The IL-1 Antagonist Anakinra Attenuates Glioblastoma Aggressiveness by Dampening Tumor-Associated Inflammation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Anakinra Dampened IL-1β-Induced Inflammatory Gene Expression in GBM and PBMC
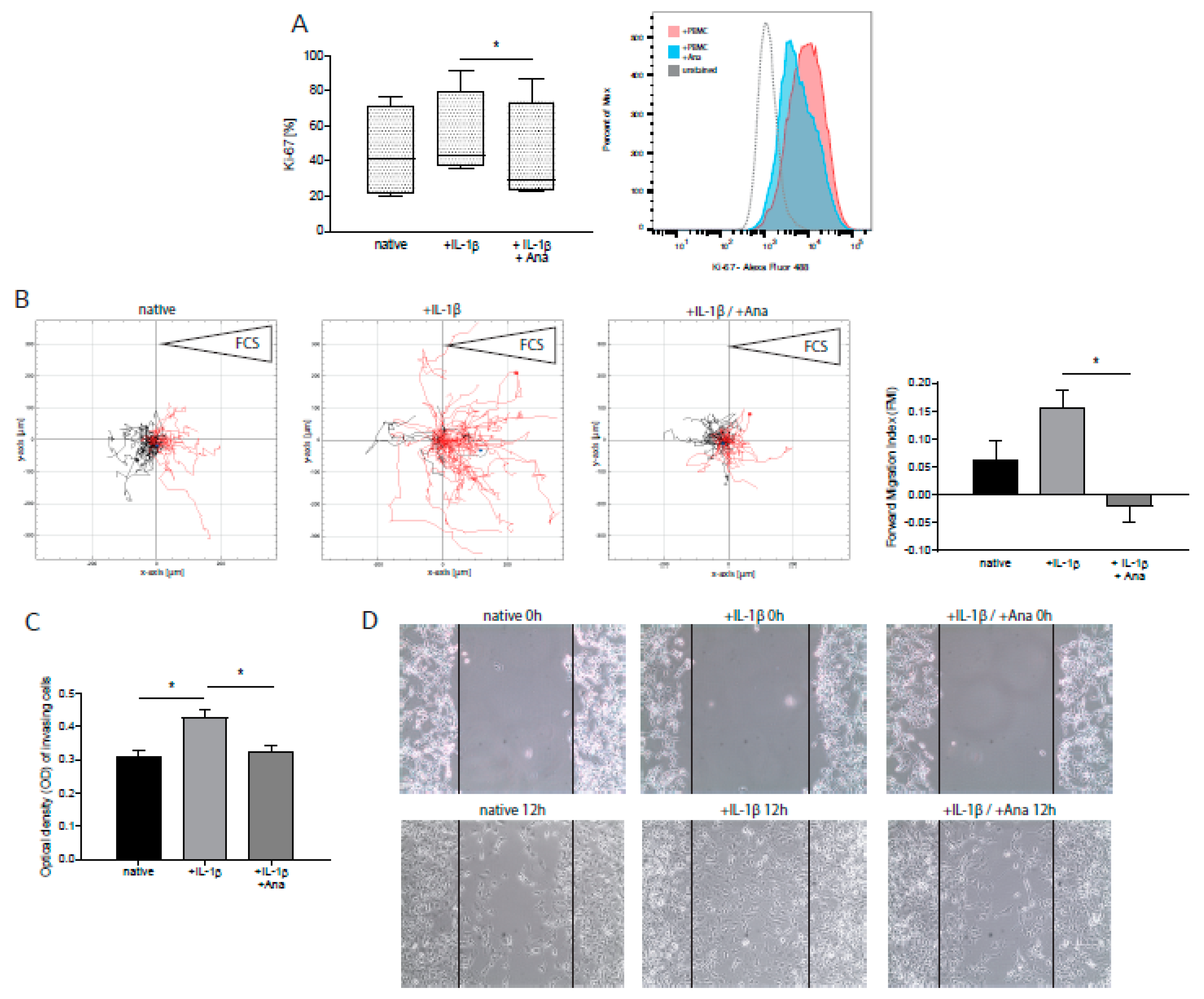
2.2. Anakinra Inhibited IL-1β-Induced Tumor Proliferation and Migration
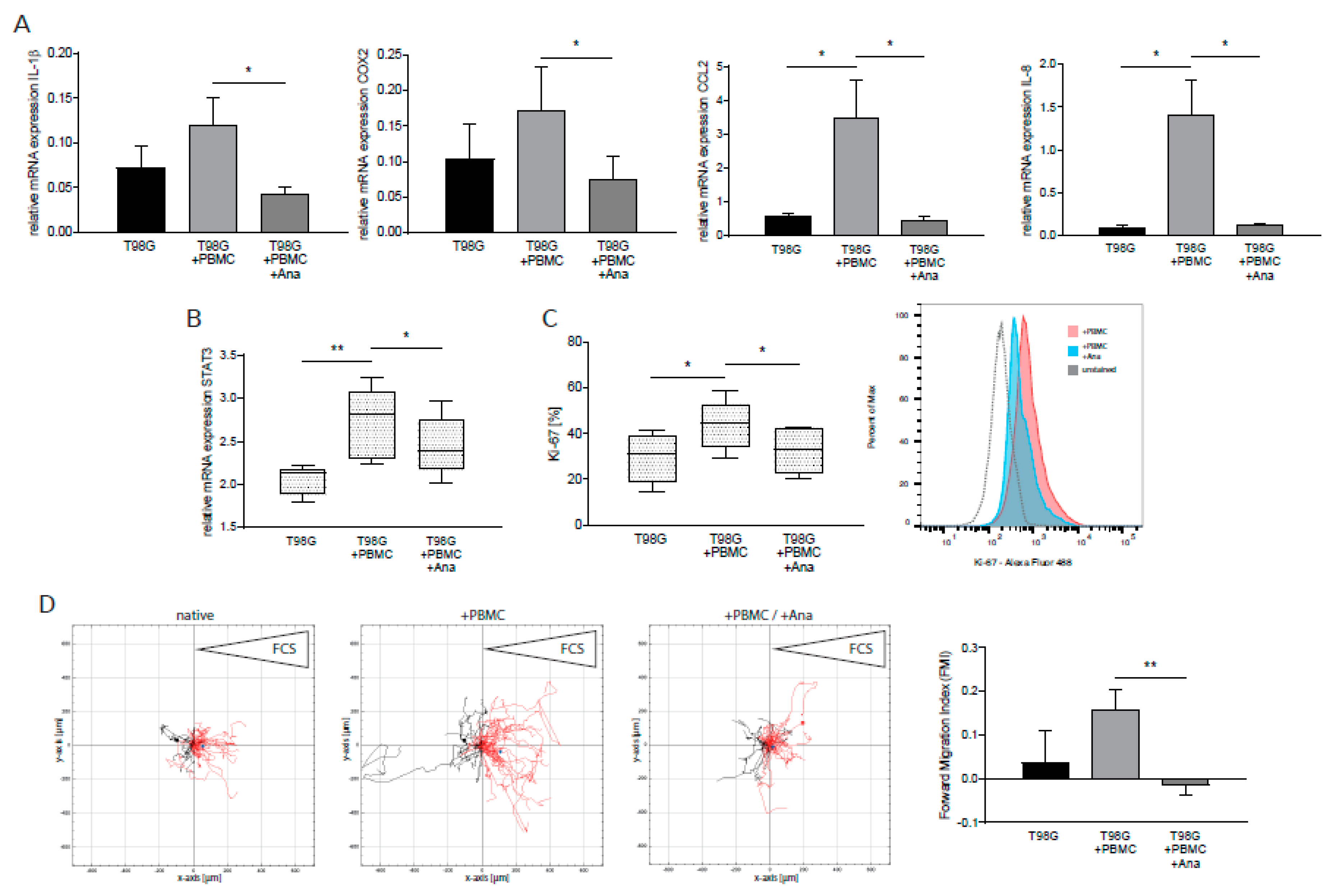
2.3. Anakinra Dampened Inflammatory Crosstalk between GBM and Immune Cells, Resulting in a Less Aggressive GBM Phenotype
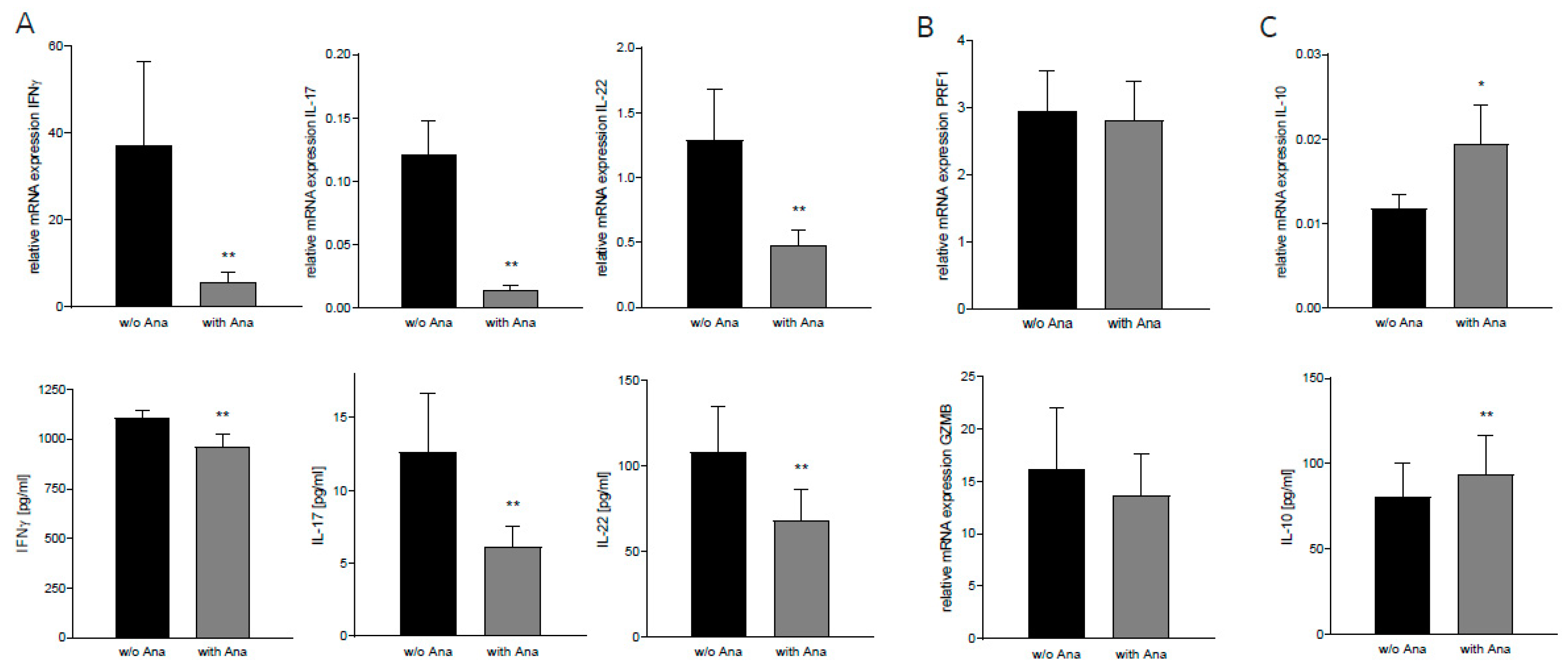
2.4. Anakinra Reduced Inflammatory Signaling in Lymphocytes Co-Cultured With GBM Cells
3. Discussion
4. Materials and Methods
4.1. Human Tissue Sample
4.2. Cell Culture and Reagents
4.3. PBMC Isolation
4.4. Co-Cultures
4.5. T-Cell Isolation
4.6. RNA Extraction and cDNA Synthesis
4.7. Quantitative RT- PCR
4.8. Flow Cytometry
4.9. Chemotaxis Assay
4.10. Enzyme-Linked Immunosorbent Assay
4.11. Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis
4.12. Transwell Invasion Assay
4.13. Wound Healing Migration Assay
4.14. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Taniguchi, K.; Karin, M. NF-B, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Shalapour, S.; Karin, M. Pas de Deux: Control of Anti-tumor Immunity by Cancer-Associated Inflammation. Immunity 2019, 51, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Multhoff, G.; Molls, M.; Radons, J. Chronic inflammation in cancer development. Front. Immunol. 2012, 2, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shalapour, S.; Karin, M. Immunity, inflammation, and cancer: An eternal fight between good and evil. J. Clin. Invest. 2015, 125, 3347–3355. [Google Scholar] [CrossRef] [Green Version]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Todoric, J.; Karin, M. The Fire within: Cell-Autonomous Mechanisms in Inflammation-Driven Cancer. Cancer Cell 2019, 35, 714–720. [Google Scholar] [CrossRef]
- Wang, D.; Du Bois, R.N. Immunosuppression associated with chronic inflammation in the tumor microenvironment. Carcinogenesis 2015, 36, 1085–1093. [Google Scholar] [CrossRef] [Green Version]
- Maimela, N.R.; Liu, S.; Zhang, Y. Fates of CD8+ T cells in Tumor Microenvironment. Comput. Struct. Biotechnol. J. 2019, 17, 1–13. [Google Scholar] [CrossRef]
- Garlanda, C.; Dinarello, C.A.; Mantovani, A. The Interleukin-1 Family: Back to the Future. Immunity 2013, 39, 1003–1018. [Google Scholar] [CrossRef] [Green Version]
- Dinarello, C.A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 2018, 281, 8–27. [Google Scholar] [CrossRef]
- Tarassishin, L.; Casper, D.; Lee, S.C. Aberrant expression of interleukin-1β and inflammasome activation in human malignant gliomas. PLoS ONE 2014, 9, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Depping, R.; Jelkmann, W. Interleukin-1β promotes hypoxia-induced apoptosis of glioblastoma cells by inhibiting hypoxia-inducible factor-1 mediated adrenomedullin production. Cell Death Dis. 2014, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeung, Y.T.; McDonald, K.L.; Grewal, T.; Munoz, L. Interleukins in glioblastoma pathophysiology: Implications for therapy. Br. J. Pharmacol. 2013, 168, 591–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavalli, G.; Dinarello, C.A. Anakinra therapy for non-cancer inflammatory diseases. Front. Pharmacol. 2018, 9, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinarello, C.A. Why not treat human cancer with interleukin-1 blockade? Cancer Metastasis Rev. 2010, 29, 317–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Litmanovich, A.; Khazim, K.; Cohen, I. The Role of Interleukin-1 in the Pathogenesis of Cancer and its Potential as a Therapeutic Target in Clinical Practice. Oncol. Ther. 2018, 6, 109–127. [Google Scholar] [CrossRef] [Green Version]
- Tarassishin, L.; Lim, J.; Weatherly, D.B.; Angeletti, R.H.; Lee, S.C. Interleukin-1-induced changes in the glioblastoma secretome suggest its role in tumor progression. J. Proteomics 2014, 99, 152–168. [Google Scholar] [CrossRef] [Green Version]
- Chahal, M.; Abdulkarim, B.; Xu, Y.; Guiot, M.C.; Easaw, J.C.; Stifani, N.; Sabri, S. O(6)-methylguanine-DNA methyltransferase is a novel negative effector of invasion in glioblastoma multiforme. Mol. Cancer Ther. 2012, 11, 2440–2450. [Google Scholar] [CrossRef] [Green Version]
- Sobecki, M.; Mrouj, K.; Camasses, A.; Parisis, N.; Nicolas, E.; Llères, D.; Gerbe, F.; Prieto, S.; Krasinska, L.; David, A.; et al. The cell proliferation antigen Ki-67 organises heterochromatin. Elife 2016, 5. [Google Scholar] [CrossRef]
- Li, N.; Grivennikov, S.I.; Karin, M. The Unholy Trinity: Inflammation, Cytokines, and STAT3 Shape The Cancer Microenvironment. Cancer Cell 2011, 19, 429–431. [Google Scholar] [CrossRef] [Green Version]
- Brantley, E.C.; Benveniste, E.N. Signal transducer and activator of transcription-3: A molecular hub for signaling pathways in gliomas. Mol. Cancer Res. 2008, 6, 675–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, N.; Hee, S.; Kong, D.; Won, H. The role of STAT3 in glioblastoma progression through dual in fl uences on tumor cells and the immune microenvironment. Mol. Cell. Endocrinol. 2017, 451, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Del Principe, M.I.; Dal Bo, M.; Bittolo, T.; Buccisano, F.; Rossi, F.M.; Zucchetto, A.; Rossi, D.; Bomben, R.; Maurillo, L.; Cefalo, M.; et al. Clinical significance of bax/bcl-2 ratio in chronic lymphocytic leukemia. Haematologica 2016, 101, 77–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, F.E.; Ironside, J.W.; Gregor, A.; Wyatt, B.B.; Stewart, M.; Rye, R.; Adams, J.; Potts, H.W.W. The prognostic influence of bcl-2 in malignant glioma. Br. J. Cancer 2002, 86, 1899–1904. [Google Scholar] [CrossRef] [Green Version]
- Voigt, C.; May, P.; Gottschlich, A.; Markota, A.; Wenk, D.; Gerlach, I.; Voigt, S.; Stathopoulos, G.T.; Arendt, K.A.M.; Heise, C.; et al. Cancer cells induce interleukin-22 production from memory CD4 + T cells via interleukin-1 to promote tumor growth. Proc. Natl. Acad. Sci. USA 2017, 114, 12994–12999. [Google Scholar] [CrossRef] [Green Version]
- Akil, H.; Abbaci, A.; Lalloué, F.; Bessette, B.; Costes, L.M.M.; Domballe, L.; Charreau, S.; Guilloteau, K.; Karayan-Tapon, L.; Bernard, F.X.; et al. IL22/IL-22R pathway induces cell survival in human glioblastoma cells. PLoS ONE 2015, 10, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Parajuli, P.; Mittal, S. Role of IL-17 in Glioma Progression. J. Spine Neurosurg. 2013, 10. [Google Scholar] [CrossRef]
- Bonnin, D.A.A.; Havrda, M.C.; Lee, M.C.; Liu, H.; Zhang, Z.; Nguyen, L.N.; Harrington, L.X.; Hassanpour, S.; Cheng, C.; Israel, M.A. Secretion-mediated STAT3 activation promotes self-renewal of glioma stem-like cells during hypoxia. Oncogene 2018, 37, 1107–1118. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Tian, L.; Han, Y.; Vogelbaum, M.; Stark, G.R. Dose-dependent cross-talk between the transforming growth factor-β and interleukin-1 signaling pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 4365–4370. [Google Scholar] [CrossRef] [Green Version]
- Fathima Hurmath, K.; Ramaswamy, P.; Nandakumar, D.N. IL-1β microenvironment promotes proliferation, migration and invasion of human glioma cells. Cell Biol. Int. 2014, 38, 1415–1422. [Google Scholar] [CrossRef]
- Gurgis, F.M.S.; Yeung, Y.T.; Tang, M.X.M.; Heng, B.; Buckland, M.; Ammit, A.J.; Haapasalo, J.; Haapasalo, H.; Guillemin, G.J.; Grewal, T.; et al. The p38-MK2-HuR pathway potentiates EGFRvIII-IL-1β-driven IL-6 secretion in glioblastoma cells. Oncogene 2015, 34, 2934–2942. [Google Scholar] [CrossRef] [PubMed]
- Christofides, A.; Kosmopoulos, M.; Piperi, C. Pathophysiological mechanisms regulated by cytokines in gliomas. Cytokine 2015, 71, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Shi, Z.; Jiang, J. Cyclooxygenase-2 in glioblastoma multiforme. Drug Discov. Today 2017, 22, 148–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, P.J.; Thomas, R.; Kingsley, P.J.; Shimizu, F.; Montrose, D.C.; Marnett, L.J.; Tabar, V.S.; Dannenberg, A.J.; Benezra, R. Cox-2-derived PGE2 induces Id1-dependent radiation resistance and self-renewal in experimental glioblastoma. Neuro. Oncol. 2016, 18, 1379–1389. [Google Scholar] [CrossRef] [Green Version]
- Xue, L.; Lu, B.; Gao, B.; Shi, Y.; Xu, J.; Yang, R.; Xu, B.; Ding, P. NLRP3 promotes glioma cell proliferation and invasion via the interleukin-1b/NF-kB p65 signals. Oncol. Res. 2019, 27, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.L.; Miska, J.; Wainwright, D.A.; Dey, M.; Rivetta, C.V.; Yu, D.; Kanojia, D.; Pituch, K.C.; Qiao, J.; Pytel, P.; et al. CCL2 produced by the glioma microenvironment is essential for the recruitment of regulatory t cells and myeloid-derived suppressor cells. Cancer Res. 2016, 76, 5671–5682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasan, T.; Caragher, S.P.; Shireman, J.M.; Park, C.H.; Atashi, F.; Baisiwala, S.; Lee, G.; Guo, D.; Wang, J.Y.; Dey, M.; et al. Interleukin-8/CXCR2 signaling regulates therapy-induced plasticity and enhances tumorigenicity in glioblastoma. Cell Death Dis. 2019, 10, 292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Infanger, D.W.; Cho, Y.J.; Lopez, B.S.; Mohanan, S.; Liu, S.C.; Gursel, D.; Boockvar, J.A.; Fischbach, C. Glioblastoma stem cells are regulated by interleukin-8 signaling in a tumoral perivascular niche. Cancer Res. 2013, 73, 7079–7089. [Google Scholar] [CrossRef] [Green Version]
- Nduom, E.K.; Wei, J.; Yaghi, N.K.; Huang, N.; Kong, L.Y.; Gabrusiewicz, K.; Ling, X.; Zhou, S.; Ivan, C.; Chen, J.Q.; et al. PD-L1 expression and prognostic impact in glioblastoma. Neuro. Oncol. 2016, 18, 195–205. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.Q.; Liu, F.; Qiu, X.Y.; Chen, X.Q. The prognostic and therapeutic value of PD-L1 in glioma. Front. Pharmacol. 2019, 9, 1503. [Google Scholar] [CrossRef] [Green Version]
- Woroniecka, K.I.; Rhodin, K.E.; Chongsathidkiet, P.; Keith, K.A.; Fecci, P.E. T-Cell dysfunction in glioblastoma: Applying a new framework. Clin. Cancer Res. 2018, 24, 3792–3802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broekman, M.L.; Maas, S.L.N.; Abels, E.R.; Mempel, T.R.; Krichevsky, A.M.; Breakefield, X.O. Multidimensional communication in the microenvirons of glioblastoma. Nat. Rev. Neurol. 2018, 14, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Tang, Y.; Hua, S. Immunological approaches towards cancer and inflammation: A cross talk. Front. Immunol. 2018, 9, 563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Q.; Diao, S.; Wang, Q.; Zhu, C.; Sun, X.; Yin, B.; Zhang, X.; Meng, X.; Wang, B. IL-17A promotes cell migration and invasion of glioblastoma cells via activation of PI3K/AKT signalling pathway. J. Cell. Mol. Med. 2018, 23, 357–369. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, P.; Gronke, K.; Diefenbach, A. A catch-22: Interleukin-22 and cancer. Eur. J. Immunol. 2018, 48, 15–31. [Google Scholar] [CrossRef] [Green Version]
- Mojic, M.; Takeda, K.; Hayakawa, Y. The dark side of IFN-γ: Its role in promoting cancer immunoevasion. Int. J. Mol. Sci. 2018, 19, 89. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Ge, J.; Xiang, B.; Wu, X.; Ma, J.; Zhou, M.; Li, X.; et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol. Cancer 2019, 18, 10. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, C.; Knight, A.; Nordström, D.; Pettersson, T.; Fransson, J.; Florin-Robertsson, E.; Pilström, B. Injection-site reactions upon Kineret (anakinra) administration: Experiences and explanations. Rheumatol. Int. 2012, 32, 295–299. [Google Scholar] [CrossRef] [Green Version]
- Nikfar, S.; Saiyarsarai, P.; Tigabu, B.M.; Abdollahi, M. Efficacy and safety of interleukin-1 antagonists in rheumatoid arthritis: a systematic review and meta-analysis. Rheumatol. Int. 2018, 38, 1363–1383. [Google Scholar] [CrossRef]
- Niu, X.; He, D.; Deng, S.; Li, W.; Xi, Y.; Xie, C.; Jiang, T.; Zhang, J.Z.; Dong, C.; Chen, G. Regulatory immune responses induced by IL-1 receptor antagonist in rheumatoid arthritis. Mol. Immunol. 2011, 49, 290–296. [Google Scholar] [CrossRef]
- Ruscitti, P.; Masedu, F.; Alvaro, S.; Airò, P.; Battafarano, N.; Cantarini, L.; Cantatore, F.P.; Carlino, G.; D’Abrosca, V.; Frassi, M.; et al. Anti-interleukin-1 treatment in patients with rheumatoid arthritis and type 2 diabetes (TRACK): A multicentre, open-label, randomised controlled trial. PLoS Med. 2019, 16. [Google Scholar] [CrossRef] [PubMed]
- Galea, J.; Ogungbenro, K.; Hulme, S.; Greenhalgh, A.; Aarons, L.; Scarth, S.; Hutchinson, P.; Grainger, S.; King, A.; Hopkins, S.J.; et al. Intravenous anakinra can achieve experimentally effective concentrations in the central nervous system within a therapeutic time window: Results of a dose-ranging study. J. Cereb. Blood Flow Metab. 2011, 31, 439–447. [Google Scholar] [CrossRef]
- Helmy, A.; Guilfoyle, M.R.; Carpenter, K.L.H.; Pickard, J.D.; Menon, D.K.; Hutchinson, P.J. Recombinant human interleukin-1 receptor antagonist in severe traumatic brain injury: A phase II randomized control trial. J. Cereb. Blood Flow Metab. 2014, 34, 845–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, S.R.; McMahon, C.J.; Gueorguieva, I.; Rowland, M.; Scarth, S.; Georgiou, R.; Tyrrell, P.J.; Hopkins, S.J.; Rothwell, N.J. Interleukin-1 receptor antagonist penetrates human brain at experimentally therapeutic concentrations. J. Cereb. Blood Flow Metab. 2008, 28, 387–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hübner, M.; Hinske, C.L.; Effinger, D.; Wu, T.; Thon, N.; Kreth, F.W.; Kreth, S. Intronic miR-744 inhibits glioblastoma migration by functionally antagonizing its host gene MAP2K4. Cancers (Basel). 2018, 10, 400. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hübner, M.; Effinger, D.; Wu, T.; Strauß, G.; Pogoda, K.; Kreth, F.-W.; Kreth, S. The IL-1 Antagonist Anakinra Attenuates Glioblastoma Aggressiveness by Dampening Tumor-Associated Inflammation. Cancers 2020, 12, 433. https://doi.org/10.3390/cancers12020433
Hübner M, Effinger D, Wu T, Strauß G, Pogoda K, Kreth F-W, Kreth S. The IL-1 Antagonist Anakinra Attenuates Glioblastoma Aggressiveness by Dampening Tumor-Associated Inflammation. Cancers. 2020; 12(2):433. https://doi.org/10.3390/cancers12020433
Chicago/Turabian StyleHübner, Max, David Effinger, Tingting Wu, Gabriele Strauß, Kristin Pogoda, Friedrich-Wilhelm Kreth, and Simone Kreth. 2020. "The IL-1 Antagonist Anakinra Attenuates Glioblastoma Aggressiveness by Dampening Tumor-Associated Inflammation" Cancers 12, no. 2: 433. https://doi.org/10.3390/cancers12020433