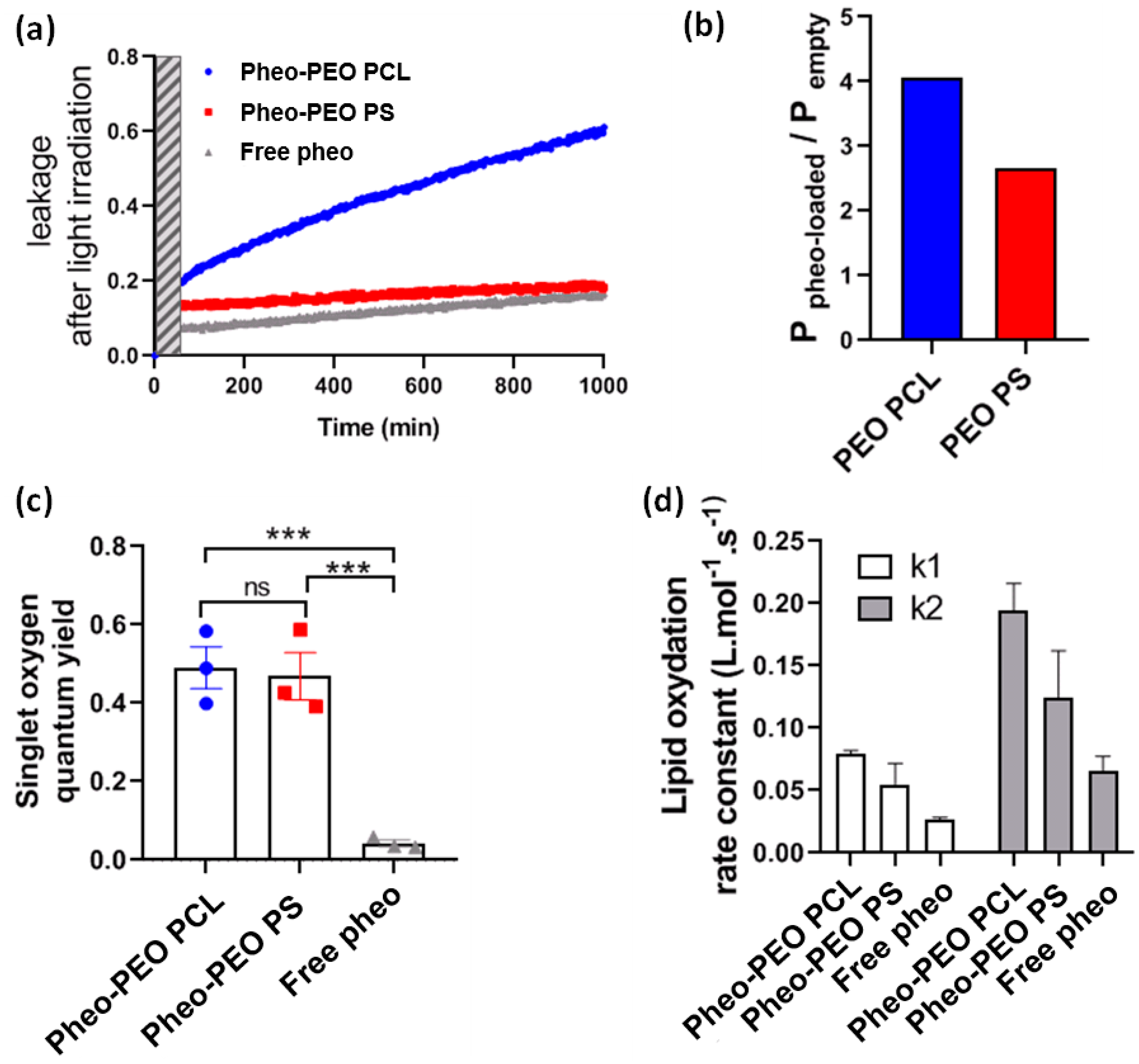
In order to study the effects of singlet oxygen production in the vicinity of lipid bilayers, we performed LUV leakage experiments under irradiation. We observed that light irradiation of Pheo loaded micelles led to an increase of LUV leakage rate, more so in the case of PEO-PCL micelles than PEO-PS micelles and free Pheo. This superior effect for PEO-PCL micelles is observed in spite of similar singlet oxygen production for both types of micelles, which is another indication of the active part played by the PEO-PCL copolymer in destabilizing the lipid membrane. Further, we showed that the rate of photo-induced peroxidation was consistently higher when Pheo was carried by PEO-PCL micelles, again confirming the active role played by the polymer. Significant peroxidation was only observed after prolonged irradiation (several hours,
Figure S12). Simple lipid peroxidations cannot lead to LUV permeabilization [
39]. Recently Bachelar et al. [
40] proposed that direct contact between photosensitizers and lipids were essential for the progress of lipid oxidation and consequently for the formation of aldehydes leading to truncated lipids and membrane permeabilization. We did not detect truncated lipids, even though Pheo encapsulated in PEO-PCL may be in direct contact with DOPC upon the intermixing of copolymer with LUVs.
Very interestingly, these studies on model membrane systems, which help us to confirm the active part played by the co-polymer carrier, are counterbalanced by cellular studies in PDT conditions. By flow cytometry, we showed that incubating cells for 30 min with Pheo loaded PEO-PCL resulted in a much greater concentration of photosensitizer within, or attached to the cells, compared to Pheo loaded PEO-PS micelles. One could expect that delivery by PEO-PCL micelles would result in greater photocytotoxic activity of Pheo. Surprisingly, on 2D cell cultures of both cancerous cell lines tested, we find similar phototoxic efficiencies irrespective of the type of nanovector, with around 50% cell viability retained after light activation of the photosensitizer.
Several hypotheses can be formulated to explain these results. Pheo delivered by PEO-PCL could enter cells via more than one pathway: mass transfer between micelles and cell where both polymer and cargo can be incorporated into the membrane (as shown in
Figure 3); direct transfer of Pheo from micelle to cell membrane, as proposed by Kerdous et al. [
27] and Till et al. [
34]; by endocytosis [
34]. In the case of Pheo delivered by PEO-PS micelles, direct jump and endocytotic mechanisms seem to be the main cell entrance pathways. Very recently Kubát et al. [
36] clearly showed that the lifetime of singlet oxygen produced by photosensitizer encapsulated in PS is higher (6.4 µs to 16 µs) than that photo-generated in PEO-PCL micelles (3.6–3.8 µs). In PEO-PS, singlet oxygen due to its longer lifetime will have time to diffuse through the nanovector in the vicinity of numerous intracellular targets and to photo-oxidize them. In PEO-PCL, the shorter singlet oxygen lifetime leads to a decrease of the amount of cellular photodamage produced, compared to those by Pheo loaded PEO-PS system. Altogether, these experiments tend to show that the microenvironment of Pheo will govern its photo-toxicity via the rate of singlet oxygen deactivation. The superior apparent efficiency of Pheo loaded PEO-PS micelles indicates that a significant part enters cells unperturbed. Finally the increased uptake of PEO-PCL might be compensated by the higher activity of the Pheo in the PEO-PS.
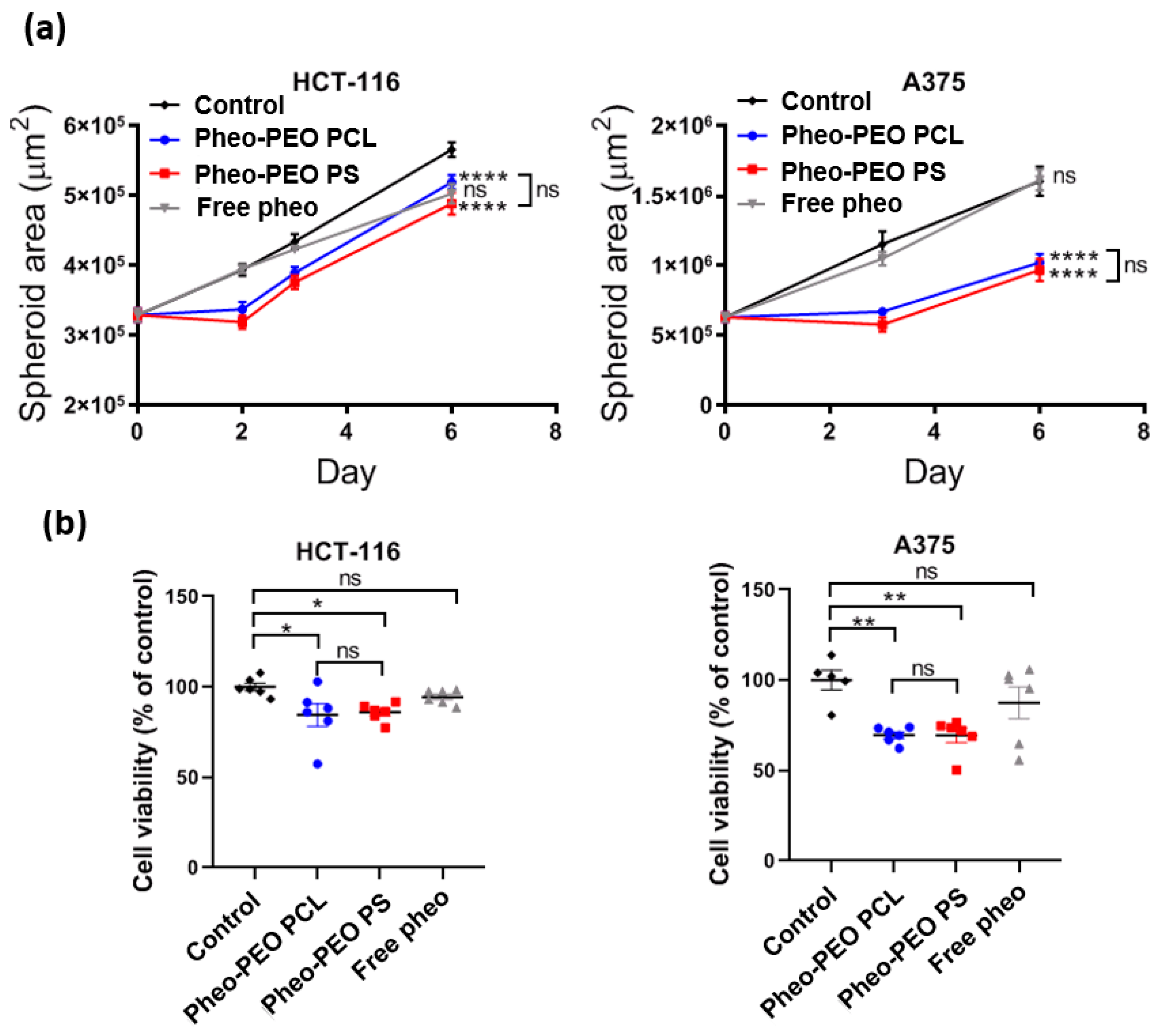
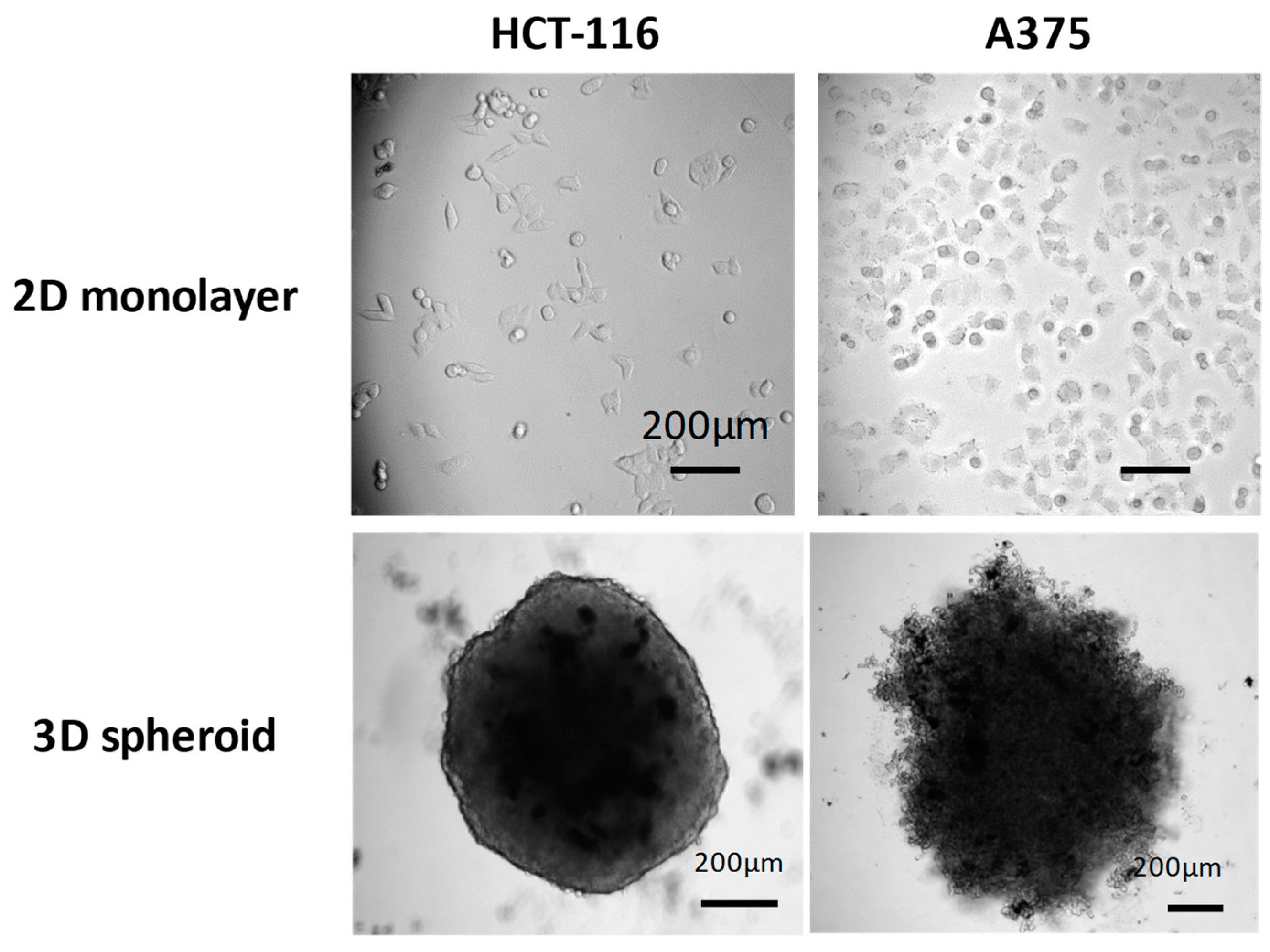
On 3D spheroid models, which are considered more relevant to in vivo situations, comparable phototoxicities were observed with both loaded PEO-PCL and PEO-PS micelles. The observed decrease in spheroid volume could be attributed to the death of the peripheral cell layer as previously described [
41]. At 2 days post irradiation, growth of the spheroids was restored. At 6 days post irradiation, spheroid cells had largely recovered their viability. This interesting result suggests that several irradiations might be necessary for a total eradication of the cancerous mass. Three-dimensional tumor spheroids, which simulate avascular tumor regions were found to be highly useful for understanding PDT mechanisms [
42]. In particular, studies on tumor spheroids have been helpful in visualizing and modeling uptake and penetration of photosensitizers through multiple cell layers. The majority of studies on free or encapsulated photosensitizer indicated nearly exclusive peripheral uptake, within the first layers of the spheroid [
21,
43,
44]. However, some photosensitizers such as EtNBS seemed to demonstrate a higher affinity for the acidic core of spheroids due to its cationic nature, which enables its efficient diffusion toward low pH gradients [
45,
46]. Since the vast majority of photosensitizers do not present this affinity for the acidic core of the spheroids, and localize preferentially on the outermost cell layers, multiples PDT treatments are useful for targeting more cell layers until the total eradication of tumor mass.
4. Materials and Methods
4.1. Materials
Poly (ethylene oxide)-b-poly (caprolactone) (PEO-PCL) 5k-4k, poly (ethylene oxide)-b-poly (styrene) (PEO-PS) 2,3k-3,1k were purchased from Polymer Source (Polymer Source, Inc., Dorval, Canada). 1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC) and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N- (lissamine rhodamine B sulphonyl) (ammonium salt) (Lys- Rhod-PE) come from Avanti Polar Lipids (Avanti Polar Lipids, Inc., USA). (N-(7-Nitrobenz-2-Oxa-1,3-Diazol-4-yl)-1,2-Dihexadecanoyl-sn-Glycero-3-Phosphoethanolamine, Triethylammonium Salt) was bought from Thermo Fisher (NBD-PE, N360, Waltham, MA, US). Pheophorbide-a was purchased from Wako (Japan). Chloroform, methanol, acetone, phosphate buffer (PBS, P4417) and carboxyfluorescein (CBF, 21877) were purchased from Sigma Aldrich (Merck, Darmstadt, Germany). The water used to prepare all PBS solutions was ultrapure water at 18 MΩ resistivity obtained with an ELGA Purelab Flex system (ELGA LabWater, High Wycombe, UK). Anthracene-9,10-dipropionic acid disodium salt (ADPA) was purchased from Santa Cruz Biotechnology (Dallas, TX, US).
4.2. Preparation of block-copolymer micelles and Large Unilamellar Vesicles
Block-copolymer micelles were prepared as reported elsewhere [
18]. A total of 20 mg of the copolymer were dispersed in 400 μL of acetone and this solution is slowly added to 5 mL of water whilst stirring. Acetone is left to evaporate during 48 h. For all samples loaded with Pheo, a solution of the photosensitizer in acetone is used to dissolve the polymer prior to its addition on the water solution. The molar ratio between the photosensitizer and the polymer was kept constant at 1/30 mol/mol to ensure the complete encapsulation. The hydrodynamic sizes of the carriers were determined by Dynamic Light Scattering (DLS) on a Malvern Zetasizer instrument, using the General Purpose, non-negative least square routine for data processing. PEO-PCL micelles had intensity-mean hydrodynamic diameter d
h = 29 ± 9 nm and PEO-PS micelles had d
h = 21 ± 7 nm.
The same protocol was used in the case of rhodamine labelled PEO-PCL. In that case, a 5mg/mL acetone stock solution of the rhodamine labelled copolymer (PEO-PCLrhod) was prepared and 400 μL of this solution were added to 20 mg of PEO-PCL to give 10% of labelled polymer in the micelles.
The Large unilamellar vesicles (LUVs) also called liposomes were prepared according to the traditional method of hydration of a dry lipid film followed by extrusion. First, the lipid is solubilized in chloroform in a haemolysis tube (20 mg·mL
−1). 1 mL of chloroform is evaporated first with a rotary evaporator followed by dynamic vacuum for a minimum of 4 h. Once the film is dry, 2 mL of filtered PBS is added to the tube to obtain a suspension of lipids at a concentration of 10 mg·mL
−1. For leakage experiments described in
Section 4.6 the dry film is hydrated with 2 mL of 60 mM CBF in PBS. After vortexing if necessary to finish peeling off the film from the bottom of the tube, the lipid suspension was put in a bath at 60 °C for 30 min and then for one h in an ultrasonic bath at 60 °C. The lipid suspension is then extruded through a 100 nm pore membrane (Nucleopore Track-Etch Membrane, Whatman, UK) using a hand extruder (Avanti Mini Extruder, Avanti Polar Lipids, Alabaster, AL, USA) to obtain a uniform LUV distribution. In the case of LUVs prepared for leakage experiments, steric exclusion separation was used and the samples were eluted twice through a column filled with Sephadex G 50 (GE Healthcare—Life Sciences, Pittsburgh, PA, USA) in order to separate the CBF loaded LUVs from the free CBF in solution. All fractions were controlled in DLS and all those containing LUVs were pooled. The consequent dilution of the samples was calculated by making the ratio of the total volume recovered and the volume of suspension initially injected into the column.
LUVs size was verified by DLS using the same routine as described above, their hydrodynamic size typically being around 100 nm in diameter. DOPC LUVs were used for the affinity constant measurements. In the case of FRET measurements DOPC LUVs contained 1% mol/mol of Liss Rhod PE or NBD-PE.
4.3. Association Constants
Association constants were derived by measuring the fluorescence emission of Pheo in the presence of different concentrations of the copolymer micelles.
For each series of measurements, a Pheo stock solution at 2 × 10−8 M in PBS was prepared by adding 30 μL of a solution of Pheo in acetone ([Pheo] = 0.01 mg·mL−1) to 25 mL of PBS. After stirring, the solution was flushed with argon for 10 min to drive off residual acetone and dissolved oxygen. Samples were prepared by adding different amounts of copolymer micelles (volumes between 5 and 500 μL of the stock copolymer solution, [copolymer] = 4 mg·mL−1), to 1 mL of the Pheo stock solution. The final total volume of 2 mL was reached by completing with PBS. Polymer concentrations varied between 1 and 100 µM and Pheo concentrations between 0.033 and 33 µM. All samples were bubbled with argon for 1–2 min to remove dissolved oxygen and then closed and incubated in the dark for a minimum of 3 h. After incubation, the fluorescence of the samples was measured by exciting them at 400 nm and measuring their emission from 600 to 780 nm. The fluorescence intensity at 675 nm was plotted as a function of the copolymers concentration in the samples. Measurements were repeated at least twice for each sample.
4.4. Förster Resonance Energy Transfer Measurements (FRET)
4.4.1. Pheo/rhodamine FRET
The samples containing a fixed rhodamine labelled liposomes concentration (0.01 mg·mL−1) and variable amounts of copolymer micelles loaded with Pheo (from 10 to 75 μL of a [copolymer] = 0.08 mg·mL−1 and from 15 to 50 μL of a [copolymer] =0.8 mg·mL−1) were used in order to change the molar ratio between copolymer and DOPC lipid from 0.03 to 1.8. In these samples, the rhodamine concentration was 0.12 μM (i.e., ~1% mol of lipid content).The samples were prepared by mixing the stock solutions in a 96-well plate (total volume 200 μL) and the fluorescence of the samples was followed for 10 h by exciting them at 560 nm and measuring their emission at 592 nm on a Synergy H1 plate reader (Biotek). For each studied system, samples at the same concentrations without LUV were measured and their fluorescence was subtracted from that of the micelles/LUV mixtures. The fluorescence values at 6 h were used for the analysis described above.
4.4.2. NBD/rhodamine FRET
Samples containing a fixed NBD-labelled LUVs concentration (0.0025 mg·mL−1) and variable amounts of micelles (from 10 to 250 μL [polymer] = 4 mg·mL−1) were prepared in order to change the molar ratio between copolymer and DOPC lipid from 0.6 to 16 and allowed to incubate for about 4 h. In these samples, the NBD concentration was 0.032 μM (i.e., ~1% mol of lipid content). After incubation, the fluorescence of the samples was measured by exciting them at 464 nm and measuring their emission from 470 to 750 nm. On these emission spectra, two peaks of interest appear: one at 578 nm belonging to the rhodamine labelled copolymer which partially overlaps to the 540 nm one relative to NBD in LUVs. For each studied system, samples at the same concentrations without liposomes were measured and their fluorescence was subtracted from that of the micelles/LUV mixtures.
We analysed the data as for the Pheo/Rhodamine FRET experiments and similarly to equations [
5] and [
6], we can write the equilibrium,
where
PEO-PCLrhod and NBD are the rhodamine and NBD at distances larger than 4 nm and
PEO-PCLrhod:NBD is the rhodamine/NBD couple undergoing to resonance energy transfer. When NBD is excited at 464 nm, the rhodamine signal appears only when the FRET occurs. In a similar way as before, the associated constant is written as,
and obtained from the plot of the rhodamine maximum fluorescence intensity (λ = 578 nm) increase as a function of copolymer concentration (see
Figure S10). As in [
7] we can express the fluorescence signal as a function of the concentration of each species and a fluorescence proportionality constant:
, are the fluorescence intensity of NBD and PEO-PCLrhod coupled to NBD at a given wavelength. and are the fluorescence proportionality constants linking NBD and PEO-PCLrhod:NBD to their concentration. is experimentally evaluated from the values of the spectrum in the absence of the fluorescent polymer. and are the fitted parameters.
4.5. Singlet Oxygen Quantum Yield
The singlet oxygen quantum yield has been measured for each system (pheo in PBS, Pheo loaded PEO-PS and Pheo loaded PEO-PCL) though an indirect method by using a water-soluble anthracene, ADPA. In order to get the singlet oxygen quantum yield () methylene blue was used as a reference photosensitizer with a known in water. [Pheo] = [methylene blue] = 1 μM, ADPA concentration was 20 μM. In the presence of singlet oxygen, ADPA is oxidized to form an endoperoxide which no longer absorbs between 300 and 420 nm. We then monitored the UV absorbance at 400 nm during irradiation with a LED at 656 nm (35 mW), the decrease of ADPA absorbance as a function of time was followed during 1 h and was modelled with an exponential function to determine its characteristic oxidation rate.
The quantum yields of singlet oxygen production,
, are calculated in the case of Pheo in PBS, and Pheo loaded in the copolymer micelles according to:
and
represent the photobleaching rates of Pheo and methylene blue, respectively;
and
are the optical densities at irradiation wavelength of Pheo and methylene blue, respectively (
).
All UV-Visible absorbance measurements (also those during irradiation) were performed using a diode array spectrometer (HP 8452A, Hewlett-Packard, Hayward, CA, USA) driven using SpectralWork software (Olis, Bogart, GA, USA).
4.6. Leakage Experiments
LUVs loaded with CBF were prepared as described above. LUVs containing CBF were mixed with Pheo loaded or not in the copolymer micelles (Pheo loaded PEO-PCL, Pheo loaded PEO-PS, and free Pheo). The total volume of the resulting PBS solution was 2 mL and [DOPC] = 120 μM, [Pheo] = 1 μM. After mixing, the solutions were incubated for 30 min in the dark. After incubation, the mixture was stirred and irradiated for 30 min using an LED at 656 nm (35 mJ·cm−²). The fluorescence of the samples was measured by exciting the CBF at 492 nm and looking at the emission between 500 and 800 nm. The fluorescence was measured before and after the addition of micelles or Pheo solution, and then for 15–20 h after irradiation. After this time, 100 μL of 10% Triton × 100 in PBS were added to the mixture to destabilize the liposomes and release all the encapsulated CBF. Fluorescence was measured again.
The leakage as a function of time is defined as the CBF fraction which exits the liposomes:
is the fluorescence intensity (λ = 518 nm) of the mixture at a given time and is proportional to the CBF concentration outside the LUVs. is normalized by the fluorescence intensity (λ = 518 nm) after addition of Triton X, , which is proportional to the total amount of CBF initially inside the LUVs.
Both and are corrected by the fluorescence intensity (λ = 518 nm) before adding the micelles or the Pheo solution so to take into account the possible presence of residual CBF outside the liposomes.
The follow-up of the leakage as a function of time allows us to calculate the permeability coefficient, P, of the liposomes using the formula,
with
where
is the system’s maximum leakage at t ∞,
is the characteristic time of leakage, and r the liposomes radius.
4.7. Oxidation Experiments
For lipid oxidation experiments, DOPC LUVs and co-polymer micelles were prepared as described above. For typical experiments, LUVs were at a concentration such that [DOPC] = 300 µM, and micelles at a concentration such that [Pheo] = 1 µM, in a total volume of 2 mL of PBS. One sample was prepared for each time of irradiation studied. Oxygen was gently bubbled in the samples throughout irradiation so as to maintain its concentration constant. The red LED, used to irradiate, was set at 35 mJ·cm−². Following irradiation, the samples were bubbled with Argon to replace O2 and stored at 4 °C in the dark. The lipids were extracted from this medium following Bilgh and Dyer. 1 mL of MeOH was added to each sample, followed by 2 mL of CHCl3 and vigorous shaking. The organic phase was collected and dried. The solid residue was then dissolved in 1 mL of MeOH and diluted until a theoretical concentration of [lipid] = 0.24 mM was obtained. A total of 3 µL of these solutions were then injected in an Acquity UPLC-MS chain (Waters, Manchester, UK) equipped with a BEH-C8 column (1.7 mm; 2.1 × 50 mm). Elution was achieved with a gradient of 8 mM ammonium acetate and methanol. Total run time was 19 min. Detection and quantification was achieved using a Xevo G2 QTOF mass spectrometer (Waters, UK). The QTof-MS was operated in the positive-ion mode with a capillary voltage of 2.5 kV, a cone voltage of 50 V, the cone gas flow of 20 L·h−1, the source temperature set to 130 °C and the desolvation temperature set to 450 °C.
4.8. Cell Culture
Human HCT-116 colorectal cancer cells (CCL-247) and A375 melanoma cancer cells (CRL-1619) were purchased from ATCC. Cells were grown in DMEM containing 4.5 g·L−1 glucose, GLUTAMax and pyruvate, supplemented with 10% of heat-inactivated fetal bovine serum and 100 U/mL penicillin, and 100 µg·mL−1 streptomycin. Cells were maintained at 37 °C in a humidified atmosphere containing 5% CO2. Cell culture media were changed three times a week. Both cell types tested negative for mycoplasma using MycoAlert mycoplasma detection kit (Lonza, Basel, Switzerland) all along the experiments.
4.9. Determination by Flow Cytometry of Photosensitizer Penetration in Cells Grown in Monolayer
The day prior to experimentation, 80,000 cells were seeded in 12-well plates. These cells were incubated for 24 h, 6 h, 4 h, 2 h, 1 h and 30 min with the two different types of Pheo loaded micelles (PEO-PCL, PEO-PS) or with free Pheo. 4 biological replicates were used in each condition. In this experiment, the final concentration of Pheo was set at 0.33 µM, which equals 10 µM of polymer. After the last incubation time, cells were washed with 2 mL of PBS, gently trypsinized and dispatched in a 96-well plate U bottom, kept on ice bed. Cells in suspension were then analyzed on a BD LSRFortessa X-20 flow cytometer, equipped with an HTS module for plate reading. At least 20,000 events were acquired in each well. Flow cytometry allows the quantification of Pheo fluorescence within the cells, meaning when localized either in the plasma membrane or intracellularly. Using this method, no speculation can be made on whether Pheo is still associated with the polymers or free in the cell.
4.10. Determination by Flow Cytometry of Rhodamine-PEO PCL Polymer Penetration in Cells Grown in Monolayer
As described in
Section 4.9, 80,000 HCT-116 or A375 cells were seeded in 12-well plates the day before the experiment. The cells were incubated for 24 h, 6 h, 4 h, 2 h, 1 h and 30 min with micelles produced with PEO-PCL, covalently-linked to rhodamine, at a ratio of 1/10 with unlabeled PEO-PCL. 4 biological replicates were used in each condition. In this experiment, the final concentration of PEO-PCL polymer was set at 10 µM, as in the previous experiment on photosensitizer penetration quantified by flow cytometry. After the last incubation time, cells were washed with 2 mL of PBS, gently trypsinized and dispatched in a 96-well plate U bottom, kept on a bed of ice. Cells in suspension were then analyzed on a BD LSRFortessa X-20 flow cytometer, equipped with an HTS module for plate reading. At least 20,000 events were acquired in each well. Flow cytometry allows the quantification of Rhodamine-PEO-PCL polymer fluorescence within the cells (either in the plasma membrane or intracellular). Using this method, no speculation can be made on whether PEO-PCL is still self-assembled as micelles or dispersed in unimer form.
4.11. Cell Viability Assessement after Photodynamic Therapy of Monolayers
The day before the experiments, 4,000 HCT-116 or A375 cells were seeded in 96-well plates. The day of experiment, cells were incubated with empty PEO-PCL micelles, Pheo loaded PEO-PCL micelles, empty PEO-PS micelles, Pheo loaded PEO-PS micelles or free Pheo for 30 min at 37 °C. Concentration of Pheo was set to 0.33 µM and polymer concentration was accordingly set to 10 µM. In light-irradiated conditions, after this 30 min incubation time with micelles, the 96-well plate was deposited on an overhead projector lamp with a band-pass filter (λ > 400 nm). Cells were photo-irradiated for a total of 4 min (2 min light on, 2 min light off and then 2 min light on). Each well received 8.2 J·cm−2. After illumination, cells were incubated for 24 h at 37 °C. Viability was then assessed using PrestoBlue reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions, and read on plate reader Synergy H1 (Biotek, Winooski, VT, USA).
4.12. Photodynamic Therapy of 3D Tumor Spheroids
HCT-116 and A375 spheroids were incubated with Pheo loaded PEO-PCL micelles, Pheo loaded PEO-PS micelles or free Pheo for 30 min at 37 °C. Concentration of Pheo was set at 3 µM as previously described for PDT in 3D spheroids [
21]. Spheroids were irradiated for 8 min (4 min light on, 4 min light off and then 4 min light on) using the same overhead projector lamp as for irradiation of monolayers. Spheroids treated by PDT were observed by optical microscopy (AxioObserver A1 equipped with a 5× plane N objective in phase contrast) and photographed over one week and their areas were determined using Image J software (NIH, Bethesda, MD, USA) as previously described [
18,
47].
4.13. Cell Viability Assessement after Photodynamic Therapy of Spheroids
The growth curve of spheroids was followed macroscopically over six days. Six days after PDT treatment, the cell viability of spheroids was assessed by quantifying intracellular ATP content using CellTiterGlo3D luminescent kit (Promega, Madison, WI, USA), according to the manufacturer protocols, as previously described [
48]. Briefly, after taking the last picture of the growth curve, cell culture medium was removed so that only 50 µL of supernatant were left with the spheroid in the well. The same volume of reagent was added and incubated under gentle shaking at room temperature for 30 min. After spheroid lysis, the whole content of each well (i.e., 100 µL) was transferred into a white-96-well plates and luminescence was read on plate reader Synergy H1 (Biotek).
4.14. Statistics
The number of biological replicates (n) and number of independent repetitions of the experiments (N) is indicated in the figure legends. Statistical differences between values were assessed either by one-way or two-way ANOVA, followed by Tukey’s multiple comparison test, which compares the mean of each condition with all other conditions. The statistical analysis were performed with GraphPad Prism 8 (GraphPad Software, San Diego, CA, USA).

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







