Cannabinoids and Hormone Receptor-Positive Breast Cancer Treatment



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Hormone-Receptor Positive Breast Cancer
2. Cannabinoids and the Endocannabinoid System
3. Cannabinoid Receptor 1
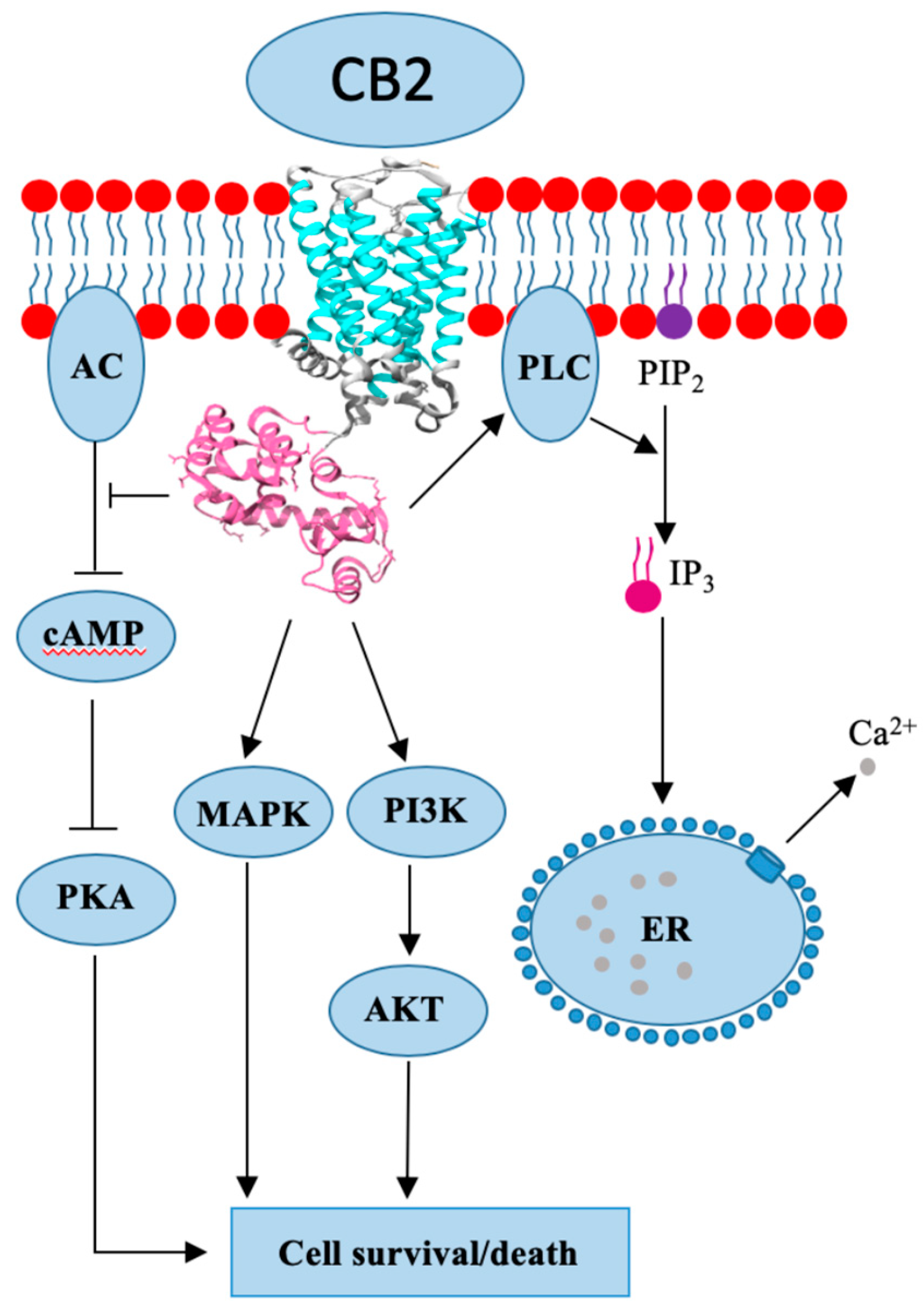
4. Cannabinoid Receptor 2
5. Cannabinoids in Connection with the Hypothalamic-Pituitary-Gonadal Axis
6. Cannabinoids and Hormone Receptor-Positive Breast Cancer (Preclinical Evidence)
7. Cannabinoids and Hormone Receptor-Positive Breast Cancer (Clinical Evidence)
8. Cannabinoids and Specific Hormone Receptor-Positive Breast Cancer Treatments
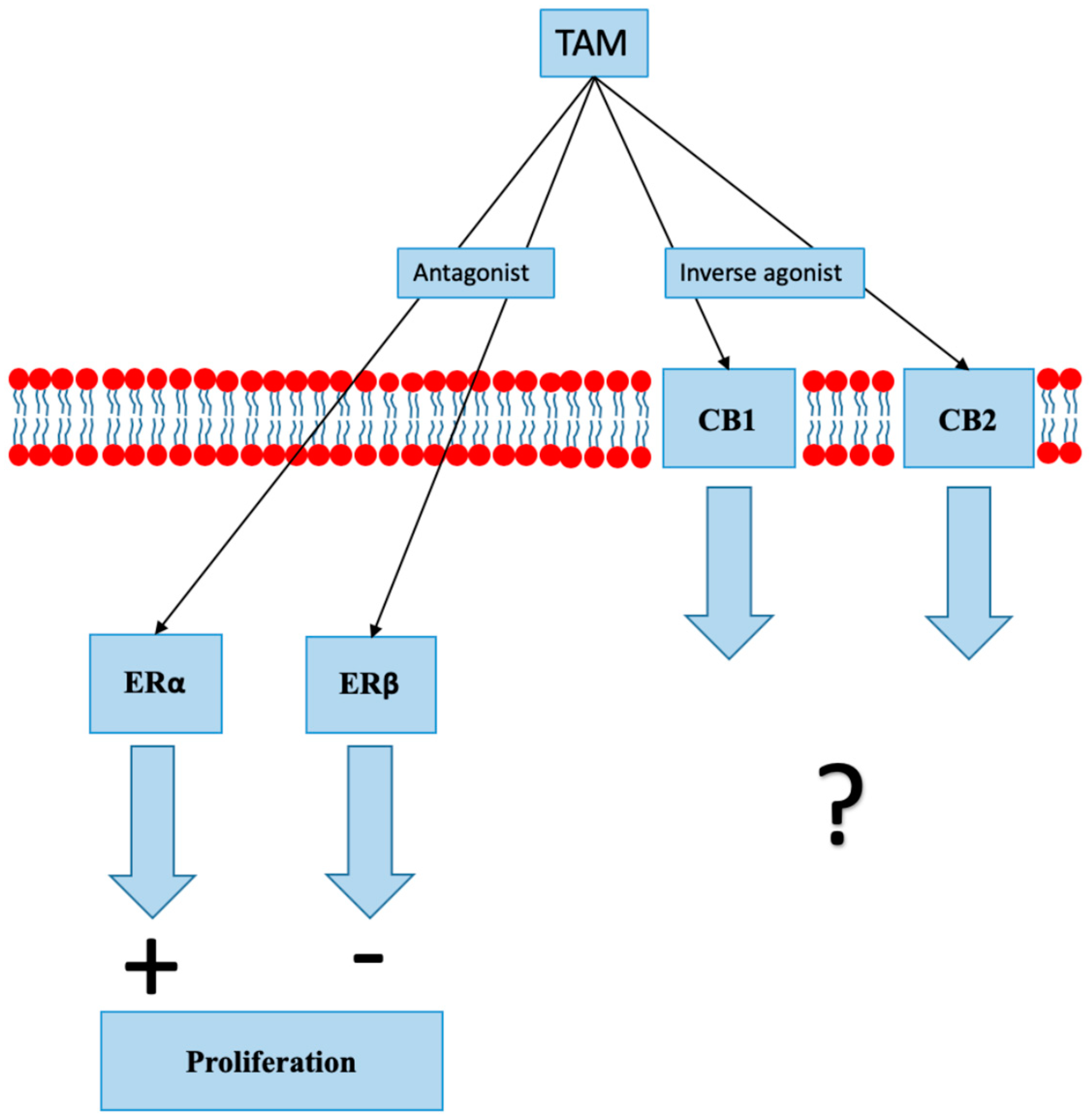
8.1. Selective Estrogen Receptor Modulators
8.2. Gonadotropin-Releasing Hormone Agonists
8.3. Aromatase Inhibitors
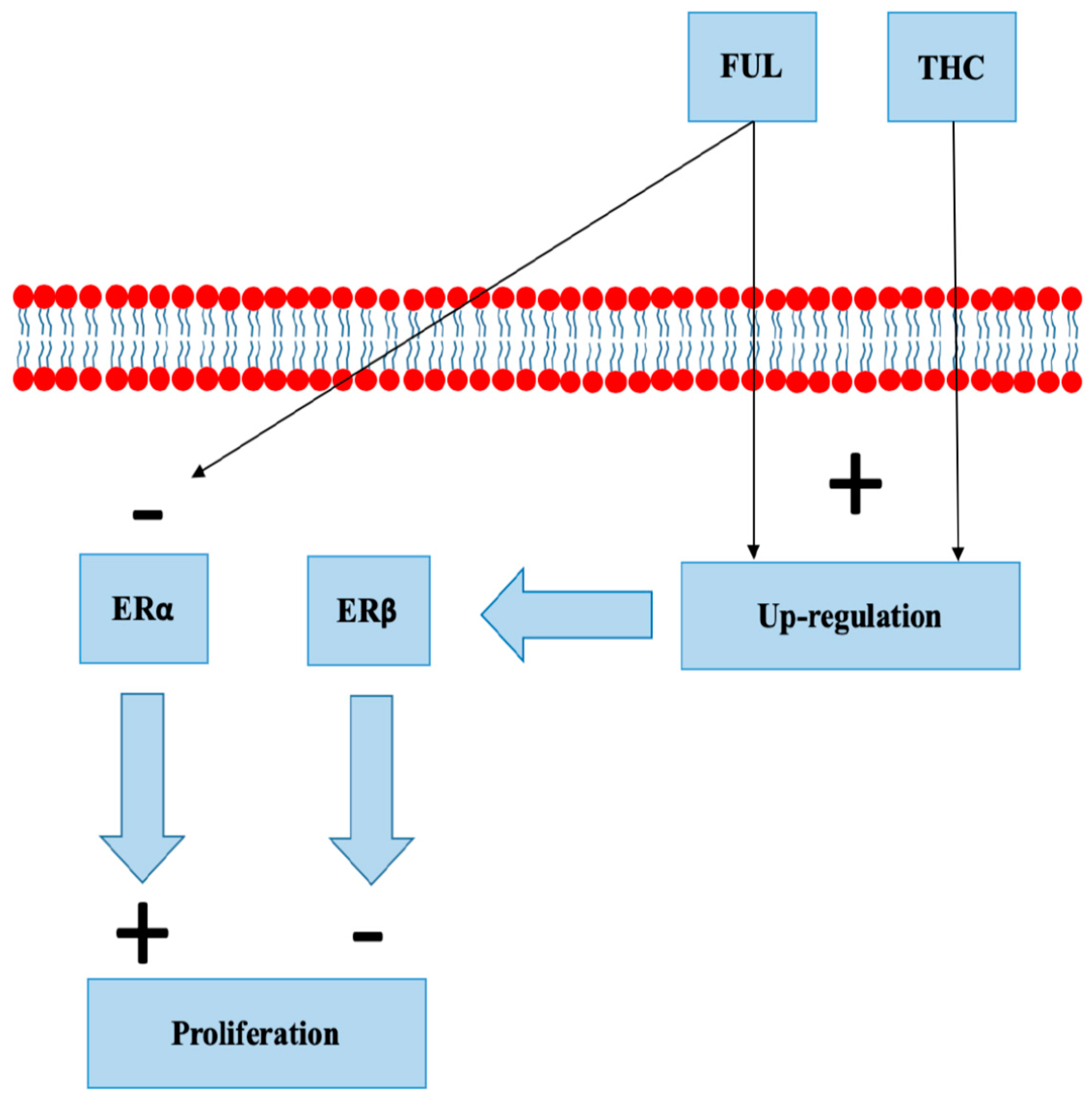
8.4. Selective Estrogen Receptor Degraders
8.5. Inhibitors of Cyclin Dependent Kinases
8.6. mTOR and PI3K Inhibitors
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ghoncheh, M.; Pournamdar, Z.; Salehiniya, H. Incidence and Mortality and Epidemiology of Breast Cancer in the World. Asian Pac. J. Cancer Prev. 2016, 17, 43–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozeboom, B.; Dey, N.; De, P. ER+ metastatic breast cancer: Past, present, and a prescription for an apoptosis-targeted future. Am. J. Cancer Res. 2019, 9, 2821–2831. [Google Scholar] [PubMed]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Mosly, D.; Turnbull, A.; Sims, A. Predictive markers of endocrine response in breast cancer. World J. Exp. Med. 2018, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Krop, I.; Ismaila, N.; Andre, F. Use of Biomarkers to Guide Decisions on Adjuvant Systemic Therapy for Women with Early-Stage Invasive Breast Cancer: American Society of Clinical Oncology Clinical Practice Guideline Focused Update. J. Clin. Oncol. 2017, 35, 2838–2847. [Google Scholar] [CrossRef] [PubMed]
- Burstein, H.J.; Curigliano, G.; Loibl, S. Estimating the benefits of therapy for early-stage breast cancer: The St. Gallen International Consensus Guidelines for the primary therapy of early breast cancer 2019. Ann. Oncol. 2019, 30, 1541–1557. [Google Scholar] [CrossRef] [Green Version]
- Szostakowska, M.; Trębińska-Stryjewska, A.; Grzybowska, E.A. Resistance to endocrine therapy in breast cancer: Molecular mechanisms and future goals. Breast Cancer Res. Treat. 2019, 173, 489–497. [Google Scholar] [CrossRef] [Green Version]
- Balfe, P.J.; McCann, A.H.; Welch, H.M. Estrogen receptor beta and breast cancer. Eur. J. Surg. Oncol. 2004, 30, 1043–1050. [Google Scholar] [CrossRef]
- Lumachi, F.; Brunello, A.; Maruzzo, M. Treatment of estrogen receptor-positive breast cancer. Curr. Med. Chem. 2013, 20, 596–604. [Google Scholar] [CrossRef]
- Yoko, O.; Hirotaka, I. Clinical significance of estrogen receptor β in breast and prostate cancer from biological aspects. Cancer Sci. 2015, 106, 337–343. [Google Scholar]
- Ryberg, E.; Larsson, N.; Sjögren, S. The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol. 2007, 152, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- McHugh, D.; Hu, S.S.; Rimmerman, N. N-arachidonoyl glycine, an abundant endogenous lipid, potently drives directed cellular migration through GPR18, the putative abnormal cannabidiol receptor. BMC Neurosci. 2010, 11, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, A.J. Novel cannabinoid receptors. Br. J. Pharmacol. 2007, 152, 567–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Marzo, V.; Piscitelli, F. The Endocannabinoid System and its Modulation by Phytocannabinoids. Neurotherapeutics 2015, 12, 692–698. [Google Scholar] [CrossRef]
- Console-Bram, L.; Marcu, J.; Abood, M.E. Cannabinoid receptors: Nomenclature and pharmacological principles. Prog. Neuropsychopharmacol. Biol. Psychiatry 2012, 38, 4–15. [Google Scholar] [CrossRef] [Green Version]
- Pisanti, S.; Malfitano, A.M.; Ciaglia, E. Cannabidiol: State of the art and new challenges for therapeutic applications. Pharmacol. Ther. 2017, 175, 133–150. [Google Scholar] [CrossRef]
- Thomas, A.; Baillie, G.L.; Phillips, A.M. Cannabidiol displays unexpectedly high potency as an antagonist of CB1 and CB2 receptor agonists in vitro. Br. J. Pharmacol. 2007, 150, 613–623. [Google Scholar] [CrossRef] [Green Version]
- Mills, B.; Yepes, A.; Nugent, K. Synthetic Cannabinoids. Am. J. Med. Sci. 2015, 350, 59–62. [Google Scholar] [CrossRef]
- Castaneto, M.S.; Gorelick, D.A.; Desrosiers, N.A. Synthetic cannabinoids: Epidemiology, pharmacodynamics, and clinical implications. Drug Alcohol Depend. 2014, 144, 12–41. [Google Scholar] [CrossRef] [Green Version]
- Badowski, M.E.; Yanful, P.K. Dronabinol oral solution in the management of anorexia and weight loss in AIDS and cancer. Ther. Clin. Risk Manag. 2018, 14, 643–651. [Google Scholar] [CrossRef] [Green Version]
- Walsh, D.; Nelson, K.A.; Mahmoud, F.A. Established and potential therapeutic applications of cannabinoids in oncology. Support Care Cancer. 2003, 11, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Arnau, B.G.; Edgar, S.-G.; Luigi, B. Version 1. F1000Res. F1000 Faculty Rev-990. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4879932/ (accessed on 22 February 2020).
- Ryberg, E.; Vu, H.K.; Larsson, N. Identification and characterisation of a novel splice variant of the human CB1 receptor. FEBS Lett. 2005, 579, 259–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, S.; Kumar, U. Cannabinoid Receptors and the Endocannabinoid System: Signaling and Function in the Central Nervous System. Int. J. Mol. Sci. 2018, 19, 833. [Google Scholar]
- Pisanti, S.; Picardi, P.; D’Alessandro, A. The endocannabinoid signaling system in cancer. Trends Pharmacol. Sci. 2013, 34, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.-R. Species differences in cannabinoid receptor 2 (CNR2 gene): Identification of novel human and rodent CB2 isoforms, differential tissue expression, and regulation by cannabinoid receptor ligands. Genes Brain Behav. 2009, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Turcotte, C.; Blanchet, M.R.; Laviolette, M. The CB2 receptor and its role as a regulator of inflammation. Cell Mol. Life Sci. 2016, 73, 4449–4470. [Google Scholar] [CrossRef] [Green Version]
- Tyrey, L. Delta-9-Tetrahydrocannabinol suppression of episodic luteinizing hormone secretion in the ovariectomized rat. Endocrinology 1978, 102, 1808–1814. [Google Scholar] [CrossRef]
- Tyrey, L. Delta 9-Tetrahydrocannabinol. A potent inhibitor of episodic luteinizing hormone secretion. J. Pharmacol. Exp. Ther. 1980, 213, 306–308. [Google Scholar]
- Kumar, M.S.; Chen, C.L. Effect of an acute dose of delta 9-THC on hypothalamic luteinizing hormone releasing hormone and met-enkephalin content and serum levels of testosterone and corticosterone in rats. Subst Alcohol Actions Misuse 1983, 4, 37–43. [Google Scholar]
- Scorticati, C.; Fernández-Solari, J.; De Laurentiis, A. The inhibitory effect of anandamide on luteinizing hormone-releasing hormone secretion is reversed by estrogen. Proc. Natl. Acad. Sci. USA 2004, 101, 11891–11896. [Google Scholar] [CrossRef] [Green Version]
- Gammon, C.M.; Freeman, G.M.J.; Xie, W. Regulation of gonadotropin-releasing hormone secretion by cannabinoids. Endocrinology 2005, 146, 4491–4499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cravatt, B.F.; Giang, D.K.; Mayfield, S.P. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 1996, 384, 83–87. [Google Scholar] [CrossRef] [PubMed]
- MacCarrone, M.; De Felici, M.; Bari, M. Downregulation of anandamide hydrolase in mouse uterus by sex hormones. Eur. J. Biochem. 2000, 267, 2991–2997. [Google Scholar] [CrossRef] [PubMed]
- El-Talatini, M.R.; Taylor, A.H.; Konje, J.C. The relationship between plasma levels of the endocannabinoid, anandamide, sex steroids, and gonadotropins during the menstrual cycle. Fertil Steril 2010, 93, 1989–1996. [Google Scholar] [CrossRef]
- Cui, N.; Wang, L.; Wang, W. The correlation of anandamide with gonadotrophin and sex steroid hormones during the menstrual cycle. Iran. J. Basic Med. Sci. 2017, 20, 1268–1274. [Google Scholar]
- Gorzalka, B.B.; Dang, S.S. Minireview: Endocannabinoids and gonadal hormones: Bidirectional interactions in physiology and behavior. Endocrinology 2012, 153, 1016–1024. [Google Scholar] [CrossRef] [Green Version]
- Dobovišek, L.; Hojnik, M.; Ferk, P. Overlapping molecular pathways between cannabinoid receptors type 1 and 2 and estrogens/androgens on the periphery and their involvement in the pathogenesis of common diseases (Review). Int. J. Mol. Med. 2016, 38, 1642–1651. [Google Scholar] [CrossRef] [Green Version]
- De Petrocellis, L.; Melck, D.; Palmisano, A. The endogenous cannabinoid anandamide inhibits human breast cancer cell proliferation. Proc. Natl. Acad. Sci. USA 1998, 95, 8375–8380. [Google Scholar] [CrossRef] [Green Version]
- Hanlon, K.E.; Lozano-Ondoua, A.N.; Umaretiya, P.J. Modulation of breast cancer cell viability by a cannabinoid receptor 2 agonist, JWH-015, is calcium dependent. Breast Cancer 2016, 8, 59–71. [Google Scholar]
- Melck, D.; Rueda, D.; Galve-Roperh, I. Involvement of the cAMP/protein kinase A pathway and of mitogen-activated protein kinase in the anti-proliferative effects of anandamide in human breast cancer cells. FEBS Lett. 1999, 463, 235–240. [Google Scholar] [CrossRef] [Green Version]
- Melck, D.; De Petrocellis, L.; Orlando, P. Suppression of nerve growth factor Trk receptors and prolactin receptors by endocannabinoids leads to inhibition of human breast and prostate cancer cell proliferation. Endocrinology 2000, 141, 118–126. [Google Scholar] [CrossRef] [PubMed]
- von Bueren, A.O.; Schlumpf, M.; Lichtensteiger, W. Delta(9)-tetrahydrocannabinol inhibits 17beta-estradiol-induced proliferation and fails to activate androgen and estrogen receptors in MCF7 human breast cancer cells. Anticancer Res. 2008, 28, 85–89. [Google Scholar] [PubMed]
- Ruh, M.F.; Taylor, J.A.; Howlett, A.C. Failure of cannabinoid compounds to stimulate estrogen receptors. Biochem. Pharmacol. 1997, 53, 35–41. [Google Scholar] [CrossRef]
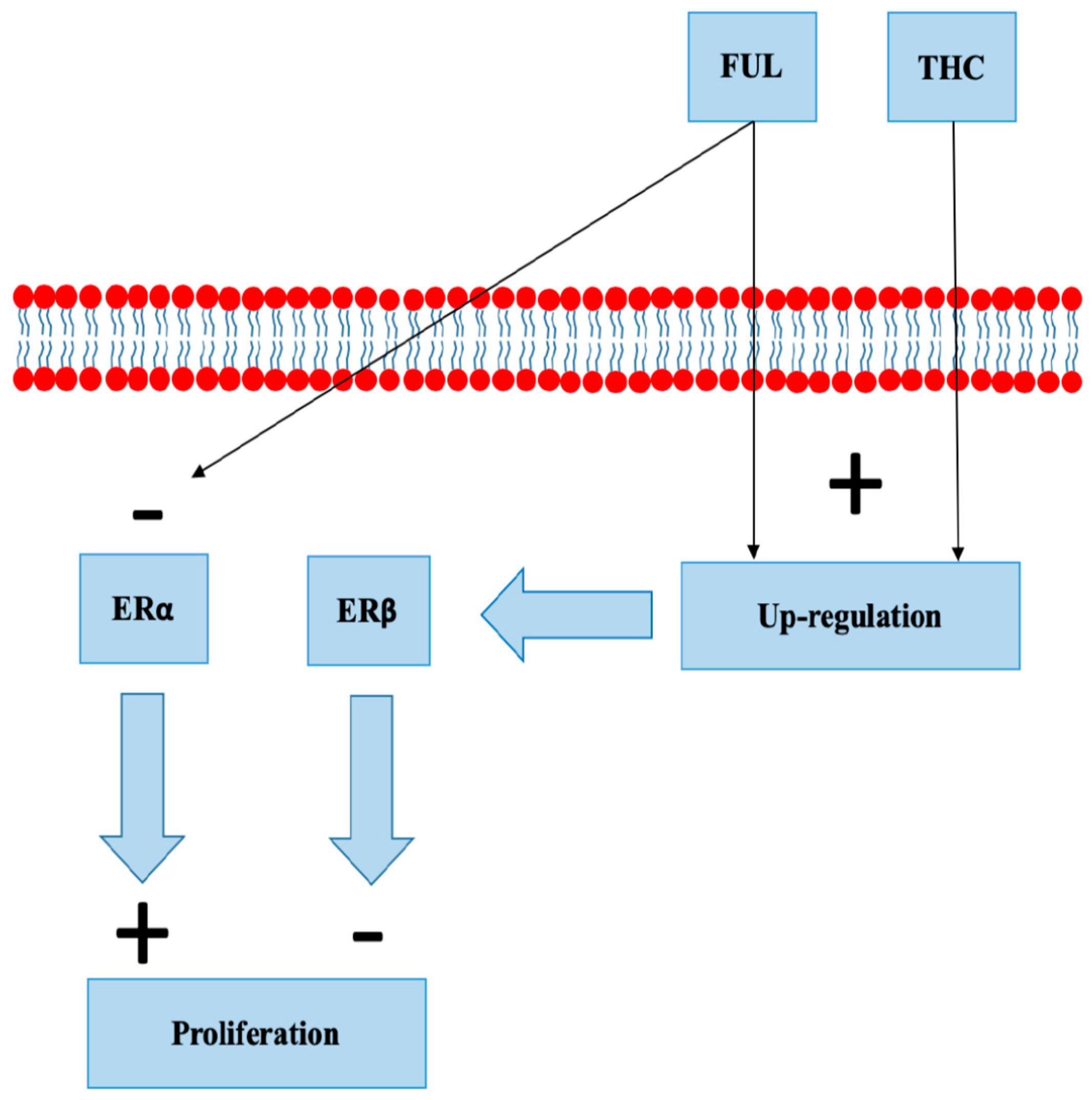
- Takeda, S.; Yoshida, K.; Nishimura, H. Δ(9)-Tetrahydrocannabinol disrupts estrogen-signaling through up-regulation of estrogen receptor β (ERβ). Chem. Res. Toxicol. 2013, 26, 1073–1079. [Google Scholar] [CrossRef] [Green Version]
- Takeda, S.; Yamamoto, I.; Watanabe, K. Modulation of Delta9-tetrahydrocannabinol-induced MCF-7 breast cancer cell growth by cyclooxygenase and aromatase. Toxicology 2009, 259, 25–32. [Google Scholar] [CrossRef]
- Sarnataro, D.; Pisanti, S.; Santoro, A.; Gazzerro, P. The cannabinoid CB1 receptor antagonist rimonabant (SR141716) inhibits human breast cancer cell proliferation through a lipid raft-mediated mechanism. Mol. Pharmacol. 2006, 70, 1298–1306. [Google Scholar] [CrossRef] [Green Version]
- Blasco-Benito, S.; Seijo-Vila, M.; Caro-Villalobos, M. Appraising the “entourage effect”: Antitumor action of a pure cannabinoid versus a botanical drug preparation in preclinical models of breast cancer. Biochem. Pharmacol. 2018, 157, 285–293. [Google Scholar] [CrossRef]
- Pérez-Gómez, E.; Andradas, C.; Blasco-Benito, S. Role of cannabinoid receptor CB2 in HER2 pro-oncogenic signaling in breast cancer. J. Natl. Cancer Inst. 2015, 107, 77. [Google Scholar] [CrossRef] [Green Version]
- Andradas, C.; Blasco-Benito, S.; Castillo-Lluva, S. Activation of the orphan receptor GPR55 by lysophosphatidylinositol promotes metastasis in triple-negative breast cancer. Oncotarget 2016, 7, 47565–47575. [Google Scholar] [CrossRef]
- International Breast Cancer Study Group; Colleoni, M.; Gelber, S. Tamoxifen after adjuvant chemotherapy for premenopausal women with lymph node-positive breast cancer: International Breast Cancer Study Group Trial 13–93. J. Clin. Oncol. 2006, 24, 1332–1341. [Google Scholar]
- Radin, D.P.; Patel, P. Delineating the molecular mechanisms of tamoxifen’s oncolytic actions in estrogen receptor-negative cancers. Eur. J. Pharmacol. 2016, 781, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Barkhem, T.; Carlsson, B.; Nilsson, Y. Differential response of estrogen receptor alpha and estrogen receptor beta to partial estrogen agonists/antagonists. Mol. Pharmacol. 1998, 54, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Viedma-Rodríguez, R.; Baiza-Gutman, L.; Salamanca-Gómez, F. Mechanisms associated with resistance to tamoxifen in estrogen receptor-positive breast cancer (review). Oncol. Rep. 2014, 32, 3–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prather, P.L.; FrancisDevaraj, F.; Dates, C.R. CB1 and CB2 receptors are novel molecular targets for Tamoxifen and 4OH-Tamoxifen. Biochem. Biophys. Res. Commun. 2013, 441, 339–343. [Google Scholar] [CrossRef] [Green Version]
- Franks, L.N.; Ford, B.M.; Prather, P.L. Selective Estrogen Receptor Modulators: Cannabinoid Receptor Inverse Agonists with Differential CB1 and CB2 Selectivity. Front Pharmacol. 2016, 7, 503. [Google Scholar] [CrossRef] [Green Version]
- Nelson, H.D.; Smith, M.E.; Griffin, J.C.; Fu, R. Use of medications to reduce risk for primary breast cancer: A systematic review for the U.S. Preventive Services Task Force. Ann. Intern. Med. 2013, 158, 604. [Google Scholar] [CrossRef] [Green Version]
- Mauri, D.; Pavlidis, N.; Polyzos, N.P.; Ioannidis, J.P. Survival with aromatase inhibitors and inactivators versus standard hormonal therapy in advanced breast cancer: Meta-analysis. J. Natl. Cancer Inst. 2006, 98, 1285. [Google Scholar] [CrossRef] [Green Version]
- Campos, S.M.; Guastalla, J.P.; Subar, M. A comparative study of exemestane versus anastrozole in patients with postmenopausal breast cancer with visceral metastases. Clin. Breast Cancer 2009, 9, 39. [Google Scholar] [CrossRef]
- Lee, C.I.; Goodwin, A.; Wilcken, N. Fulvestrant for hormone-sensitive metastatic breast cancer. Cochrane Database Syst. Rev. 2017, 1, CD011093. [Google Scholar] [CrossRef]
- DeFriend, D.J.; Howell, A.; Nicholson, R.I. Investigation of a new pure antiestrogen (ICI 182780) in women with primary breast cancer. Cancer Res. 1994, 54, 408. [Google Scholar]
- Howell, A.; Robertson, J.F.; Abram, P. Comparison of fulvestrant versus tamoxifen for the treatment of advanced breast cancer in postmenopausal women previously untreated with endocrine therapy: A multinational, double-blind, randomized trial. J. Clin. Oncol. 2004, 22, 1605. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.K.; Abrahamsson, A.; Dabrosin, C. Fulvestrant inhibits growth of triple negative breast cancer and synergizes with tamoxifen in ERα positive breast cancer by up-regulation of ERβ. Oncotarget 2016, 7, 56876–56888. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S. The Cyclin-Dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): A randomised phase 2 study. Lancet Oncol. 2015, 16, 25–35. [Google Scholar] [CrossRef]
- Tripathy, D. Ribociclib plus endocrine therapy for premenopausal women with hormone-receptor-positive, advanced breast cancer (MONALEESA-7): A randomised phase 3 trial. Lancet Oncol. 2018, 18, 904–915. [Google Scholar] [CrossRef]
- Johnston, S. MONARCH 3 final PFS: A randomized study of abemaciclib as initial therapy for advanced breast cancer. NPJ Breast Cancer 2019, 17, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matutino, A.; Joy, A.A.; Brezden-Masley, C. Hormone receptor–positive, HER2-negative metastatic breast cancer: Redrawing the lines. Curr. Oncol. 2018, 25, S131–S141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laezza, C.; Pisanti, S.; Crescenzi, E. Anandamide inhibits Cdk2 and activates Chk1 leading to cell cycle arrest in human breast cancer cells. FEBS Lett. 2006, 580, 6076–6082. [Google Scholar] [CrossRef] [Green Version]
- Caffarel, M.M.; Sarrió, D.; Palacios, J. Delta9-tetrahydrocannabinol inhibits cell cycle progression in human breast cancer cells through Cdc2 regulation. Cancer Res. 2006, 66, 6615–6621. [Google Scholar] [CrossRef] [Green Version]
- Mayer, I.A.; Abramson, V.G.; Formisano, L. A Phase Ib Study of Alpelisib (BYL719), a PI3Kα-Specific Inhibitor, with Letrozole in ER+/HER2-Metastatic Breast Cancer. Clin. Cancer Res. 2017, 23, 26–34. [Google Scholar] [CrossRef] [Green Version]
- Hasskarl, J. Everolimus. Recent Results Cancer Res. 2018, 211, 101–123. [Google Scholar]
- André, F.; Ciruelos, E.; Rubovszky, G. Alpelisib for PIK3CA-Mutated, Hormone Receptor–Positive Advanced Breast Cancer. N. Engl. J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, A.; Kuzontkoski, P.M.; Groopman, J.E. Cannabidiol induces programmed cell death in breast cancer cells by coordinating the cross-talk between apoptosis and autophagy. Mol. Cancer Ther. 2011, 10, 1161–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elbaz, M.; Nasser, M.W.; Ravi, J. Modulation of the tumor microenvironment and inhibition of EGF/EGFR pathway: Novel anti-tumor mechanisms of Cannabidiol in breast cancer. Mol. Oncol. 2015, 9, 906–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caffarel, M.M.; Andradas, C.; Mira, E. Cannabinoids reduce ErbB2-driven breast cancer progression through Akt inhibition. Mol. Cancer 2010, 9, 196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kisková, T.; Mungenast, F.; Suváková, M. Future Aspects for Cannabinoids in Breast Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 1673. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dobovišek, L.; Krstanović, F.; Borštnar, S.; Debeljak, N. Cannabinoids and Hormone Receptor-Positive Breast Cancer Treatment. Cancers 2020, 12, 525. https://doi.org/10.3390/cancers12030525
Dobovišek L, Krstanović F, Borštnar S, Debeljak N. Cannabinoids and Hormone Receptor-Positive Breast Cancer Treatment. Cancers. 2020; 12(3):525. https://doi.org/10.3390/cancers12030525
Chicago/Turabian StyleDobovišek, Luka, Fran Krstanović, Simona Borštnar, and Nataša Debeljak. 2020. "Cannabinoids and Hormone Receptor-Positive Breast Cancer Treatment" Cancers 12, no. 3: 525. https://doi.org/10.3390/cancers12030525
APA StyleDobovišek, L., Krstanović, F., Borštnar, S., & Debeljak, N. (2020). Cannabinoids and Hormone Receptor-Positive Breast Cancer Treatment. Cancers, 12(3), 525. https://doi.org/10.3390/cancers12030525