P-REX1-Independent, Calcium-Dependent RAC1 Hyperactivation in Prostate Cancer





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
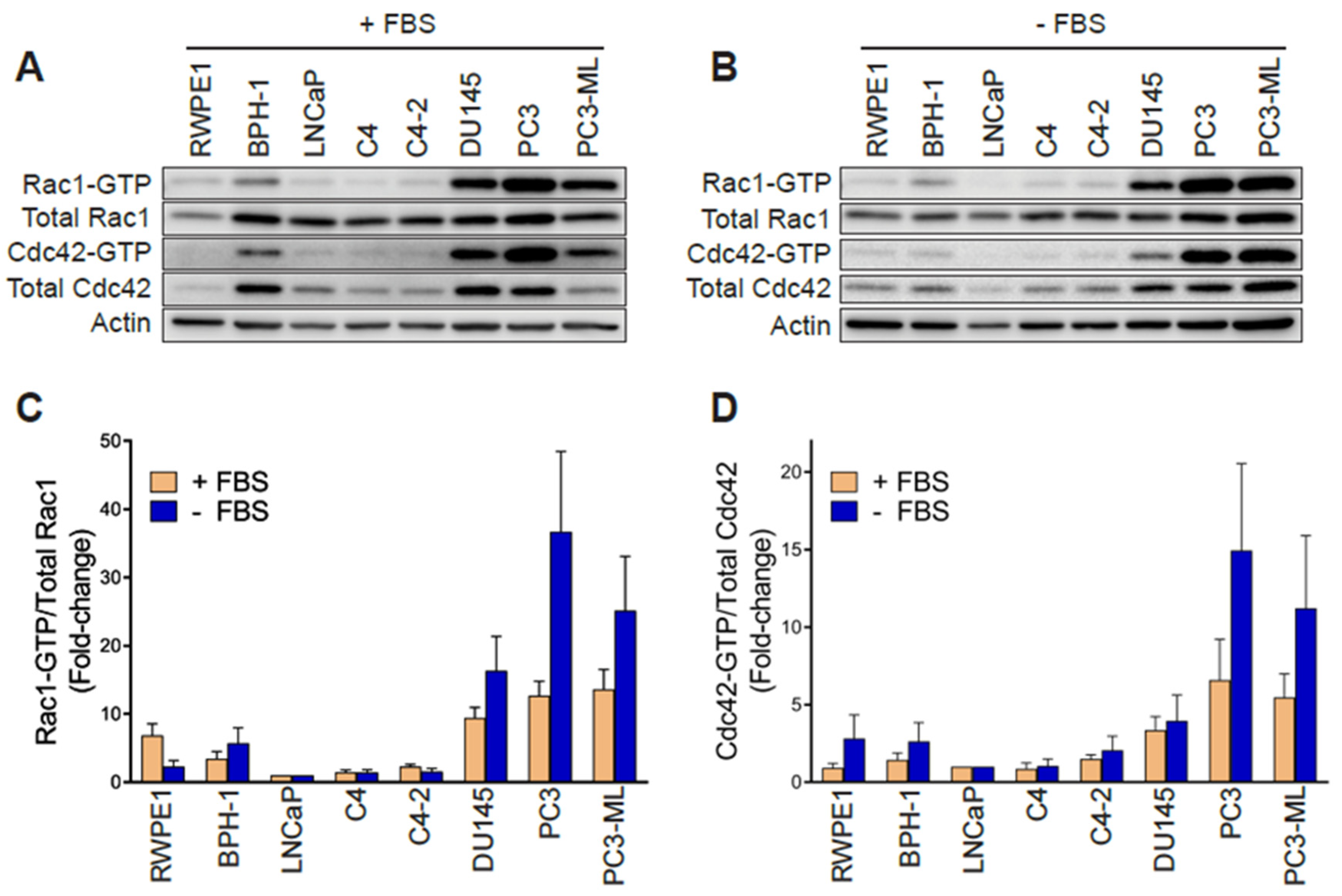
2.1. Aggressive Androgen-Independent Prostate Cancer Cells Display Elevated Rac1-GTP Levels
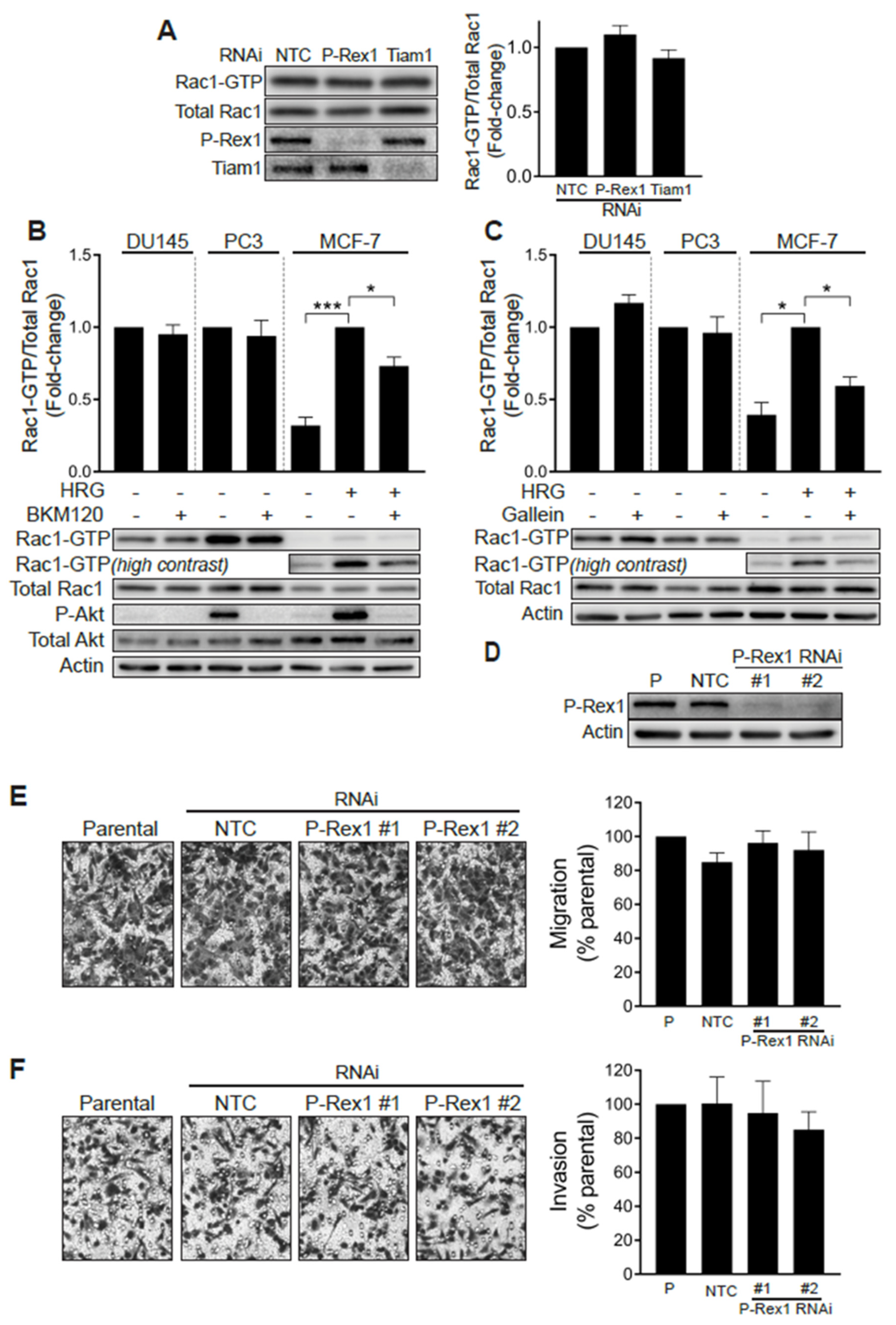
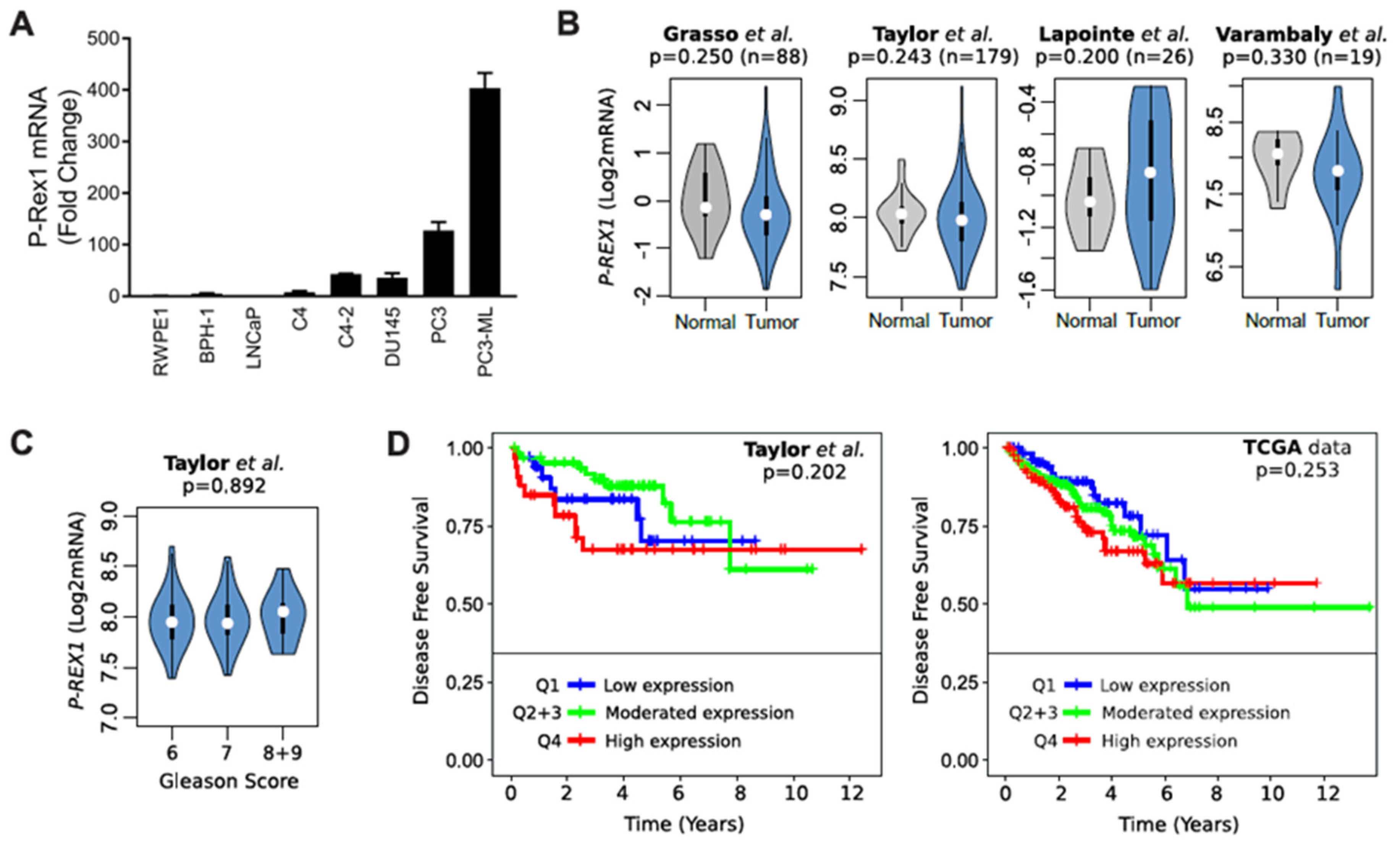
2.2. Elevated Rac1-GTP in Prostate Cancer Is Independent of P-Rex1
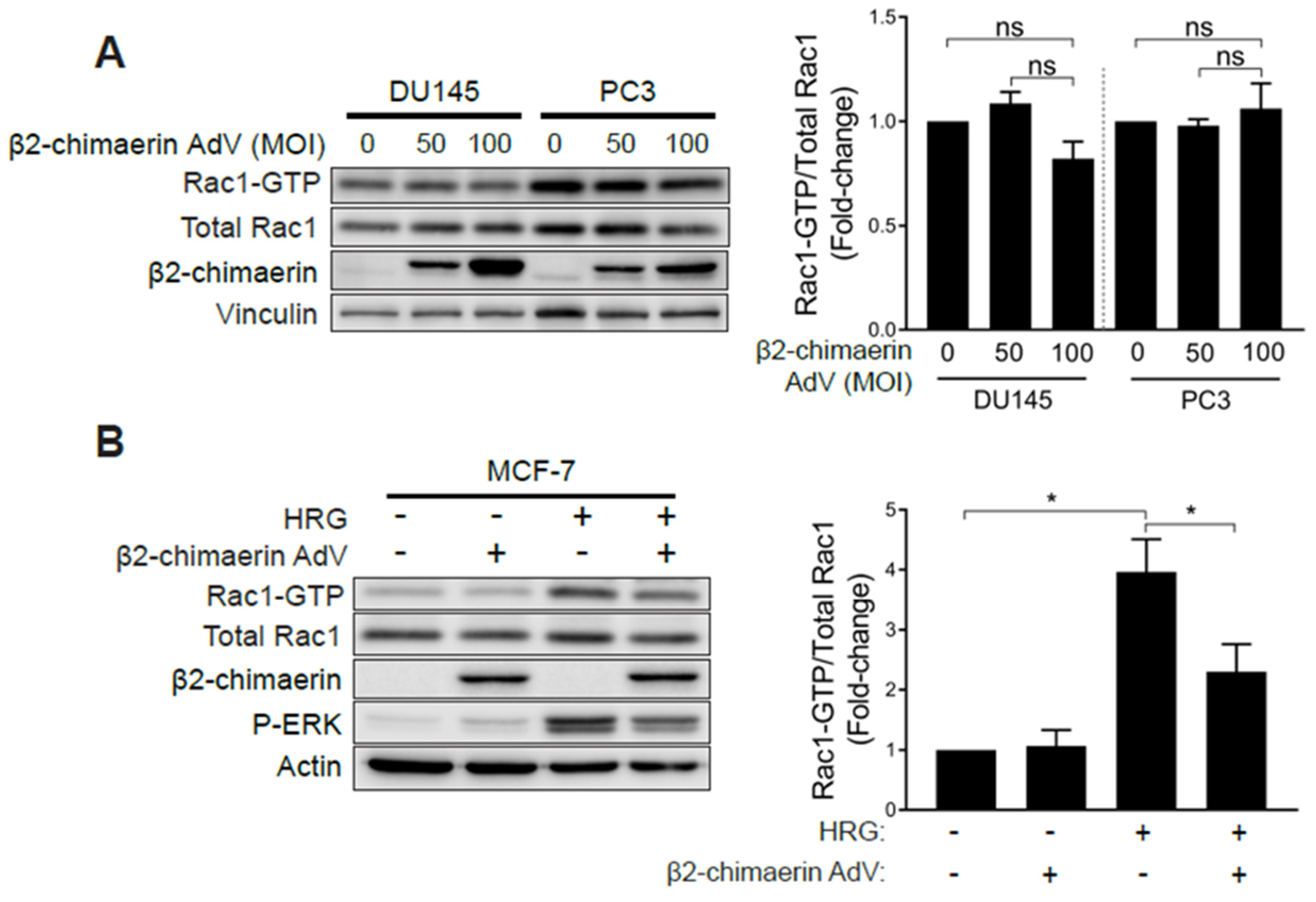
2.3. Elevated Rac1 Activation in Androgen-Independent Prostate Cancer Cells Is Insensitive to the Rac-GAP β2-Chimaerin
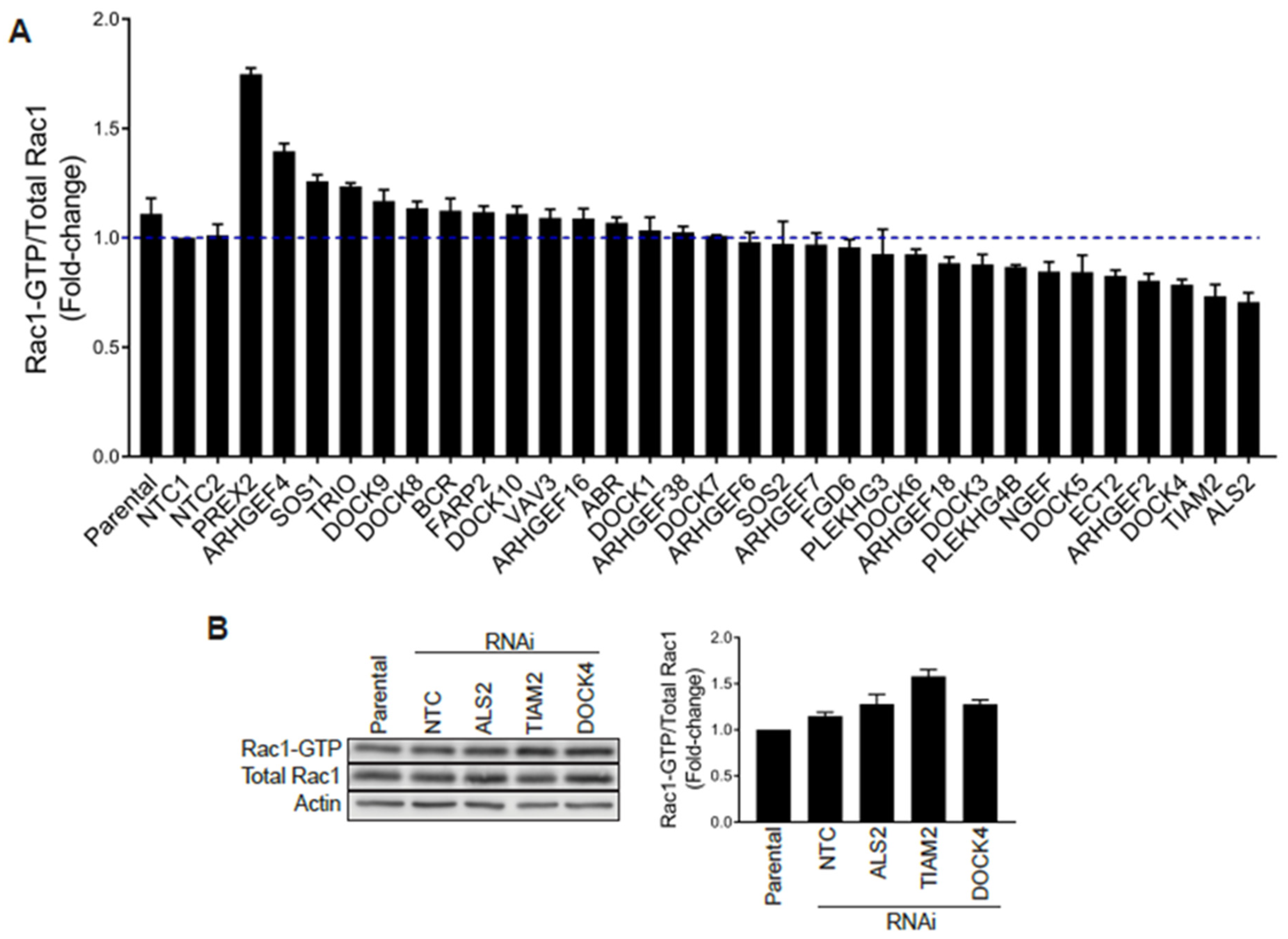
2.4. Silencing Rac-GEFs in PC3 Cells Did Not Impact Rac1-GTP Levels
2.5. Identification of Rac1 Interacting Partners in Prostate Cancer Cells Using Mass Spectrometry
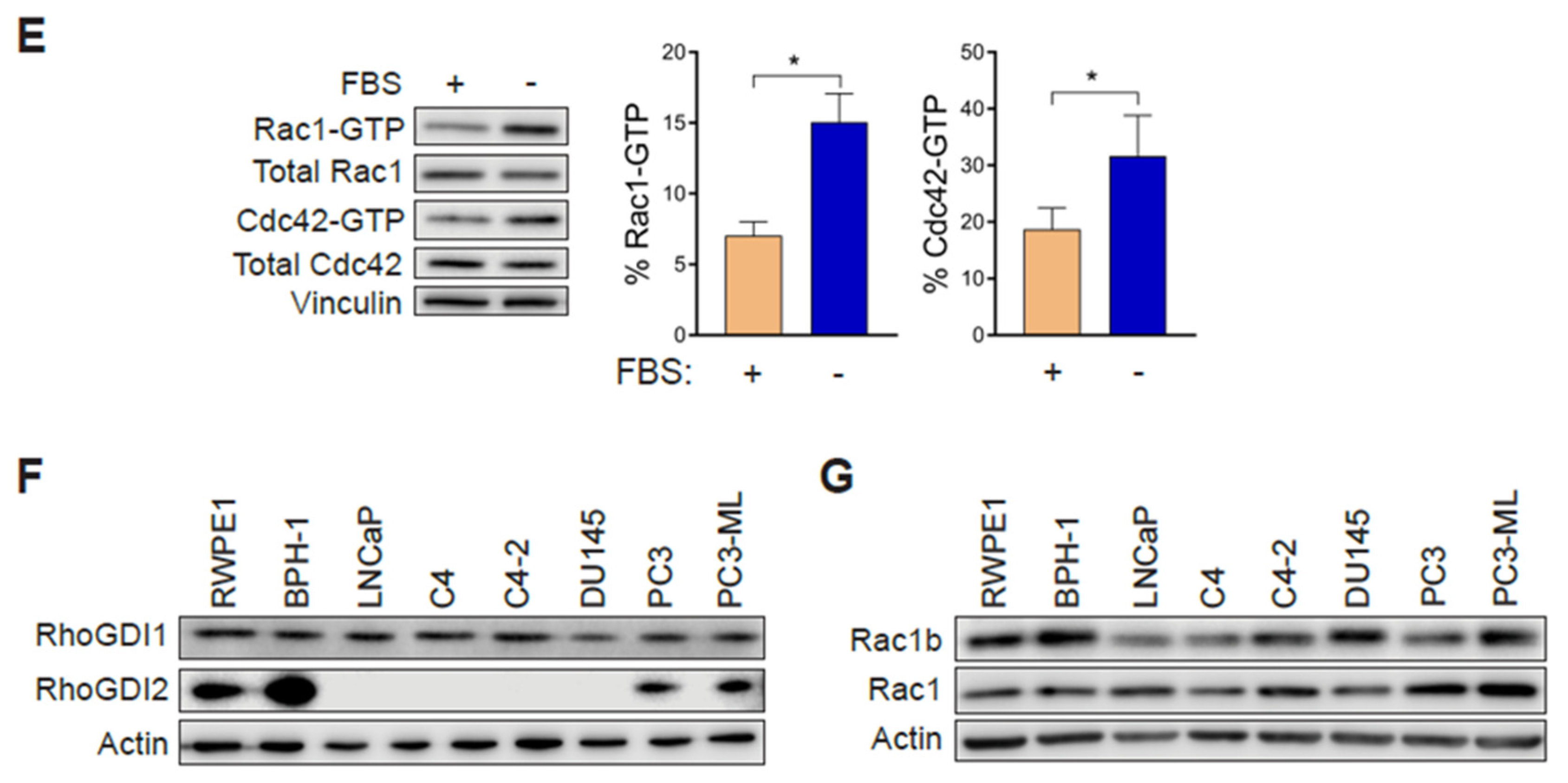
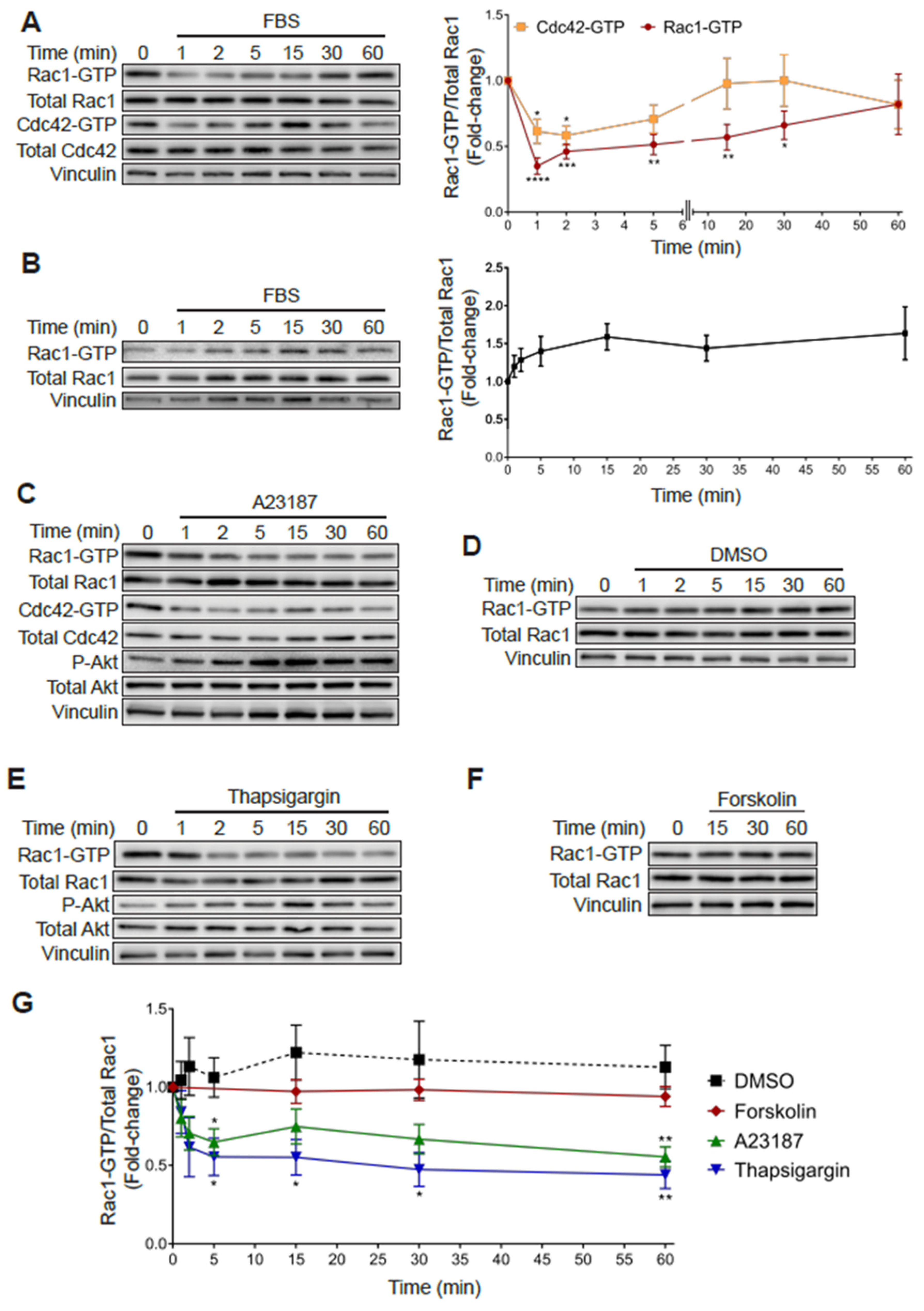
2.6. Rac1 Hyperactivation in PC3 Cells Is Reduced in Response to Serum and Elevations in Intracellular Calcium
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Cell Culture, and Reagents
4.2. RNA Interference (RNAi)
4.3. Generation of Stable GEF-Depleted Cell Lines
4.4. Rac1-GTP/Cdc42-GTP Pull-Down (PBD) Assays
4.5. G-LISA Assay for Rac1-GTP
4.6. Adenoviral Expression of β2-Chimaerin
4.7. Western Blot Analysis
4.8. Analysis of mRNA Expression by qPCR
4.9. Migration and Invasion Assays
4.10. In silico Analysis of P-REX1 mRNA Expression among Normal and Prostate Cancer Tissues
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Seruga, B.; Ocana, A.; Tannock, I.F. Drug resistance in metastatic castration-resistant prostate cancer. Nat. Rev. Clin. Oncol. 2011, 8, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Casado-Medrano, V.; Baker, M.J.; Lopez-Haber, C.; Cooke, M.; Wang, S.; Caloca, M.J.; Kazanietz, M.G. The role of Rac in tumor susceptibility and disease progression: From biochemistry to the clinic. Biochem. Soc. Trans. 2018, 46, 1003–1012. [Google Scholar] [CrossRef] [PubMed]
- De, P.; Aske, J.C.; Dey, N. RAC1 Takes the Lead in Solid Tumors. Cells 2019, 8, 382. [Google Scholar] [CrossRef] [PubMed]
- Svensmark, J.H.; Brakebusch, C. Rho GTPases in cancer: Friend or foe? Oncogene 2019, 38, 7447–7456. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.R.; Rossman, K.L.; Der, C.J. Rho guanine nucleotide exchange factors: Regulators of Rho GTPase activity in development and disease. Oncogene 2014, 33, 4021–4035. [Google Scholar] [CrossRef]
- Michaelson, D.; Silletti, J.; Murphy, G.; D’Eustachio, P.; Rush, M.; Philips, M.R. Differential Localization of Rho Gtpases in Live Cells. J. Cell Biol. 2001, 152, 111–126. [Google Scholar] [CrossRef]
- Schaks, M.; Giannone, G.; Rottner, K. Actin dynamics in cell migration. Essays Biochem. 2019, 63, 483–495. [Google Scholar]
- Myant, K.B.; Cammareri, P.; McGhee, E.J.; Ridgway, R.A.; Huels, D.J.; Cordero, J.B.; Schwitalla, S.; Kalna, G.; Ogg, E.-L.; Athineos, D.; et al. ROS production and NF-κB activation triggered by RAC1 facilitate WNT-driven intestinal stem cell proliferation and colorectal cancer initiation. Cell Stem Cell 2013, 12, 761–773. [Google Scholar] [CrossRef]
- Acevedo, A.; González-Billault, C. Crosstalk between Rac1-mediated actin regulation and ROS production. Free Radic. Biol. Med. 2018, 116, 101–113. [Google Scholar] [CrossRef]
- Cuadrado, A.; Martín-Moldes, Z.; Ye, J.; Lastres-Becker, I. Transcription factors NRF2 and NF-κB are coordinated effectors of the Rho family, GTP-binding protein RAC1 during inflammation. J. Biol. Chem. 2014, 289, 15244–15258. [Google Scholar] [CrossRef]
- Joyce, D.; Bouzahzah, B.; Fu, M.; Albanese, C.; D’Amico, M.; Steer, J.; Klein, J.U.; Lee, R.J.; Segall, J.E.; Westwick, J.K.; et al. Integration of Rac-dependent regulation of cyclin D1 transcription through a nuclear factor-kappaB-dependent pathway. J. Biol. Chem. 1999, 274, 25245–25249. [Google Scholar] [CrossRef]
- Justilien, V.; Ali, S.A.; Jamieson, L.; Yin, N.; Cox, A.D.; Der, C.J.; Murray, N.R.; Fields, A.P. Ect2-Dependent rRNA Synthesis Is Required for KRAS-TRP53-Driven Lung Adenocarcinoma. Cancer Cell 2017, 31, 256–269. [Google Scholar] [CrossRef]
- Baker, M.J.; Cooke, M.; Kazanietz, M.G.M.G. Nuclear PKCι-ECT2-Rac1 and Ribosome Biogenesis: A Novel Axis in Lung Tumorigenesis. Cancer Cell 2017, 31, 167–169. [Google Scholar] [CrossRef]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.-P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A Landscape of Driver Mutations in Melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef]
- Krauthammer, M.; Kong, Y.; Ha, B.H.; Evans, P.; Bacchiocchi, A.; McCusker, J.P.; Cheng, E.; Davis, M.J.; Goh, G.; Choi, M.; et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet. 2012, 44, 1006–1014. [Google Scholar] [CrossRef]
- Porter, A.P.; Papaioannou, A.; Malliri, A. Deregulation of Rho GTPases in cancer. Small GTPases 2016, 7, 123–138. [Google Scholar] [CrossRef]
- Kazanietz, M.G.; Caloca, M.J. The Rac GTPase in Cancer: From Old Concepts to New Paradigms. Cancer Res. 2017, 77, 5445–5451. [Google Scholar] [CrossRef]
- Kobayashi, T.; Inoue, T.; Shimizu, Y.; Terada, N.; Maeno, A.; Kajita, Y.; Yamasaki, T.; Kamba, T.; Toda, Y.; Mikami, Y.; et al. Activation of Rac1 Is Closely Related to Androgen-Independent Cell Proliferation of Prostate Cancer Cells Both in Vitro and in Vivo. Mol. Endocrinol. 2010, 24, 722–734. [Google Scholar] [CrossRef]
- Knight-Krajewski, S.; Welsh, C.F.; Liu, Y.; Lyons, L.S.; Faysal, J.M.; Yang, E.S.; Burnstein, K.L. Deregulation of the Rho GTPase, Rac1, suppresses cyclin-dependent kinase inhibitor p21CIP1 levels in androgen-independent human prostate cancer cells. Oncogene 2004, 23, 5513–5522. [Google Scholar] [CrossRef][Green Version]
- Chen, X.; Cheng, H.; Pan, T.; Liu, Y.; Su, Y.; Ren, C.; Huang, D.; Zha, X.; Liang, C. mTOR regulate EMT through RhoA and Rac1 pathway in prostate cancer. Mol. Carcinog. 2015, 54, 1086–1095. [Google Scholar] [CrossRef]
- Henderson, V.; Smith, B.; Burton, L.J.; Randle, D.; Morris, M.; Odero-Marah, V.A. Snail promotes cell migration through PI3K/AKT-dependent Rac1 activation as well as PI3K/AKT-independent pathways during prostate cancer progression. Cell Adh. Migr. 2015, 9, 255–264. [Google Scholar] [CrossRef]
- Tajadura-Ortega, V.; Garg, R.; Allen, R.; Owczarek, C.; Bright, M.D.; Kean, S.; Mohd-Noor, A.; Grigoriadis, A.; Elston, T.C.; Hahn, K.M.; et al. An RNAi screen of Rho signalling networks identifies RhoH as a regulator of Rac1 in prostate cancer cell migration. BMC Biol. 2018, 16, 29. [Google Scholar] [CrossRef]
- Welch, H.C.E.; Coadwell, W.J.; Ellson, C.D.; Ferguson, G.J.; Andrews, S.R.; Erdjument-Bromage, H.; Tempst, P.; Hawkins, P.T.; Stephens, L.R. P-Rex1, a PtdIns(3,4,5)P3- and Gβγ-Regulated Guanine-Nucleotide Exchange Factor for Rac. Cell 2002, 108, 809–821. [Google Scholar] [CrossRef]
- Welch, H.C.E. Regulation and function of P-Rex family Rac-GEFs. Small GTPases 2015, 6, 49–70. [Google Scholar] [CrossRef]
- Sosa, M.S.; Lopez-Haber, C.; Yang, C.; Wang, H.; Lemmon, M.A.; Busillo, J.M.; Luo, J.; Benovic, J.L.; Klein-Szanto, A.; Yagi, H.; et al. Identification of the Rac-GEF P-Rex1 as an Essential Mediator of ErbB Signaling in Breast Cancer. Mol. Cell 2010, 40, 877–892. [Google Scholar] [CrossRef]
- Lindsay, C.R.; Lawn, S.; Campbell, A.D.; Faller, W.J.; Rambow, F.; Mort, R.L.; Timpson, P.; Li, A.; Cammareri, P.; Ridgway, R.A.; et al. P-Rex1 is required for efficient melanoblast migration and melanoma metastasis. Nat. Commun. 2011, 2, 555. [Google Scholar] [CrossRef]
- Qin, J.; Xie, Y.; Wang, B.; Hoshino, M.; Wolff, D.W.; Zhao, J.; Scofield, M.A.; Dowd, F.J.; Lin, M.-F.; Tu, Y. Upregulation of PIP3-dependent Rac exchanger 1 (P-Rex1) promotes prostate cancer metastasis. Oncogene 2009, 28, 1853–1863. [Google Scholar] [CrossRef]
- Welch, H.C.E.; Condliffe, A.M.; Milne, L.J.; Ferguson, G.J.; Hill, K.; Webb, L.M.C.; Okkenhaug, K.; Coadwell, W.J.; Andrews, S.R.; Thelen, M.; et al. P-Rex1 Regulates Neutrophil Function. Cur. Biol. 2005, 15, 1867–1873. [Google Scholar]
- Montero, J.C.; Seoane, S.; Ocaña, A.; Pandiella, A. P-Rex1 participates in Neuregulin-ErbB signal transduction and its expression correlates with patient outcome in breast cancer. Oncogene 2011, 30, 1059–1071. [Google Scholar] [CrossRef]
- Grasso, C.S.; Wu, Y.-M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef]
- Lapointe, J.; Li, C.; Higgins, J.P.; Van De Rijn, M.; Bair, E.; Montgomery, K.; Ferrari, M.; Egevad, L.; Rayford, W.; Bergerheim, U.; et al. Gene expression profiling identifies clinically relevant subtypes of prostate cancer. Proc. Natl. Acad. Sci. USA 2004, 101, 811–816. [Google Scholar] [CrossRef]
- Varambally, S.; Yu, J.; Laxman, B.; Rhodes, D.R.; Mehra, R.; Tomlins, S.A.; Shah, R.B.; Chandran, U.; Monzon, F.A.; Becich, M.J.; et al. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell 2005, 8, 393–406. [Google Scholar] [CrossRef]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef]
- Menna, P.L.; Skilton, G.; Leskow, F.C.; Alonso, D.F.; Gomez, D.E.; Kazanietz, M.G. Inhibition of Aggressiveness of Metastatic Mouse Mammary Carcinoma Cells by the β2-Chimaerin GAP Domain. Cancer Res. 2003, 55, 3456–3461. [Google Scholar]
- Caloca, M.J.; Wang, H.; Kazanietz, M.G. Characterization of the Rac-GAP (Rac-GTPase-activating protein) activity of beta2-chimaerin, a “non-protein kinase C” phorbol ester receptor. Biochem. J. 2003, 375, 313–321. [Google Scholar] [CrossRef]
- Wang, H.; Yang, C.; Leskow, F.C.; Sun, J.; Canagarajah, B.; Hurley, J.H.; Kazanietz, M.G. Phospholipase Cγ/diacylglycerol-dependent activation of β2-chimaerin restricts EGF-induced Rac signaling. EMBO J. 2006, 25, 2062–2074. [Google Scholar] [CrossRef]
- Yang, C.; Liu, Y.; Lemmon, M.A.; Kazanietz, M.G. Essential role for Rac in heregulin beta1 mitogenic signaling: A mechanism that involves epidermal growth factor receptor and is independent of ErbB4. Mol. Cell. Biol. 2006, 26, 831–842. [Google Scholar] [CrossRef]
- Ohta, Y.; Hartwig, J.H.; Stossel, T.P. FilGAP, a Rho- and ROCK-regulated GAP for Rac binds filamin A to control actin remodelling. Nat. Cell Biol. 2006, 8, 803–814. [Google Scholar] [CrossRef]
- Flatau, G.; Lemichez, E.; Gauthier, M.; Chardin, P.; Paris, S.; Fiorentini, C.; Boquet, P. Toxin-induced activation of the G protein p21 Rho by deamidation of glutamine. Nature 1997, 387, 729–733. [Google Scholar] [CrossRef]
- García-Mata, R.; Wennerberg, K.; Arthur, W.T.; Noren, N.K.; Ellerbroek, S.M.; Burridge, K. Analysis of Activated GAPs and GEFs in Cell Lysates. Meth. Enzymol. 2006, 406, 425–437. [Google Scholar]
- Shin, E.-Y.; Lee, C.-S.; Cho, T.G.; Kim, Y.G.; Song, S.; Juhnn, Y.-S.; Park, S.C.; Manser, E.; Kim, E.-G. betaPak-interacting exchange factor-mediated Rac1 activation requires smgGDS guanine nucleotide exchange factor in basic fibroblast growth factor-induced neurite outgrowth. J. Biol. Chem. 2006, 281, 35954–35964. [Google Scholar] [CrossRef]
- Johmura, S.; Oh-hora, M.; Inabe, K.; Nishikawa, Y.; Hayashi, K.; Vigorito, E.; Kitamura, D.; Turner, M.; Shingu, K.; Hikida, M.; et al. Regulation of Vav Localization in Membrane Rafts by Adaptor Molecules Grb2 and BLNK. Immunity 2003, 18, 777–787. [Google Scholar] [CrossRef]
- Thastrup, O.; Cullen, P.J.; Drøbak, B.K.; Hanley, M.R.; Dawson, A.P. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2(+)-ATPase. Proc. Natl. Acad. Sci. USA 1990, 87, 2466–2470. [Google Scholar] [CrossRef]
- Barrio-Real, L.; Benedetti, L.G.; Engel, N.; Tu, Y.; Cho, S.; Sukumar, S.; Kazanietz, M.G. Subtype-specific overexpression of the Rac-GEF P-REX1 in breast cancer is associated with promoter hypomethylation. Breast Cancer Res. 2014, 16, 441. [Google Scholar] [CrossRef]
- Berg, T.J.; Gastonguay, A.J.; Lorimer, E.L.; Kuhnmuench, J.R.; Li, R.; Fields, A.P.; Williams, C.L. Splice Variants of SmgGDS Control Small GTPase Prenylation and Membrane Localization. J. Biol. Chem. 2010, 285, 35255–35266. [Google Scholar] [CrossRef]
- Briggs, M.W.; Sacks, D.B. IQGAP proteins are integral components of cytoskeletal regulation. EMBO Rep. 2003, 4, 571–574. [Google Scholar] [CrossRef]
- Caino, M.C.; Lopez-Haber, C.; Kissil, J.L.; Kazanietz, M.G. Non-small cell lung carcinoma cell motility, rac activation and metastatic dissemination are mediated by protein kinase C epsilon. PLoS ONE 2012, 7, e31714. [Google Scholar] [CrossRef]
- Yang, C.; Liu, Y.; Leskow, F.C.; Weaver, V.M.; Kazanietz, M.G. Rac-GAP-dependent inhibition of breast cancer cell proliferation by β2-chimerin. J. Biol. Chem. 2005, 280, 24363–24370. [Google Scholar] [CrossRef]
- Schlam, D.; Canton, J. Every day I’m rufflin’: Calcium sensing and actin dynamics in the growth factor-independent membrane ruffling of professional phagocytes. Small GTPases 2017, 8, 65–70. [Google Scholar] [CrossRef]
- Noren, N.K.; Niessen, C.M.; Gumbiner, B.M.; Burridge, K. Cadherin engagement regulates Rho family GTPases. J. Biol. Chem. 2001, 276, 33305–33308. [Google Scholar] [CrossRef]
- Castillo-Lluva, S.; Tatham, M.H.; Jones, R.C.; Jaffray, E.G.; Edmondson, R.D.; Hay, R.T.; Malliri, A. SUMOylation of the GTPase Rac1 is required for optimal cell migration. Nat. Cell Biol. 2010, 12, 1078–1085. [Google Scholar] [CrossRef]
- Gutierrez-Uzquiza, A.; Lopez-Haber, C.; Jernigan, D.L.; Fatatis, A.; Kazanietz, M.G. PKC Is an Essential Mediator of Prostate Cancer Bone Metastasis. Mol. Cancer Res. 2015, 13, 1336–1346. [Google Scholar] [CrossRef]
- Benard, V.; Bohl, B.P.; Bokoch, G.M. Characterization of rac and cdc42 activation in chemoattractant-stimulated human neutrophils using a novel assay for active GTPases. J. Biol. Chem. 1999, 274, 13198–13204. [Google Scholar] [CrossRef]
- Lopez-Haber, C.; Kazanietz, M.G. Cucurbitacin I inhibits Rac1 activation in breast cancer cells by a reactive oxygen species-mediated mechanism and independently of Janus tyrosine kinase 2 and P-Rex1. Mol. Pharmacol. 2013, 83, 1141–1154. [Google Scholar] [CrossRef]
- Cortazar, A.R.; Torrano, V.; Martín-Martín, N.; Caro-Maldonado, A.; Camacho, L.; Hermanova, I.; Guruceaga, E.; Lorenzo-Martín, L.F.; Caloto, R.; Gomis, R.R.; et al. Cancertool: A visualization and representation interface to exploit cancer datasets. Cancer Res. 2018, 78, 6320–6328. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baker, M.J.; Abba, M.C.; Garcia-Mata, R.; Kazanietz, M.G. P-REX1-Independent, Calcium-Dependent RAC1 Hyperactivation in Prostate Cancer. Cancers 2020, 12, 480. https://doi.org/10.3390/cancers12020480
Baker MJ, Abba MC, Garcia-Mata R, Kazanietz MG. P-REX1-Independent, Calcium-Dependent RAC1 Hyperactivation in Prostate Cancer. Cancers. 2020; 12(2):480. https://doi.org/10.3390/cancers12020480
Chicago/Turabian StyleBaker, Martin J., Martín C. Abba, Rafael Garcia-Mata, and Marcelo G. Kazanietz. 2020. "P-REX1-Independent, Calcium-Dependent RAC1 Hyperactivation in Prostate Cancer" Cancers 12, no. 2: 480. https://doi.org/10.3390/cancers12020480
APA StyleBaker, M. J., Abba, M. C., Garcia-Mata, R., & Kazanietz, M. G. (2020). P-REX1-Independent, Calcium-Dependent RAC1 Hyperactivation in Prostate Cancer. Cancers, 12(2), 480. https://doi.org/10.3390/cancers12020480