Monitoring Immune Responses in Neuroblastoma Patients during Therapy


Abstract
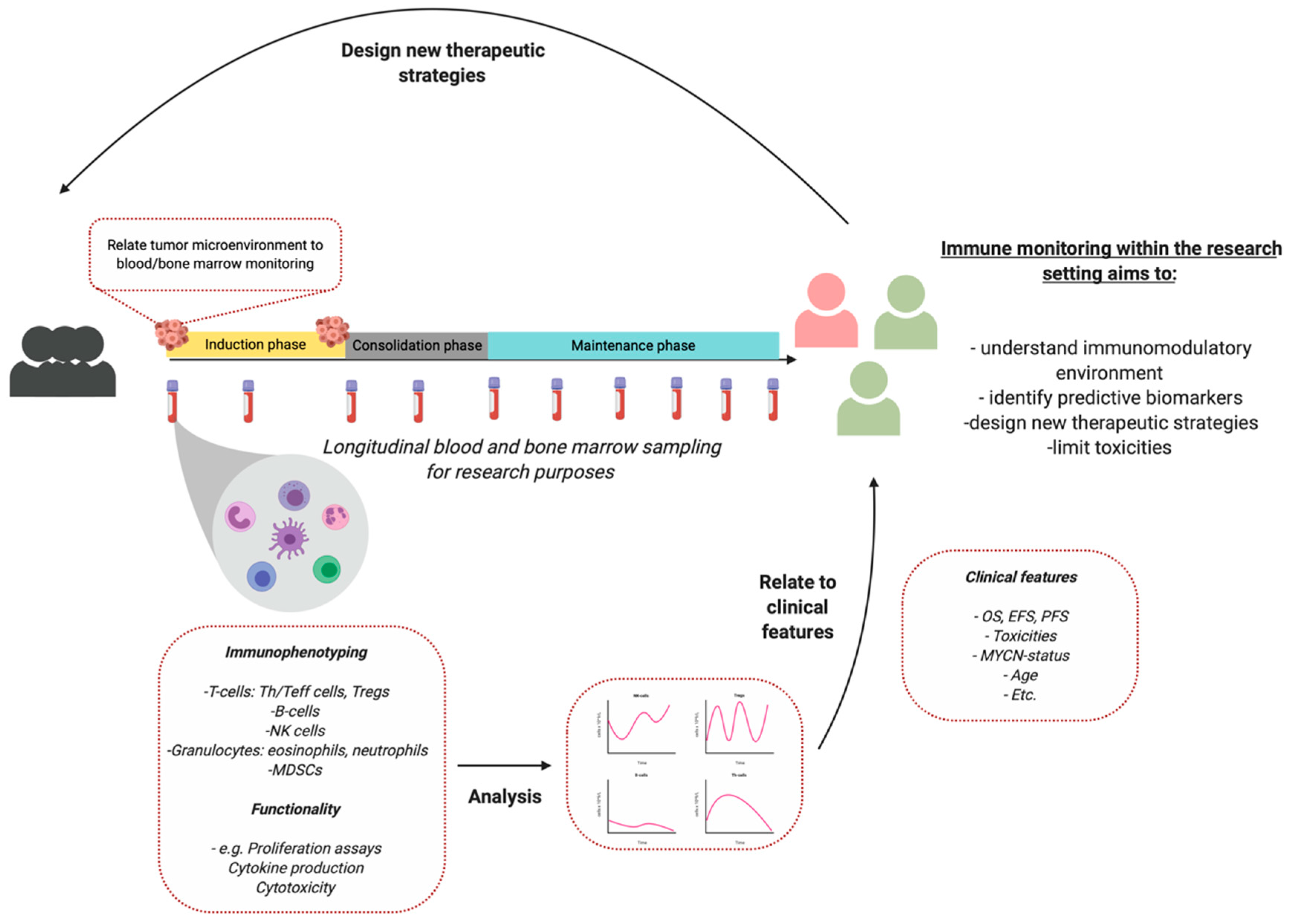
:1. Introduction
2. Immune Monitoring at Diagnosis: Correlates of Outcome?
2.1. Immune Markers at the Tumor Site
2.2. Circulatory Immune Markers
2.2.1. Cytokines and Soluble Molecules in Plasma/Serum
2.2.2. Immune Cells in Peripheral Blood
3. Immune Monitoring during Therapy
3.1. Chemotherapy
3.2. Monoclonal Antibody Therapy
3.3. Adoptive Cell Therapy
3.4. Checkpoint Inhibitors
4. Discussion
5. Conclusions
Funding
Conflicts of Interest
References
- Matthay, K.K.; Maris, J.M.; Schleiermacher, G.; Nakagawara, A.; Mackall, C.L.; Diller, L.; Weiss, W.A. Neuroblastoma. Nat. Rev. Dis. Prim. 2016, 2, 16078. [Google Scholar] [CrossRef]
- Park, J.R.; Eggert, A.; Caron, H. Neuroblastoma: Biology, Prognosis, and Treatment. Hematol. Oncol. Clin. N. Am. 2010, 24, 65–86. [Google Scholar] [CrossRef] [PubMed]
- Maris, J.M. Recent Advances in Neuroblastoma. N. Engl. J. Med. 2010, 362, 2202–2211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidoff, A.M. Neuroblastoma. Semin. Pediatr. Surg. 2012, 21, 2–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanichapol, T.; Chutipongtanate, S.; Anurathapan, U.; Hongeng, S. Immune Escape Mechanisms and Future Prospects for Immunotherapy in Neuroblastoma. Biomed. Res. Int. 2018, 2018, 1812535. [Google Scholar] [CrossRef]
- Mina, M.; Boldrini, R.; Citti, A.; Romania, P.; D’Alicandro, V.; De Ioris, M.; Castellano, A.; Furlanello, C.; Locatelli, F.; Fruci, D. Tumor-infiltrating T lymphocytes improve clinical outcome of therapy-resistant neuroblastoma. Oncoimmunology 2015, 4, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagasawa, M.; Kawamoto, H.; Tsuji, Y.; Mizutani, S. Transient increase of serum granulysin in a stage IVs neuroblastoma patient during spontaneous regression: Case report. Int. J. Hematol. 2005, 82, 456–457. [Google Scholar] [CrossRef]
- Yu, A.L.; Gilman, A.L.; Ozkaynak, M.F.; London, W.B.; Kreissman, S.G.; Chen, H.X.; Smith, M.; Anderson, B.; Villablanca, J.G.; Matthay, K.K.; et al. Anti-GD2 Antibody with GM-CSF, Interleukin-2, and Isotretinoin for Neuroblastoma. N. Engl. J. Med. 2010, 363, 1324–1334. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Y.; Fest, S.; Kunert, R.; Katinger, H.; Pistoia, V.; Michon, J.; Lewis, G.; Ladenstein, R.; Lode, H.N. Anti-neuroblastoma effect of ch14.18 antibody produced in CHO cells is mediated by NK-cells in mice. Mol. Immunol. 2005, 42, 1311–1319. [Google Scholar] [CrossRef] [PubMed]
- Modak, S.; Cheung, N.-K. V Neuroblastoma: Therapeutic strategies for a clinical enigma. Cancer Treat. Rev. 2010, 36, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.; Coppola, D.; Wang, E.; Chang, Y.D.; Kim, Y.; Anaya, D.; Kim, D.W. Prognostic value of CD8CD45RO tumor infiltrating lymphocytes in patients with extrahepatic cholangiocarcinoma. Oncotarget 2018, 9, 23366–23372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parra, E.R.; Villalobos, P.; Behrens, C.; Jiang, M.; Pataer, A.; Swisher, S.G.; William, W.N.; Zhang, J.; Lee, J.; Cascone, T.; et al. Effect of neoadjuvant chemotherapy on the immune microenvironment in non-small cell lung carcinomas as determined by multiplex immunofluorescence and image analysis approaches. J. Immunother. Cancer 2018, 6, 48. [Google Scholar] [CrossRef]
- Lo, C.S.; Sanii, S.; Kroeger, D.R.; Milne, K.; Talhouk, A.; Chiu, D.S.; Rahimi, K.; Shaw, P.A.; Clarke, B.A.; Nelson, B.H. Neoadjuvant chemotherapy of ovarian cancer results in three patterns of tumor-infiltrating lymphocyte response with distinct implications for immunotherapy. Clin. Cancer Res. 2017, 23, 925–934. [Google Scholar] [CrossRef] [Green Version]
- Hegde, P.S.; Karanikas, V.; Evers, S. The where, the when, and the how of immune monitoring for cancer immunotherapies in the era of checkpoint inhibition. Clin. Cancer Res. 2016, 22, 1865–1874. [Google Scholar] [CrossRef] [Green Version]
- Martin, R.F.; Bruce Beckwith, J. Lymphoid infiltrates in neuroblastomas: Their occurrence and prognostic significance. J. Pediatr. Surg. 1968, 3, 161–164. [Google Scholar] [CrossRef]
- Lauder, I. The significance of lymphocytic infiltration in neuroblastoma. Br. J. Cancer 1972, 26, 321–330. [Google Scholar] [CrossRef] [Green Version]
- Hellstrom, I.E.; Hellstrom, K.E.; Pierce, G.E.; Bill, A.H. Demonstration of cell-bound and humoral immunity against neuroblastoma cells. Proc. Natl. Acad. Sci. USA 1968, 60, 1231–1238. [Google Scholar] [CrossRef] [Green Version]
- Ollé Hurtado, M.; Wolbert, J.; Fisher, J.; Flutter, B.; Stafford, S.; Barton, J.; Jain, N.; Barone, G.; Majani, Y.; Anderson, J. Tumor infiltrating lymphocytes expanded from pediatric neuroblastoma display heterogeneity of phenotype and function. PLoS ONE 2019, 14, e0216373. [Google Scholar] [CrossRef] [Green Version]
- Fridman, W.H.; Zitvogel, L.; Sautès-Fridman, C.; Kroemer, G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717–734. [Google Scholar] [CrossRef] [PubMed]
- Berghoff, A.S.; Fuchs, E.; Ricken, G.; Mlecnik, B.; Bindea, G.; Spanberger, T.; Hackl, M.; Widhalm, G.; Dieckmann, K.; Prayer, D.; et al. Density of tumor-infiltrating lymphocytes correlates with extent of brain edema and overall survival time in patients with brain metastases. Oncoimmunology 2016, 5, e1057388. [Google Scholar] [CrossRef] [PubMed]
- Layer, J.P.; Kronmüller, M.T.; Quast, T.; Van Den Boorn, D.; Effern, M.; Hinze, D.; Althoff, K.; Westermann, F.; Peifer, M.; Hartmann, G.; et al. Amplification of N-Myc is associated with a T-cell-poor microenvironment in metastatic neuroblastoma restraining interferon pathway activity and chemokine expression. Oncoimmunology 2017, 6, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mussai, F.; Egan, S.; Hunter, S.; Webber, H.; Fisher, J.; Wheat, R.; McConville, C.; Sbirkov, Y.; Wheeler, K.; Bendle, G.; et al. Neuroblastoma arginase activity creates an immunosuppressive microenvironment that impairs autologous and engineered immunity. Cancer Res. 2015, 75, 3043–3053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Wu, X.; Basu, M.; Dong, C.; Zheng, P.; Liu, Y.; Sandler, A.D. MYCN amplification is associated with repressed cellular immunity in neuroblastoma: An in silico immunological analysis of TARGET database. Front. Immunol. 2017, 8, 1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, L.; Asgharzadeh, S.; Salo, J.; Engell, K.; Wu, H.; Sposto, R.; Ara, T.; Silverman, A.M.; Declerck, Y.A.; Seeger, R.C.; et al. V α 24-invariant NKT cells mediate antitumor activity via killing of tumor-associated macrophages. J. Clin. Invest. 2009, 119, 1524–1536. [Google Scholar] [CrossRef] [Green Version]
- Asgharzadeh, S.; Salo, J.A.; Ji, L.; Oberthuer, A.; Fischer, M.; Berthold, F.; Hadjidaniel, M.; Liu, C.W.Y.; Metelitsa, L.S.; Pique-Regi, R.; et al. Clinical significance of tumor-associated inflammatory cells in metastatic neuroblastoma. J. Clin. Oncol. 2012, 30, 3525–3532. [Google Scholar] [CrossRef] [Green Version]
- Qian, B.Z.; Pollard, J.W. Macrophage Diversity Enhances Tumor Progression and Metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Morandi, F.; Croce, M.; Cangemi, G.; Barco, S.; Rigo, V.; Carlini, B.; Amoroso, L.; Pistoia, V.; Ferrini, S.; Corrias, M.V. IL-10 and ARG-1 concentrations in bone marrow and peripheral blood of metastatic neuroblastoma patients do not associate with clinical outcome. J. Immunol. Res. 2015, 2015, 718975. [Google Scholar] [CrossRef] [Green Version]
- Scarpa, S.; Coppa, A.; Ragano-Caracciolo, M.; Mincione, G.; Giuffrida, A.; Modesti, A.; Colletta, G. Transforming growth factor β regulates differentiation and proliferation of human neuroblastoma. Exp. Cell Res. 1996, 229, 147–154. [Google Scholar] [CrossRef]
- Oliveira, F.B.; Magalhães, L.M.; Passos, L.S.; Neto, J.C.A.; Dutra, Á.P.; Martins, P.R.; Salles, P.G.O.; Dutra, W.O.; Gollob, K.J. Abstract 707: Circulating immune profile in childhood neuroblastoma displays an activated response with simultaneous expression of checkpoint proteins by T cells and monocytes. In Proceedings of the AACR Annual Meeting 2018, Chicago, IL, USA, 14–18 April 2018; p. 707. [Google Scholar] [CrossRef]
- Egler, R.A.; Li, Y.; Dang, T.A.T.; Peters, T.L.; Leung, E.; Huang, S.; Russell, H.V.; Liu, H.; Man, T.K. An integrated proteomic approach to identifying circulating biomarkers in high-risk neuroblastoma and their potential in relapse monitoring. Proteomics Clin. Appl. 2011, 5, 532–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morandi, F.; Cangemi, G.; Barco, S.; Amoroso, L.; Giuliano, M.; Gigliotti, A.R.; Pistoia, V.; Corrias, M.V. Plasma levels of soluble HLA-E and HLA-F at diagnosis may predict overall survival of neuroblastoma patients. Biomed. Res. Int. 2013, 2013, 956878. [Google Scholar] [CrossRef] [PubMed]
- Gogali, A.; Charalabopoulos, K.; Zampira, I.; Konstantinidis, A.K.; Tachmazoglou, F.; Daskalopoulos, G.; Constantopoulos, S.H.; Dalavanga, Y. Soluble Adhesion Molecules E-Cadherin, Intercellular Adhesion Molecule-1, and E-Selectin as Lung Cancer Biomarkers. Chest 2010, 138, 1173–1179. [Google Scholar] [CrossRef] [PubMed]
- Mirabelli, P.; Incoronato, M. Usefulness of traditional serum biomarkers for management of breast cancer patients. Biomed. Res. Int. 2013, 2013, 685641. [Google Scholar] [CrossRef] [Green Version]
- Bassani-Sternberg, M.; Barnea, E.; Beer, I.; Avivi, I.; Katz, T.; Admon, A. Soluble plasma HLA peptidome as a potential source for cancer biomarkers. Proc. Natl. Acad. Sci. USA 2010, 107, 18769. [Google Scholar] [CrossRef] [Green Version]
- Baron, A.T.; Cora, E.M.; Lafky, J.M.; Boardman, C.H.; Buenafe, M.C.; Rademaker, A.; Liu, D.; Fishman, D.A.; Podratz, K.C.; Maihle, N.J. Soluble Epidermal Growth Factor Receptor (sEGFR/sErbB1) as a Potential Risk, Screening, and Diagnostic Serum Biomarker of Epithelial Ovarian Cancer. Cancer Epidemiol. Biomarkers Prev. 2003, 12, 103–113. [Google Scholar] [PubMed]
- Morandi, F.; Barco, S.; Stigliani, S.; Croce, M.; Persico, L.; Lagazio, C.; Scuderi, F.; Belli, M.L.; Montera, M.; Cangemi, G.; et al. Altered erythropoiesis and decreased number of erythrocytes in children with neuroblastoma. Oncotarget 2017, 8, 53194–53209. [Google Scholar] [CrossRef] [PubMed]
- Semeraro, M.; Rusakiewicz, S.; Minard-colin, V.; Delahaye, N.F.; Enot, D.; Vély, F.; Marabelle, A.; Papoular, B.; Piperoglou, C.; Ponzoni, M.; et al. Clinical impact of the NKp30 / B7-H6 axis in high-risk neuroblastoma patients. Sci. Transl. Med. 2015, 7, 283ra55. [Google Scholar] [CrossRef]
- Spel, L.; Boelens, J.-J.; van der Steen, D.M.; Blokland, N.J.G.; van Noesel, M.M.; Molenaar, J.J.; Heemskerk, M.H.M.; Boes, M.; Nierkens, S. Natural killer cells facilitate PRAME-specific T-cell reactivity against neuroblastoma. Oncotarget 2015, 6, 35770–35781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morandi, F.; Pozzi, S.; Barco, S.; Cangemi, G.; Amoroso, L.; Carlini, B.; Pistoia, V.; Corrias, M.V. CD4+CD25hiCD127− Treg and CD4+CD45R0+CD49b+LAG3+ Tr1 cells in bone marrow and peripheral blood samples from children with neuroblastoma. Oncoimmunology 2016, 5, e1249553. [Google Scholar] [CrossRef] [Green Version]
- Mao, Y.; Eissler, N.; Le Blanc, K.; Johnsen, J.I.; Kogner, P.; Kiessling, R. Targeting suppressive myeloid cells potentiates checkpoint inhibitors to control spontaneous neuroblastoma. Clin. Cancer Res. 2016, 22, 3849–3859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlson, L.-M.; De Geer, A.; Sveinbjørnsson, B.; Orrego, A.; Martinsson, T.; Kogner, P.; Levitskaya, J. The microenvironment of human neuroblastoma supports the activation of tumor-associated T lymphocytes. Oncoimmunology 2013, 2, e23618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gowda, M.; Payne, K.; Godder, K.; Manjili, M.H. HLA-DR expression on myeloid cells is a potential prognostic factor in patients with high-risk neuroblastoma. Oncoimmunology 2013, 2, e26616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apps, J.R.; Hasan, F.; Campus, O.; Behjati, S.; Jacques, T.S.J.; Sebire, N.; Anderson, J. The immune environment of paediatric solid malignancies: Evidence from an immunohistochemical study of clinical cases. Fetal Pediatr. Pathol. 2013, 32, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Egler, R.A.; Burlingame, S.M.; Nuchtern, J.G.; Russell, H.V. Interleukin-6 and soluble interleukin-6 receptor levels as markers of disease extent and prognosis in neuroblastoma. Clin. Cancer Res. 2008, 14, 7028–7034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, V.; Foster, J. High-Risk Neuroblastoma Treatment Review. Child 2018, 5, 114. [Google Scholar] [CrossRef] [Green Version]
- Behl, D.; Porrata, L.F.; Markovic, S.N.; Letendre, L.; Pruthi, R.K.; Hook, C.C.; Tefferi, A.; Elliot, M.A.; Kaufmann, S.H.; Mesa, R.A.; et al. Absolute lymphocyte count recovery after induction chemotherapy predicts superior survival in acute myelogenous leukemia. Leukemia 2006, 20, 29–34. [Google Scholar] [CrossRef]
- Siddiqui, M.; Ristow, K.; Markovic, S.N.; Witzig, T.E.; Habermann, T.M.; Colgan, J.P.; Inwards, D.J.; White, W.L.; Ansell, S.M.; Micallef, I.N.; et al. Absolute lymphocyte count predicts overall survival in follicular lymphomas. Br. J. Haematol. 2006, 134, 596–601. [Google Scholar] [CrossRef]
- Thoma, M.D.; Huneke, T.J.; DeCook, L.J.; Johnson, N.D.; Wiegand, R.A.; Litzow, M.R.; Hogan, W.J.; Porrata, L.F.; Holtan, S.G. Peripheral blood lymphocyte and monocyte recovery and survival in acute leukemia postmyeloablative allogeneic hematopoietic stem cell transplant. Biol. Blood Marrow Transplant. 2012, 18, 600–607. [Google Scholar] [CrossRef] [Green Version]
- Galvez-Silva, J.; Maher, O.M.; Park, M.; Liu, D.; Hernandez, F.; Tewari, P.; Nieto, Y. Prognostic Analysis of Absolute Lymphocyte and Monocyte Counts after Autologous Stem Cell Transplantation in Children, Adolescents, and Young Adults with Refractory or Relapsed Hodgkin Lymphoma. Biol. Blood Marrow Transplant. 2017, 23, 1276–1281. [Google Scholar] [CrossRef]
- Nayak, A.; McDowell, D.T.; Kellie, S.J.; Karpelowsky, J. Elevated Preoperative Neutrophil–Lymphocyte Ratio is Predictive of a Poorer Prognosis for Pediatric Patients with Solid Tumors. Ann. Surg. Oncol. 2017, 23, 1276–1281. [Google Scholar] [CrossRef]
- Tilak, T.; Sherawat, S.; Agarwala, S.; Gupta, R.; Vishnubhatla, S.; Bakhshi, S. Circulating T-regulatory cells in neuroblastoma: A pilot prospective study. Pediatr. Hematol. Oncol. 2014, 31, 717–722. [Google Scholar] [CrossRef]
- Mackall, C.L.; Fleisher, T.A.; Brown, M.R.; Magrath, I.T.; Shad, A.T.; Horowitz, M.E.; Wexler, L.H.; Adde, M.A.; McClure, L.L.; Gress, R.E. Lymphocyte depletion during treatment with intensive chemotherapy for cancer. Blood 1994, 84, 2221–2228. [Google Scholar] [CrossRef] [Green Version]
- Delahaye, N.F.; Rusakiewicz, S.; Martins, I.; Ménard, C.; Roux, S.; Lyonnet, L.; Paul, P.; Sarabi, M.; Chaput, N.; Semeraro, M.; et al. Alternatively spliced NKp30 isoforms affect the prognosis of gastrointestinal stromal tumors. Nat. Med. 2011, 17, 700–707. [Google Scholar] [CrossRef]
- Webb, M.W.; Sun, J.; Sheard, M.A.; Liu, W.-Y.; Wu, H.-W.; Jackson, J.R.; Malvar, J.; Sposto, R.; Daniel, D.; Seeger, R.C. Colony stimulating factor 1 receptor blockade improves the efficacy of chemotherapy against human neuroblastoma in the absence of T lymphocytes. Int. J. Cancer 2018, 143, 1483–1493. [Google Scholar] [CrossRef]
- Cheung, N.-K.V.; Guo, H.; Hu, J.; Tassev, D.V.; Cheung, I.Y. Humanizing murine IgG3 anti-GD2 antibody m3F8 substantially improves antibody-dependent cell-mediated cytotoxicity while retaining targeting in vivo. Oncoimmunology 2012, 1, 477–486. [Google Scholar] [CrossRef] [Green Version]
- Shusterman, S.; London, W.B.; Gillies, S.D.; Hank, J.A.; Voss, S.D.; Seeger, R.C.; Reynolds, C.P.; Kimball, J.; Albertini, M.R.; Wagner, B.; et al. Antitumor activity of hu14.18-IL2 in patients with relapsed/refractory neuroblastoma: A Children’s Oncology Group (COG) phase II study. J. Clin. Oncol. 2010, 28, 4969–4975. [Google Scholar] [CrossRef] [Green Version]
- Metelitsa, L.S.; Gillies, S.D.; Super, M.; Shimada, H.; Reynolds, C.P.; Seeger, R.C. Antidisialoganglioside/granulocyte macrophage-colony-stimulating factor fusion protein facilitates neutrophil antibody-dependent cellular cytotoxicity and depends on FcgammaRII (CD32) and Mac-1 (CD11b/CD18) for enhanced effector cell adhesion and azurophi. Blood 2002, 99, 4166–4173. [Google Scholar] [CrossRef] [Green Version]
- Siebert, N.; Eger, C.; Seidel, D.; Jüttner, M.; Zumpe, M.; Wegner, D.; Kietz, S.; Ehlert, K.; Veal, G.J.; Siegmund, W.; et al. Pharmacokinetics and pharmacodynamics of ch14.18/CHO in relapsed/refractory high-risk neuroblastoma patients treated by long-term infusion in combination with IL-2. MAbs 2016, 8, 604–616. [Google Scholar] [CrossRef]
- Nassin, M.L.; Nicolaou, E.; Gurbuxani, S.; Cohn, S.L.; Cunningham, J.M.; LaBelle, J.L. Immune Reconstitution Following Autologous Stem Cell Transplantation in Patients with High-Risk Neuroblastoma at the Time of Immunotherapy. Biol. Blood Marrow Transplant. 2017, 24, 452–459. [Google Scholar] [CrossRef] [Green Version]
- Siebert, N.; Troschke-Meurer, S.; Marx, M.; Zumpe, M.; Ehlert, K.; Gray, J.; Garaventa, A.; Manzitti, C.; Ash, S.; Klingebiel, T.; et al. Impact of HACA on Immunomodulation and Treatment Toxicity Following ch14.18/CHO Long-Term Infusion with Interleukin-2: Results from a SIOPEN Phase 2 Trial. Cancers 2018, 10, 387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, N.-K.V.; Guo, H.; Heller, G.; Cheung, I.Y. Induction of Ab3 and Ab3′ Antibody Was Associated with Long-Term Survival after Anti-GD2 Antibody Therapy of Stage 4 Neuroblastoma. Clin. Cancer Res. 2000, 6, 2653–2660. [Google Scholar] [PubMed]
- Kushner, B.H.; Ostrovnaya, I.; Cheung, I.Y.; Kuk, D.; Kramer, K.; Modak, S.; Yataghene, K.; Cheung, N.K. Prolonged progression-free survival after consolidating second or later remissions of neuroblastoma with Anti-G(D2) immunotherapy and isotretinoin: A prospective Phase II study. Oncoimmunology 2015, 4, e1016704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, N.K.; Kushner, B.H.; Yeh, S.D.; Larson, S.M. 3F8 monoclonal antibody treatment of patients with stage 4 neuroblastoma: A phase II study. Int. J. Oncol. 1998, 12, 1299–1306. [Google Scholar] [CrossRef]
- Chowdhury, F.; Lode, H.N.; Cragg, M.S.; Glennie, M.J.; Gray, J.C. Development of immunomonitoring of antibody-dependent cellular cytotoxicity against neuroblastoma cells using whole blood. Cancer Immunol. Immunother. 2014, 63, 559–569. [Google Scholar] [CrossRef]
- Siebert, N.; Jensen, C.; Troschke-Meurer, S.; Zumpe, M.; Jüttner, M.; Ehlert, K.; Kietz, S.; Müller, I.; Lode, H.N. Neuroblastoma patients with high-affinity FCGR2A, -3A and stimulatory KIR 2DS2 treated by long-term infusion of anti-GD2 antibody ch14.18/CHO show higher ADCC levels and improved event-free survival. Oncoimmunology 2016, 5, e1235108. [Google Scholar] [CrossRef] [Green Version]
- Tarek, N.; Le Luduec, J.-B.; Gallagher, M.M.; Zheng, J.; Venstrom, J.M.; Chamberlain, E.; Modak, S.; Heller, G.; Dupont, B.; Cheung, N.-K.V.; et al. Unlicensed NK cells target neuroblastoma following anti-GD2 antibody treatment. J. Clin. Invest. 2012, 122, 3260–3270. [Google Scholar] [CrossRef] [Green Version]
- Scheid, C.; Pettengell, R.; Ghielmini, M.; Radford, J.A.; Morgenstern, G.R.; Stern, P.L.; Crowther, D. Time-course of the recovery of cellular immune function after high-dose chemotherapy and peripheral blood progenitor cell transplantation for high-grade non-Hodgkin’s lymphoma. Bone Marrow Transplant. 1995, 15, 901–906. [Google Scholar]
- Perez-Garcia, A.; Cabezudo, E.; Lopez-Jimenez, J.; Marugan, I.; Peralta, T.; Arnan, M.; Ramos-Oliva, P.; Benet, I.; Lopez, S.; Mestre, M.; et al. Immune Reconstitution of Regulatory T-Cells Following Autologous Hematopoietic Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2009, 15, 140. [Google Scholar] [CrossRef] [Green Version]
- Ladenstein, R.; Potschger, U.; Valteau-Couanet, D.; Luksch, R.; Castel, V.; Yaniv, I.; Laureys, G.; Brock, P.; Michon, J.M.; Owens, C.; et al. Interleukin 2 with anti-GD2 antibody ch14.18/CHO (dinutuximab beta) in patients with high-risk neuroblastoma (HR-NBL1/SIOPEN): A multicentre, randomised, phase 3 trial. Lancet. Oncol. 2018, 19, 1617–1629. [Google Scholar] [CrossRef]
- Ye, C.; Brand, D.; Zheng, S.G. Targeting IL-2: An unexpected effect in treating immunological diseases. Signal Transduct. Target. Ther. 2018, 3, 2. [Google Scholar] [CrossRef] [Green Version]
- Van Haelst Pisani, C.; Kovach, J.S.; Kita, H.; Leiferman, K.M.; Gleich, G.J.; Silver, J.E.; Dennin, R.; Abrams, J.S. Administration of interleukin-2 (IL-2) results in increased plasma concentrations of IL-5 and eosinophilia in patients with cancer. Blood 1991, 78, 1538–1544. [Google Scholar] [CrossRef] [Green Version]
- Ozkaynak, M.F.; Gilman, A.L.; London, W.B.; Naranjo, A.; Diccianni, M.B.; Tenney, S.C.; Smith, M.; Messer, K.S.; Seeger, R.; Reynolds, C.P.; et al. A Comprehensive Safety Trial of Chimeric Antibody 14.18 With GM-CSF, IL-2, and Isotretinoin in High-Risk Neuroblastoma Patients Following Myeloablative Therapy: Children’s Oncology Group Study ANBL0931. Front. Immunol. 2018, 9, 1355. [Google Scholar] [CrossRef]
- Goldmann, O.; Beineke, A.; Medina, E. Identifcation of a novel subset of myeloid-derived suppressor cells during chronic staphylococcal infection tat resembles immature eosinophils. J. Infect. Dis. 2017, 216, 1444–1451. [Google Scholar] [CrossRef] [Green Version]
- Kushner, B.H.; Cheung, I.Y.; Modak, S.; Kramer, K.; Ragupathi, G.; Cheung, N.-K. V Phase I trial of a bivalent gangliosides vaccine in combination with β-glucan for high-risk neuroblastoma in second or later remission. Clin. Cancer Res. 2014, 20, 1375–1382. [Google Scholar] [CrossRef] [Green Version]
- Qasim, W.; Zhan, H.; Samarasinghe, S.; Adams, S.; Amrolia, P.; Stafford, S.; Butler, K.; Rivat, C.; Wright, G.; Somana, K.; et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl. Med. 2017, 9, eaaj2013. [Google Scholar] [CrossRef]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Somerville, R.P.T.; Lu, T.; Yang, J.C.; Sherry, R.M.; Feldman, S.A.; McIntyre, L.; Bot, A.; Rossi, J.; Lam, N.; et al. Long-Duration Complete Remissions of Diffuse Large B Cell Lymphoma after Anti-CD19 Chimeric Antigen Receptor T Cell Therapy. Mol. Ther. 2017, 25, 2245–2253. [Google Scholar] [CrossRef] [Green Version]
- Tawara, I.; Kageyama, S.; Miyahara, Y.; Fujiwara, H.; Nishida, T.; Akatsuka, Y.; Ikeda, H.; Tanimoto, K.; Terakura, S.; Murata, M.; et al. Safety and persistence of WT1-specific T-cell receptor gene2transduced lymphocytes in patients with AML and MDS. Blood 2017, 130, 1985–1994. [Google Scholar] [CrossRef] [Green Version]
- Tesfaye, M.; Savoldo, B. Adoptive Cell Therapy in Treating Pediatric Solid Tumors. Curr. Oncol. Rep. 2018, 20, 73. [Google Scholar] [CrossRef]
- Grupp, S.A.; Prak, E.L.; Boyer, J.; McDonald, K.R.; Shusterman, S.; Thompson, E.; Callahan, C.; Jawad, A.F.; Levine, B.L.; June, C.H.; et al. Adoptive transfer of autologous T cells improves T-cell repertoire diversity and long-term B-cell function in pediatric patients with neuroblastoma. Clin. Cancer Res. 2012, 18, 6732–6741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanold, J.; Paillard, C.; Tchirkov, A.; Lang, P.; Kelly, A.; Halle, P.; Isfan, F.; Merlin, E.; Marabelle, A.; Rochette, E.; et al. NK Cell immunotherapy for high-risk neuroblastoma relapse after haploidentical HSCT. Pediatr. Blood Cancer 2012, 59, 739–742. [Google Scholar] [CrossRef] [PubMed]
- Federico, S.M.; McCarville, M.B.; Shulkin, B.L.; Sondel, P.M.; Hank, J.A.; Hutson, P.; Meagher, M.; Shafer, A.; Ng, C.Y.; Leung, W.; et al. A pilot trial of humanized anti-GD2 monoclonal antibody (hu14.18K322A) with chemotherapy and natural killer cells in children with recurrent/refractory neuroblastoma. Clin. Cancer Res. 2017, 23, 5441–6449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modak, S.; Le Luduec, J.B.; Cheung, I.Y.; Goldman, D.A.; Ostrovnaya, I.; Doubrovina, E.; Basu, E.; Kushner, B.H.; Kramer, K.; Roberts, S.S.; et al. Adoptive immunotherapy with haploidentical natural killer cells and Anti-GD2 monoclonal antibody m3F8 for resistant neuroblastoma: Results of a phase I study. Oncoimmunology 2018, 7, e1461305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, N.; Kulikovskaya, I.; Barrett, D.M.; Binder-Scholl, G.; Jakobsen, B.; Martinez, D.; Pawel, B.; June, C.H.; Kalos, M.D.; Grupp, S.A. T cells targeting NY-ESO-1 demonstrate efficacy against disseminated neuroblastoma. Oncoimmunology 2016, 5, e1040216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintarelli, C.; Orlando, D.; Boffa, I.; Guercio, M.; Polito, V.A.; Petretto, A.; Lavarello, C.; Sinibaldi, M.; Weber, G.; Del Bufalo, F.; et al. Choice of costimulatory domains and of cytokines determines CAR T-cell activity in neuroblastoma. Oncoimmunology 2018, 7, e1433518. [Google Scholar] [CrossRef]
- Heczey, A.; Liu, D.; Tian, G.; Courtney, A.N.; Wei, J.; Marinova, E.; Gao, X.; Guo, L.; Yvon, E.; Hicks, J.; et al. Invariant NKT cells with chimeric antigen receptor provide a novel platform for safe and effective cancer immunotherapy. Blood 2014, 124, 2824–2833. [Google Scholar] [CrossRef] [Green Version]
- Heczey, A.; Louis, C.U.; Savoldo, B.; Dakhova, O.; Durett, A.; Grilley, B.; Liu, H.; Wu, M.F.; Mei, Z.; Gee, A.; et al. CAR T Cells Administered in Combination with Lymphodepletion and PD-1 Inhibition to Patients with Neuroblastoma. Mol. Ther. 2017, 25, 2214–2224. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Ma, X.; Liu, Y.-C.; Zhao, W.; Yu, L.; Qin, M.; Zhu, G.; Wang, K.; Shi, X.; Zhang, Z.; et al. Chimeric Antigen Receptor 4SCAR-GD2-Modified T Cells Targeting High-Risk and Recurrent Neuroblastoma: A Phase II Multi-Center Trial in China. Blood 2017, 130, 3335. [Google Scholar]
- Künkele, A.; Taraseviciute, A.; Finn, L.S.; Johnson, A.J.; Berger, C.; Finney, O.; Chang, C.A.; Rolczynski, L.S.; Brown, C.; Mgebroff, S.; et al. Preclinical assessment of CD171-directed CAR T-cell adoptive therapy for childhood neuroblastoma: CE7 epitope target safety and product manufacturing feasibility. Clin. Cancer Res. 2017, 23, 466–477. [Google Scholar] [CrossRef] [Green Version]
- Walker, A.J.; Majzner, R.G.; Zhang, L.; Wanhainen, K.; Long, A.H.; Nguyen, S.M.; Lopomo, P.; Vigny, M.; Fry, T.J.; Orentas, R.J.; et al. Tumor Antigen and Receptor Densities Regulate Efficacy of a Chimeric Antigen Receptor Targeting Anaplastic Lymphoma Kinase. Mol. Ther. 2017, 25, 2189–2201. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Sun, C.; Landoni, E.; Metelitsa, L.; Dotti, G.; Savoldo, B. Eradication of neuroblastoma by T cells redirected with an optimized GD2-specific chimeric antigen receptor and interleukin-15. Clin. Cancer Res. 2019, 25, 2915–2924. [Google Scholar] [CrossRef] [Green Version]
- Du, H.; Hirabayashi, K.; Ahn, S.; Kren, N.P.; Montgomery, S.A.; Wang, X.; Tiruthani, K.; Mirlekar, B.; Michaud, D.; Greene, K.; et al. Antitumor Responses in the Absence of Toxicity in Solid Tumors by Targeting B7-H3 via Chimeric Antigen Receptor T Cells. Cancer Cell 2019, 35, 221–237. [Google Scholar] [CrossRef] [Green Version]
- Majzner, R.G.; Theruvath, J.L.; Nellan, A.; Heitzeneder, S.; Cui, Y.; Mount, C.W.; Rietberg, S.P.; Linde, M.H.; Xu, P.; Rota, C.; et al. CAR T cells targeting B7-H3, a pan-cancer antigen, demonstrate potent preclinical activity against pediatric solid tumors and brain tumors. Clin. Cancer Res. 2019, 25, 2560–2574. [Google Scholar] [CrossRef]
- Yankelevich, M.; Kondadasula, S.V.; Thakur, A.; Buck, S.; Cheung, N.K.V.; Lum, L.G. Anti-CD3×anti-GD2 bispecific antibody redirects T-cell cytolytic activity to neuroblastoma targets. Pediatr. Blood Cancer 2012, 59, 1198–1205. [Google Scholar] [CrossRef] [Green Version]
- Yankelevich, M.; Modak, S.; Chu, R.; Lee, D.W.; Thakur, A.; Cheung, N.-K.V.; Lum, L.G. Phase I study of OKT3 x hu3F8 bispecific antibody (GD2Bi) armed T cells (GD2BATs) in GD2-positive tumors. J. Clin. Oncol. 2019, 14, 2533. [Google Scholar] [CrossRef]
- Le, T.P.; Thai, T.H. The state of cellular adoptive immunotherapy for neuroblastoma and other pediatric solid tumors. Front. Immunol. 2017, 8, 1640. [Google Scholar] [CrossRef] [Green Version]
- Zage, P. Novel Therapies for Relapsed and Refractory Neuroblastoma. Children 2018, 5, 148. [Google Scholar] [CrossRef] [Green Version]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef] [Green Version]
- McLellan, A.D.; Ali Hosseini Rad, S.M. Chimeric antigen receptor T cell persistence and memory cell formation. Immunol. Cell Biol. 2019, 97, 664–674. [Google Scholar] [CrossRef]
- Martinez, M.; Moon, E.K. CAR T cells for solid tumors: New strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef] [Green Version]
- Gattinoni, L.; Zhong, X.S.; Palmer, D.C.; Ji, Y.; Hinrichs, C.S.; Yu, Z.; Wrzesinski, C.; Boni, A.; Cassard, L.; Garvin, L.M.; et al. Wnt signaling arrests effector T cell differentiation and generates CD8 + memory stem cells. Nat. Med. 2009, 15, 808–813. [Google Scholar] [CrossRef]
- Louis, C.U.; Savoldo, B.; Dotti, G.; Pule, M.; Yvon, E.; Myers, G.D.; Rossig, C.; Russell, H.V.; Diouf, O.; Liu, E.; et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011, 118, 6050–6056. [Google Scholar] [CrossRef]
- Sommermeyer, D.; Hudecek, M.; Kosasih, P.L.; Gogishvili, T.; Maloney, D.G.; Turtle, C.J.; Riddell, S.R. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 2016, 30, 492–500. [Google Scholar] [CrossRef] [Green Version]
- Turtle, C.J.; Hanafi, L.A.; Berger, C.; Hudecek, M.; Pender, B.; Robinson, E.; Hawkins, R.; Chaney, C.; Cherian, S.; Chen, X.; et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci. Transl. Med. 2016, 30, 492–500. [Google Scholar] [CrossRef] [Green Version]
- Chong, E.A.; Melenhorst, J.J.; Lacey, S.F.; Ambrose, D.E.; Gonzalez, V.; Levine, B.L.; June, C.H.; Schuster, S.J. PD-1 blockade modulates chimeric antigen receptor (CAR)-modified T cells: Refueling the CAR. Blood 2017, 129, 1039–1041. [Google Scholar] [CrossRef] [Green Version]
- Long, A.H.; Highfill, S.L.; Cui, Y.; Smith, J.P.; Walker, A.J.; Ramakrishna, S.; El-Etriby, R.; Galli, S.; Tsokos, M.; Orentas, R.J.; et al. Reduction of MDSCs with all-trans retinoic acid improves CAR therapy efficacy for sarcomas. Cancer Immunol. Res. 2016, 4, 869–880. [Google Scholar] [CrossRef] [Green Version]
- Kotecki, N.; Awada, A. Checkpoints inhibitors in the (neo)adjuvant setting of solid tumors. Curr. Opin. Oncol. 2019, 31, 429–444. [Google Scholar] [CrossRef]
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol. 2018, 62, 29–39. [Google Scholar] [CrossRef]
- Rigo, V.; Emionite, L.; Daga, A.; Astigiano, S.; Corrias, M.V.; Quintarelli, C.; Locatelli, F.; Ferrini, S.; Croce, M. Combined immunotherapy with anti-PDL-1/PD-1 and anti-CD4 antibodies cures syngeneic disseminated neuroblastoma. Sci. Rep. 2017, 25, 14049. [Google Scholar] [CrossRef] [Green Version]
- Siebert, N.; Zumpe, M.; Jüttner, M.; Troschke-Meurer, S.; Lode, H.N. PD-1 blockade augments anti-neuroblastoma immune response induced by anti-GD2 antibody ch14.18/CHO. Oncoimmunology 2017, 6, e1343775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivasan, P.; Wu, X.; Basu, M.; Rossi, C.; Sandler, A.D. PD-L1 checkpoint inhibition and anti-CTLA-4 whole tumor cell vaccination counter adaptive immune resistance: A mouse neuroblastoma model that mimics human disease. PLoS Med. 2018, 15, e1002497. [Google Scholar] [CrossRef] [PubMed]
- Merchant, M.S.; Wright, M.; Baird, K.; Wexler, L.H.; Rodriguez-Galindo, C.; Bernstein, D.; Delbrook, C.; Lodish, M.; Bishop, R.; Wolchok, J.D.; et al. Phase i clinical trial of ipilimumab in pediatric patients with advanced solid tumors. Clin. Cancer Res. 2016, 22, 1364–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, R.; Fleming, V.; Hu, X.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Myeloid-derived suppressor cells hinder the anti-cancer activity of immune checkpoint inhibitors. Front. Immunol. 2018, 9, 1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eissler, N.; Mao, Y.; Brodin, D.; Reuterswärd, P.; Andersson Svahn, H.; Johnsen, J.I.; Kiessling, R.; Kogner, P. Regulation of myeloid cells by activated T cells determines the efficacy of PD-1 blockade. Oncoimmunology 2016, 5, e1232222. [Google Scholar] [CrossRef] [Green Version]
- Eranki, A.; Srinivasan, P.; Ries, M.; Kim, A.; Lazarski, C.A.; Rossi, C.T.; Khokhlova, T.D.; Wilson, E.; Knoblach, S.; Sharma, K.V.; et al. High Intensity Focused Ultrasound (HIFU) Triggers Immune Sensitization of Refractory Murine Neuroblastoma to Checkpoint Inhibitor Therapy. Clin. Cancer Res. 2019. [Google Scholar] [CrossRef] [Green Version]
- Matthay, K.K. Interleukin 2 plus anti-GD2 immunotherapy: Helpful or harmful? Lancet. Oncol. 2018, 19, 1549–1551. [Google Scholar] [CrossRef]
- Nierkens, S.; Lankester, A.C.; Egeler, R.M.; Bader, P.; Locatelli, F.; Pulsipher, M.A.; Bollard, C.M.; Boelens, J.J. Challenges in the harmonization of immune monitoring studies and trial design for cell-based therapies in the context of hematopoietic cell transplantation for pediatric cancer patients. Cytotherapy 2015, 17, 1667–1674. [Google Scholar] [CrossRef]
- Bate-Eya, L.T.; Ebus, M.E.; Koster, J.; den Hartog, I.J.M.; Zwijnenburg, D.A.; Schild, L.; van der Ploeg, I.; Dolman, M.E.M.; Caron, H.N.; Versteeg, R.; et al. Newly-derived neuroblastoma cell lines propagated in serum-free media recapitulate the genotype and phenotype of primary neuroblastoma tumours. Eur. J. Cancer 2014, 50, 628–637. [Google Scholar] [CrossRef]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernández-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Wu, X.; Wang, H.-Y.; Chen, L. Immune contexture defined by single cell technology for prognosis prediction and immunotherapy guidance in cancer. Cancer Commun. 2019, 39, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
| Flow Cytometry | ||||
|---|---|---|---|---|
| Number of Unique Patient Samples Measured Including Material | Tumor Characteristics | Markers | Reference | |
| 8 | Primary Tumor | I (4x) + II (1x) + III (2x) + IV (1x) | CD3, CD4, CD8, CD25, CD45RA, CCR7 | Carlson et al. 2013 [43] |
| PB | ||||
| 26 | Primary Tumor | II (2x) + III (2x) + IV (21x) + IVs (1x) 15 MYCN amp, 11 non MYCN amp | GD2 | Mussai et al. 2015 [24] |
| PB | CD15, CD14, CD11b | |||
| 20 | BM | IV (20x) | CD45, CD33, CD14, GD2, CD56 | Song et al. 2009 [26] |
| 41 | PB | 13 MYCN amp, 28 non MYCN amp | CD4, CD25, CD127, CD45RO, CD49b, LAG-3 | Morandi et al. 2015 [29] |
| 5 | PB | High-Risk Patients | HLA-DR, CD33, CD11b | Gowda et al. 2013 [44] |
| 21 | PB | 7 MYCN amp, 20 non MYCN amp (14 localized, 13 metastatic) | CD4, CD25, CD127, CD45RO, CD49b, LAG-3 | Morandi et al. 2016 [41] |
| 27 | BM | |||
| 59 | PB | 23 localized, 36 metastatic tumors | CD8, NKp46, CD4, CD16, CD56, NKp30, DNAM-1, CD127, CD25, CD14, CD45, CD15, GD2, CD235a, CD9, CD81, TCRgd, NKp44, NKp80, CD3e, CD158a/h, CD158b, CD158e/k, CD158i, FoxP3 | Semeraro et al. 2015 [39] |
| BM | ||||
| Immunohistochemistry | ||||
| 24 | Primary Tumor | 7 MYCN amp, 26 non MYCN amp | CD68 | Apps et al. 2013 [45] |
| 21 | CD3 | |||
| 19 | pSTAT3 | |||
| 8 | Primary Tumor | I (4x) + II (1x) + III (2x) + IV(1x) | Ki67, CD3 | Carlson et al. 2013 [43] |
| 15 | Primary Tumor | 3 low risk, 6 intermediate risk, 6 high risk | CD4, CD45 | Zhang et al. 2017 [25] |
| 129 | Primary Tumor | IV (129x) | CD1d, Vα24-Jα18inv, TCRα6β11 | Song et al. 2017 [26] |
| 71 | Primary Tumor | stage I–III (n = 29), stage IV (n = 31), stage IVS (n = 11) | CD163, AIF1 | Asgharzadeh et al. 2012 [27] |
| 84 | Primary Tumor | I (34x) + II (19x) + III (5x) + IV (20x) + IVS (6x) | CD3, CD4, CD8, CD25, FOXP3, Ki67, β2m-free MHC1 heavy chain | Mina et al. 2015 [8] |
| Elisa | ||||
| 57 | Plasma | IV (49x) + non IV (8x) | IL-10, ARG-1 (57) | Morandi et al. 2015 [29] |
| 53 | PB | PB: I (8x) + II (8x) + III (6x) + IV (28x) + IVS (3x) | IL-6 | Egler et al. 2008 [46] |
| 18 | BM | BM: I (1x) + II (3x) + III (2x) + IV (11x) + IVS (1x) | ||
| 35 | PB | PB: I (5x) + II (5x) + III (3x) + IV (20x) + IVS (2x) | sIL-6R | |
| 16 | BM | BM: I (1x) + II (2x) + III (2x) + IV (10x) + IVS (1x) | ||
| 84 | Primary Tumor | I (7x) + II (8x) + III (22x) + IV (42x) + 4S (5x) 27 MYCN amp, 57 non MYCN amp | sHLA-F, sB7H3, sHLA-E | Morandi et al. 2013 [33] |
| Luminex | ||||
| 55 | Plasma | 20 low-risk, 35 high-risk In addition, 28 HR blood samples from 7 patients at various timepoints during treatment | GM-CSF, G-CSF, IFNγ, IL-1a, IL-1ra, IL-1b, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-12p70, IL-12p40, IL-13, IL-15, IL-17, MCP-1, MCP-3, MDC, TNFα, TNFβ, TGFα, Eotaxin, IFNα2, IP-10, MIP-1a, MIP-1b, EGF, FGF-2, FLT3L, Fractalkine, GRO, VEGF, sCD40L, sIL-2Ra | Egler et al. 2011 [32] |
| PB | ||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szanto, C.L.; Cornel, A.M.; Vijver, S.V.; Nierkens, S. Monitoring Immune Responses in Neuroblastoma Patients during Therapy. Cancers 2020, 12, 519. https://doi.org/10.3390/cancers12020519
Szanto CL, Cornel AM, Vijver SV, Nierkens S. Monitoring Immune Responses in Neuroblastoma Patients during Therapy. Cancers. 2020; 12(2):519. https://doi.org/10.3390/cancers12020519
Chicago/Turabian StyleSzanto, Celina L., Annelisa M. Cornel, Saskia V. Vijver, and Stefan Nierkens. 2020. "Monitoring Immune Responses in Neuroblastoma Patients during Therapy" Cancers 12, no. 2: 519. https://doi.org/10.3390/cancers12020519
APA StyleSzanto, C. L., Cornel, A. M., Vijver, S. V., & Nierkens, S. (2020). Monitoring Immune Responses in Neuroblastoma Patients during Therapy. Cancers, 12(2), 519. https://doi.org/10.3390/cancers12020519