The Disturbed Iron Phenotype of Tumor Cells and Macrophages in Renal Cell Carcinoma Influences Tumor Growth

 , , , and
, , , and
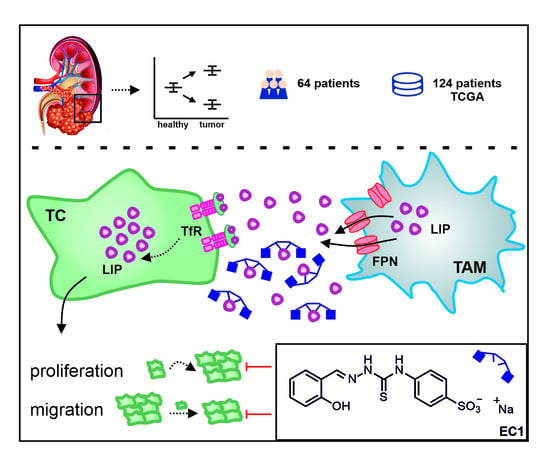
Abstract
:
1. Introduction
2. Results
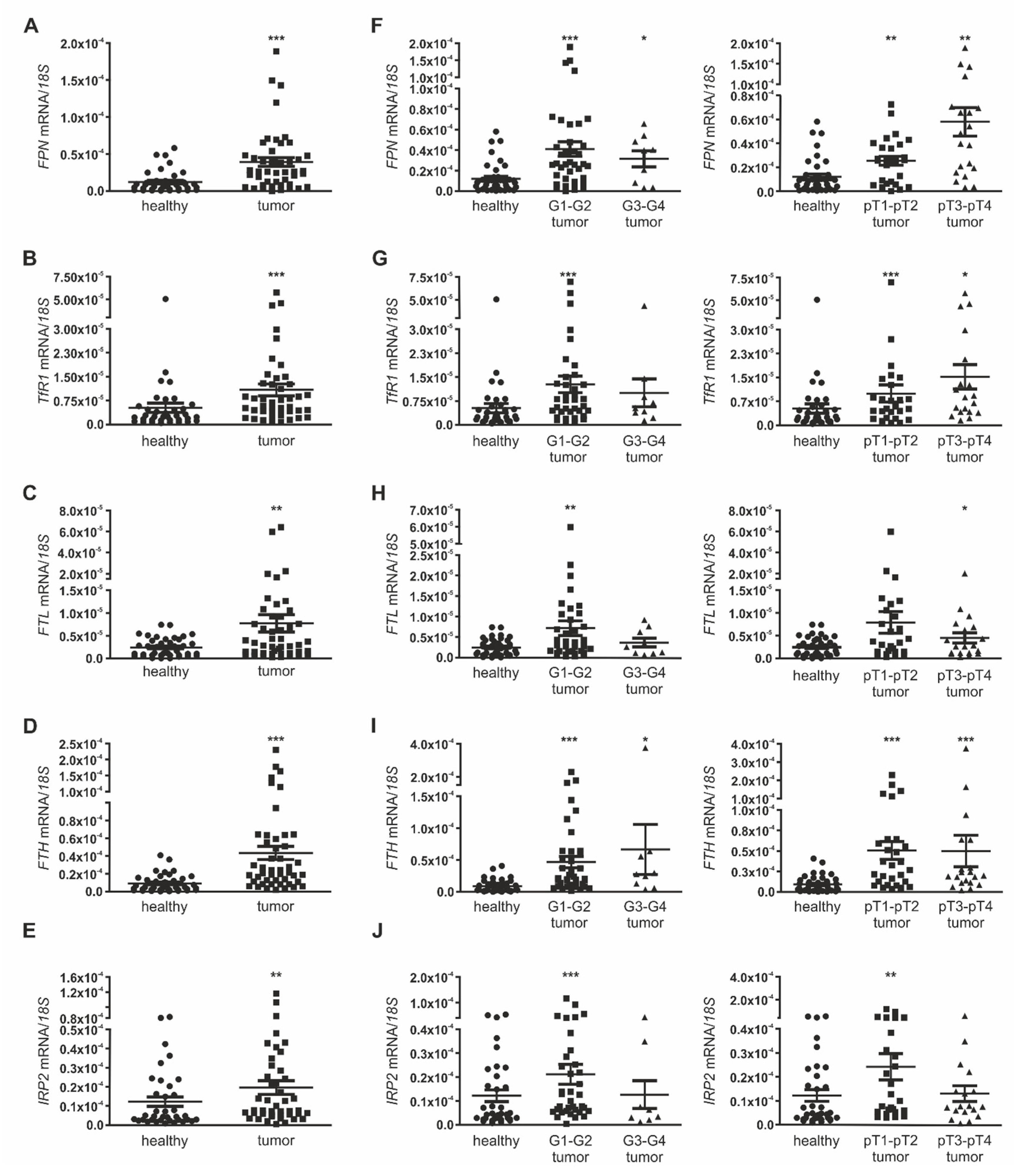
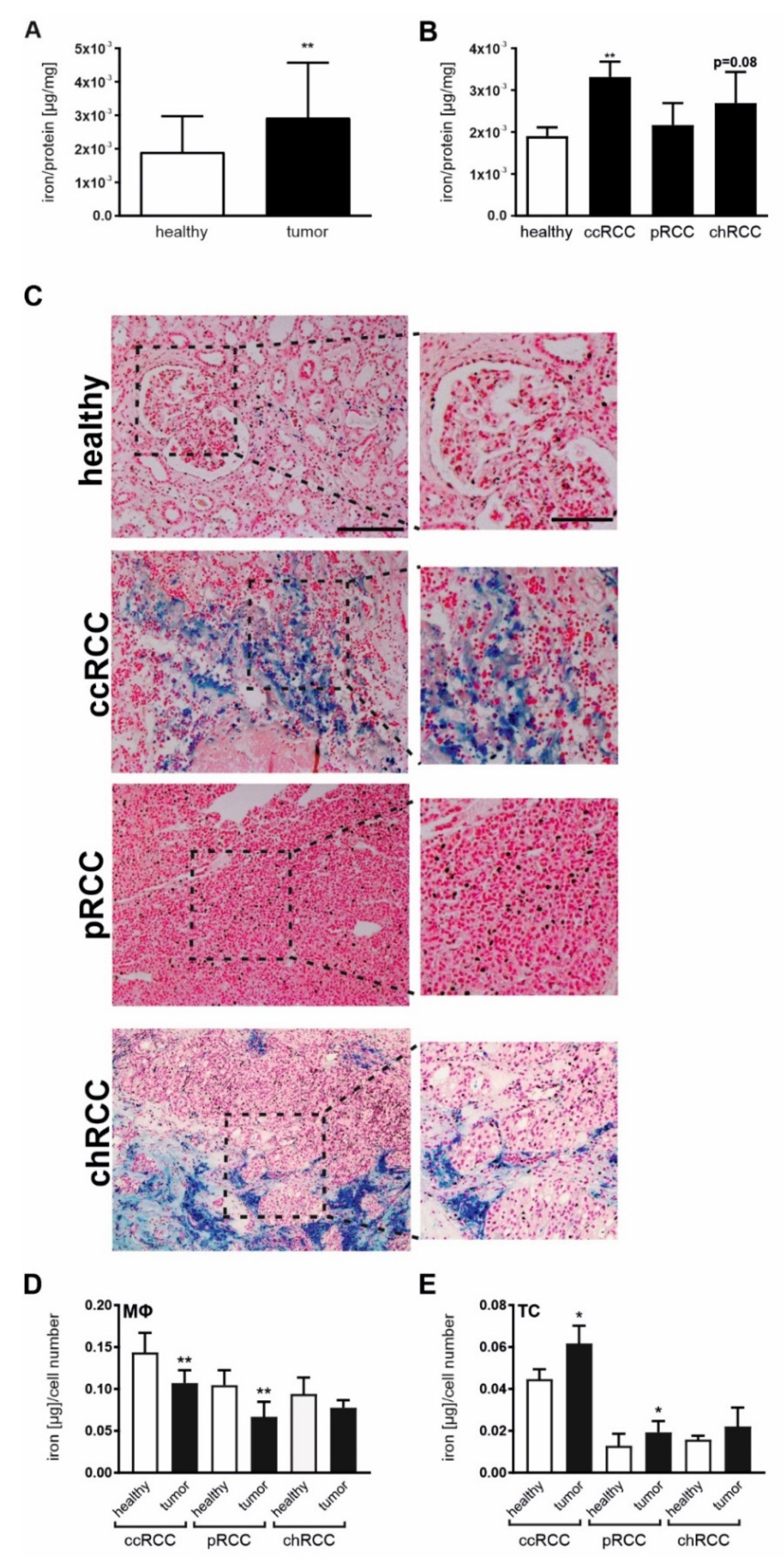
2.1. Iron Homeostasis Is Altered in RCC
2.2. Iron Promotes Renal Tumor Cell Growth
2.3. Tumor Proliferation by Macrophage-Secreted Iron Is Suppressed by EC1
2.4. Macrophage-Derived Iron Is Exported Via the Iron Exporter FPN, Which Is Positively Associated with Poor Patient Outcome
3. Discussion
4. Materials and Methods
4.1. Ethics
4.2. Participants
4.3. RNA Extraction and Quantitative Real-Time PCR (qRT-PCR)
4.4. Data baSe Analysis
4.5. Atomic Absorption Spectroscopy (AAS)
4.6. Perl’s Stain
4.7. Flow Cytometric Analyses
4.8. FACS Sorting and Processing of Sorted Cells
4.9. Cell Culture
4.10. Tumor Tubular Epithelial Cell Isolation
4.11. EC Fluids Generation
4.12. Generation of Conditioned Medium (CM) from Human MΦ
4.13. Proliferation and Migration Assays
4.14. Immunohistochemistry
4.15. Hematoxylin and Eosin Stain
4.16. XTT
4.17. Synthesis and Chemical Characterization of the Extracellular Chelator EC1
4.18. Chelator Solutions
4.19. Statistical Analysis
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- White, M.F.; Dillingham, M.S. Iron-sulphur clusters in nucleic acid processing enzymes. Curr. Opin. Struct. Biol. 2012, 22, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yu, L.; Ding, J.; Chen, Y. Iron Metabolism in Cancer. Int. J. Mol. Sci. 2018, 20, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantopoulos, K.; Porwal, S.K.; Tartakoff, A.; Devireddy, L. Mechanisms of mammalian iron homeostasis. Biochemistry 2012, 51, 5705–5724. [Google Scholar] [CrossRef] [PubMed]
- Haase, V.H. Hypoxic regulation of erythropoiesis and iron metabolism. Am. J. Physiol. Ren. Physiol. 2010, 299, F1–F13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seminog, O.O.; Ogunlaja, O.I.; Yeates, D.; Goldacre, M.J. Risk of individual malignant neoplasms in patients with sickle cell disease: English national record linkage study. J. R. Soc. Med. 2016, 109, 303–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vargas-Olvera, C.Y.; Sánchez-González, D.J.; Solano, J.D.; Aguilar-Alonso, F.A.; Montalvo-Muñoz, F.; Martínez-Martínez, C.M.; Medina-Campos, O.N.; Ibarra-Rubio, M.E. Characterization of N-diethylnitrosamine-initiated and ferric nitrilotriacetate-promoted renal cell carcinoma experimental model and effect of a tamarind seed extract against acute nephrotoxicity and carcinogenesis. Mol. Cell. Biochem. 2012, 369, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Greene, C.J.; Attwood, K.; Sharma, N.J.; Gross, K.W.; Smith, G.J.; Xu, B.; Kauffman, E.C. Transferrin receptor 1 upregulation in primary tumor and downregulation in benign kidney is associated with progression and mortality in renal cell carcinoma patients. Oncotarget 2017, 8, 107052–107075. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018. [Google Scholar] [CrossRef] [Green Version]
- Makhov, P.; Joshi, S.; Ghatalia, P.; Kutikov, A.; Uzzo, R.G.; Kolenko, V.M. Resistance to Systemic Therapies in Clear Cell Renal Cell Carcinoma: Mechanisms and Management Strategies. Mol. Cancer Ther. 2018, 17, 1355–1364. [Google Scholar] [CrossRef] [Green Version]
- Gill, D.M.; Agarwal, N.; Vaishampayan, U. Evolving Treatment Paradigm in Metastatic Renal Cell Carcinoma. Am. Soc. Clin. Oncol. Educ. Book 2017, 37, 319–329. [Google Scholar] [CrossRef]
- Amin, A.; Hammers, H. The Evolving Landscape of Immunotherapy-Based Combinations for Frontline Treatment of Advanced Renal Cell Carcinoma. Front. Immunol. 2018, 9, 3120. [Google Scholar] [CrossRef] [PubMed]
- Cella, D.; Grünwald, V.; Escudier, B.; Hammers, H.J.; George, S.; Nathan, P.; Grimm, M.-O.; Rini, B.I.; Doan, J.; Ivanescu, C.; et al. Patient-reported outcomes of patients with advanced renal cell carcinoma treated with nivolumab plus ipilimumab versus sunitinib (CheckMate 214): A randomised, phase 3 trial. Lancet Oncol. 2019, 20, 297–310. [Google Scholar] [CrossRef]
- Kohgo, Y.; Ikuta, K.; Ohtake, T.; Torimoto, Y.; Kato, J. Body iron metabolism and pathophysiology of iron overload. Int. J. Hematol. 2008, 88, 7–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torti, S.V.; Torti, F.M. Iron and cancer: More ore to be mined. Nat. Rev. Cancer 2013, 13, 342–355. [Google Scholar] [CrossRef] [Green Version]
- Kirkali, Z.; Esen, A.A.; Kirkali, G.; Güner, G. Ferritin: A tumor marker expressed by renal cell carcinoma. Eur. Urol. 1995, 28, 131–134. [Google Scholar] [CrossRef]
- Jézéquel, P.; Campion, L.; Spyratos, F.; Loussouarn, D.; Campone, M.; Guérin-Charbonnel, C.; Joalland, M.-P.; André, J.; Descotes, F.; Grenot, C.; et al. Validation of tumor-associated macrophage ferritin light chain as a prognostic biomarker in node-negative breast cancer tumors: A multicentric 2004 national PHRC study. Int. J. Cancer 2012, 131, 426–437. [Google Scholar] [CrossRef]
- Brookes, M.J.; Hughes, S.; Turner, F.E.; Reynolds, G.; Sharma, N.; Ismail, T.; Berx, G.; McKie, A.T.; Hotchin, N.; Anderson, G.J.; et al. Modulation of iron transport proteins in human colorectal carcinogenesis. Gut 2006, 55, 1449–1460. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Deng, Z.; Hatcher, H.; Miller, L.D.; Di, X.; Tesfay, L.; Sui, G.; D’Agostino, R.B.; Torti, F.M.; Torti, S.V. IRP2 regulates breast tumor growth. Cancer Res. 2014, 74, 497–507. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, H.; Müldür, E.; Endler, G.; Hübl, W. Prevalence of iron deficiency across different tumors and its association with poor performance status, disease status and anemia. Ann. Oncol. 2013, 24, 1886–1892. [Google Scholar] [CrossRef]
- Corna, G.; Campana, L.; Pignatti, E.; Castiglioni, A.; Tagliafico, E.; Bosurgi, L.; Campanella, A.; Brunelli, S.; Manfredi, A.A.; Apostoli, P.; et al. Polarization dictates iron handling by inflammatory and alternatively activated macrophages. Haematologica 2010, 95, 1814–1822. [Google Scholar] [CrossRef] [Green Version]
- Cairo, G.; Recalcati, S.; Mantovani, A.; Locati, M. Iron trafficking and metabolism in macrophages: Contribution to the polarized phenotype. Trends Immunol. 2011, 32, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Recalcati, S.; Locati, M.; Marini, A.; Santambrogio, P.; Zaninotto, F.; De Pizzol, M.; Zammataro, L.; Girelli, D.; Cairo, G. Differential regulation of iron homeostasis during human macrophage polarized activation. Eur. J. Immunol. 2010, 40, 824–835. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Chevrier, S.; Levine, J.H.; Zanotelli, V.R.T.; Silina, K.; Schulz, D.; Bacac, M.; Ries, C.H.; Ailles, L.; Jewett, M.A.S.; Moch, H.; et al. An Immune Atlas of Clear Cell Renal Cell Carcinoma. Cell 2017, 169, 736–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chanmee, T.; Ontong, P.; Konno, K.; Itano, N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers 2014, 6, 1670–1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikemoto, S.; Yoshida, N.; Narita, K.; Wada, S.; Kishimoto, T.; Sugimura, K.; Nakatani, T. Role of tumor-associated macrophages in renal cell carcinoma. Oncol. Rep. 2003. [Google Scholar] [CrossRef]
- Toge, H.; Inagaki, T.; Kojimoto, Y.; Shinka, T.; Hara, I. Angiogenesis in renal cell carcinoma: The role of tumor-associated macrophages. Int. J. Urol. 2009, 16, 801–807. [Google Scholar] [CrossRef]
- Lewis, C.E.; Pollard, J.W. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006, 66, 605–612. [Google Scholar] [CrossRef] [Green Version]
- Komohara, Y.; Hasita, H.; Ohnishi, K.; Fujiwara, Y.; Suzu, S.; Eto, M.; Takeya, M. Macrophage infiltration and its prognostic relevance in clear cell renal cell carcinoma. Cancer Sci. 2011, 102, 1424–1431. [Google Scholar] [CrossRef]
- Xu, L.; Zhu, Y.; Chen, L.; An, H.; Zhang, W.; Wang, G.; Lin, Z.; Xu, J. Prognostic value of diametrically polarized tumor-associated macrophages in renal cell carcinoma. Ann. Surg. Oncol. 2014, 21, 3142–3150. [Google Scholar] [CrossRef]
- Sangaletti, S.; Di Carlo, E.; Gariboldi, S.; Miotti, S.; Cappetti, B.; Parenza, M.; Rumio, C.; Brekken, R.A.; Chiodoni, C.; Colombo, M.P. Macrophage-derived SPARC bridges tumor cell-extracellular matrix interactions toward metastasis. Cancer Res. 2008, 68, 9050–9059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandlund, J.; Oosterwijk, E.; Grankvist, K.; Oosterwijk-Wakka, J.; Ljungberg, B.; Rasmuson, T. Prognostic impact of carbonic anhydrase IX expression in human renal cell carcinoma. BJU Int. 2007, 100, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.P.; Vertosick, E.A.; Corradi, R.B.; Vilaseca, A.; Benfante, N.E.; Touijer, K.A.; Sjoberg, D.D.; Russo, P. Histological subtype of renal cell carcinoma significantly affects survival in the era of partial nephrectomy. Urol. Oncol. 2016, 34, 259.e1–259.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques, O.; Porto, G.; Rêma, A.; Faria, F.; Cruz Paula, A.; Gomez-Lazaro, M.; Silva, P.; Martins da Silva, B.; Lopes, C. Local iron homeostasis in the breast ductal carcinoma microenvironment. BMC Cancer 2016, 16, 187. [Google Scholar] [CrossRef] [Green Version]
- Mertens, C.; Mora, J.; Ören, B.; Grein, S.; Winslow, S.; Scholich, K.; Weigert, A.; Malmström, P.; Forsare, C.; Fernö, M.; et al. Macrophage-derived lipocalin-2 transports iron in the tumor microenvironment. Oncoimmunology 2018, 7, e1408751. [Google Scholar] [CrossRef] [Green Version]
- Jung, M.; Weigert, A.; Tausendschön, M.; Mora, J.; Ören, B.; Sola, A.; Hotter, G.; Muta, T.; Brüne, B. Interleukin-10-induced neutrophil gelatinase-associated lipocalin production in macrophages with consequences for tumor growth. Mol. Cell. Biol. 2012, 32, 3938–3948. [Google Scholar] [CrossRef] [Green Version]
- Mertens, C.; Akam, E.A.; Rehwald, C.; Brüne, B.; Tomat, E.; Jung, M. Intracellular Iron Chelation Modulates the Macrophage Iron Phenotype with Consequences on Tumor Progression. PLoS ONE 2016, 11, e0166164. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Kalinowski, D.S.; Kovacevic, Z.; Siafakas, A.R.; Jansson, P.J.; Stefani, C.; Lovejoy, D.B.; Sharpe, P.C.; Bernhardt, P.V.; Des Richardson, R. Thiosemicarbazones from the old to new: Iron chelators that are more than just ribonucleotide reductase inhibitors. J. Med. Chem. 2009, 52, 5271–5294. [Google Scholar] [CrossRef]
- Utterback, R.D.; Tomat, E. Developing Ligands to Target Transition Metals in Cancer. Encycl. Inorg. Bioinorg. Chem. 2019. [Google Scholar] [CrossRef]
- Akam, E.A.; Chang, T.M.; Astashkin, A.V.; Tomat, E. Intracellular reduction/activation of a disulfide switch in thiosemicarbazone iron chelators. Metallomics 2014, 6, 1905–1912. [Google Scholar] [CrossRef] [Green Version]
- Akam, E.A.; Utterback, R.D.; Marcero, J.R.; Dailey, H.A.; Tomat, E. Disulfide-masked iron prochelators: Effects on cell death, proliferation, and hemoglobin production. J. Inorg. Biochem. 2018, 180, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Greene, B.T.; Thorburn, J.; Willingham, M.C.; Thorburn, A.; Planalp, R.P.; Brechbiel, M.W.; Jennings-Gee, J.; Wilkinson, J.; Torti, F.M.; Torti, S.V. Activation of caspase pathways during iron chelator-mediated apoptosis. J. Biol. Chem. 2002, 277, 25568–25575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, M.; Mertens, C.; Tomat, E.; Brüne, B. Iron as a Central Player and Promising Target in Cancer Progression. Int. J. Mol. Sci. 2019, 20, 273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alkhateeb, A.A.; Han, B.; Connor, J.R. Ferritin stimulates breast cancer cells through an iron-independent mechanism and is localized within tumor-associated macrophages. Breast Cancer Res. Treat. 2013, 137, 733–744. [Google Scholar] [CrossRef]
- Kukulj, S.; Jaganjac, M.; Boranic, M.; Krizanac, S.; Santic, Z.; Poljak-Blazi, M. Altered iron metabolism, inflammation, transferrin receptors, and ferritin expression in non-small-cell lung cancer. Med. Oncol. 2010, 27, 268–277. [Google Scholar] [CrossRef]
- Nseyo, U.O.; Williams, P.D.; Murphy, G.E. Clinical significance of erythropoietin levels in renal carcinoma. Urology 1986, 28, 301–306. [Google Scholar] [CrossRef]
- Dowd, A.A.; Ibrahim, F.I.; Mohammed, M.M. Renal cell carcinoma as a cause of iron deficiency anemia. Afr. J. Urol. 2014, 20, 25–27. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.P.; Elliott, R.L.; Head, J.F. Manipulation of iron transporter genes results in the suppression of human and mouse mammary adenocarcinomas. Anticancer Res. 2010, 30, 759–765. [Google Scholar]
- Coombs, G.S.; Schmitt, A.A.; Canning, C.A.; Alok, A.; Low, I.C.C.; Banerjee, N.; Kaur, S.; Utomo, V.; Jones, C.M.; Pervaiz, S.; et al. Modulation of Wnt/β-catenin signaling and proliferation by a ferrous iron chelator with therapeutic efficacy in genetically engineered mouse models of cancer. Oncogene 2012, 31, 213–225. [Google Scholar] [CrossRef]
- Hrabinski, D.; Hertz, J.L.; Tantillo, C.; Berger, V.; Sherman, A.R. Iron repletion attenuates the protective effects of iron deficiency in DMBA-induced mammary tumors in rats. Nutr. Cancer 1995, 24, 133–142. [Google Scholar] [CrossRef]
- Brierley, J.; Gospodarowicz, M.K.; Wittekind, C. TNM Classification of Malignant Tumours, 8th ed.; Brierley, J., Gospodarowicz, M.K., Wittekind, C., O’Sullivan, B., Eds.; John Wiley & Sons, Inc: Oxford, UK, 2017; ISBN 9781119263562. [Google Scholar]
- Baer, P.C.; Nockher, W.A.; Haase, W.; Scherberich, J.E. Isolation of proximal and distal tubule cells from human kidney by immunomagnetic separation. Technical note. Kidney Int. 1997, 52, 1321–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, M.; Ören, B.; Mora, J.; Mertens, C.; Dziumbla, S.; Popp, R.; Weigert, A.; Grossmann, N.; Fleming, I.; Brüne, B. Lipocalin 2 from macrophages stimulated by tumor cell-derived sphingosine 1-phosphate promotes lymphangiogenesis and tumor metastasis. Sci. Signal. 2016, 9, ra64. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.M.; Tomat, E. Disulfide/thiol switches in thiosemicarbazone ligands for redox-directed iron chelation. Dalton Trans. 2013, 42, 7846–7849. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Patients | ccRCC | pRCC | chRCC |
|---|---|---|---|
| 56 | 7 | 7 | |
| Age (years) | |||
| mean | 64 ± 10 | 68 ± 11 | 63 ± 10 |
| median | 64 ± 10 | 71 ± 11 | 62 ± 10 |
| range | 44–85 | 48–79 | 48–75 |
| sex | |||
| female | 24% | 29% | 80% |
| male | 76% | 71% | 20% |
| pT-stage | |||
| pT1-pT2 | 55% | ||
| pT3-pT4 | 45% | ||
| Grade | |||
| G1-G2 | 84% | ||
| G3-G4 | 16% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schnetz, M.; Meier, J.K.; Rehwald, C.; Mertens, C.; Urbschat, A.; Tomat, E.; Akam, E.A.; Baer, P.; Roos, F.C.; Brüne, B.; et al. The Disturbed Iron Phenotype of Tumor Cells and Macrophages in Renal Cell Carcinoma Influences Tumor Growth. Cancers 2020, 12, 530. https://doi.org/10.3390/cancers12030530
Schnetz M, Meier JK, Rehwald C, Mertens C, Urbschat A, Tomat E, Akam EA, Baer P, Roos FC, Brüne B, et al. The Disturbed Iron Phenotype of Tumor Cells and Macrophages in Renal Cell Carcinoma Influences Tumor Growth. Cancers. 2020; 12(3):530. https://doi.org/10.3390/cancers12030530
Chicago/Turabian StyleSchnetz, Matthias, Julia K. Meier, Claudia Rehwald, Christina Mertens, Anja Urbschat, Elisa Tomat, Eman A. Akam, Patrick Baer, Frederik C. Roos, Bernhard Brüne, and et al. 2020. "The Disturbed Iron Phenotype of Tumor Cells and Macrophages in Renal Cell Carcinoma Influences Tumor Growth" Cancers 12, no. 3: 530. https://doi.org/10.3390/cancers12030530