NUDT7 Loss Promotes KrasG12D CRC Development


 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
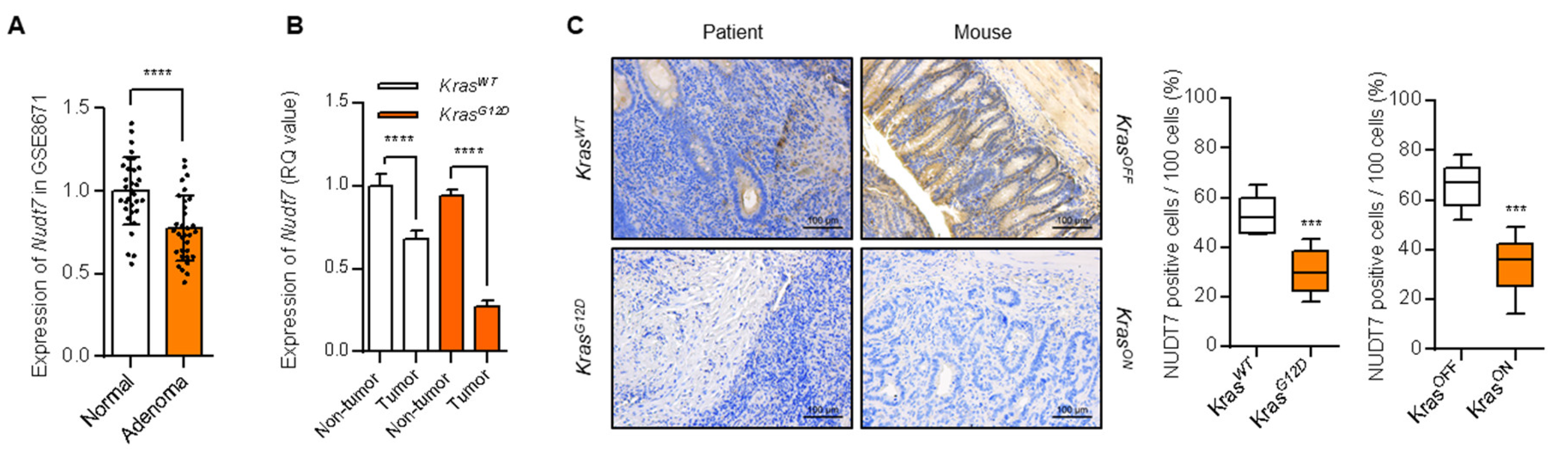
2.1. Peroxisomal Dysfunction Is Responsible for Dysregulation of Lipid Metabolism in KrasG12D CRC
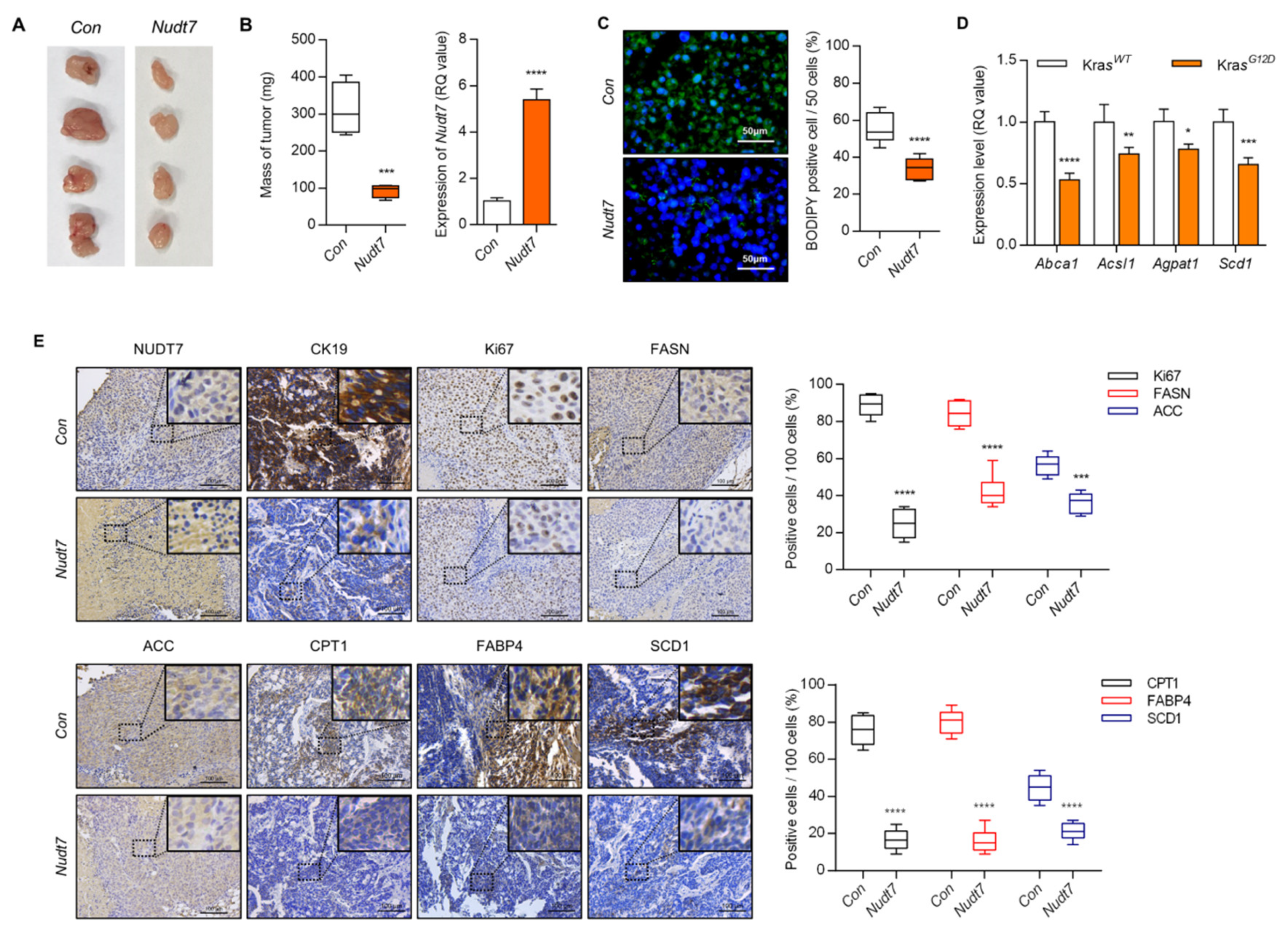
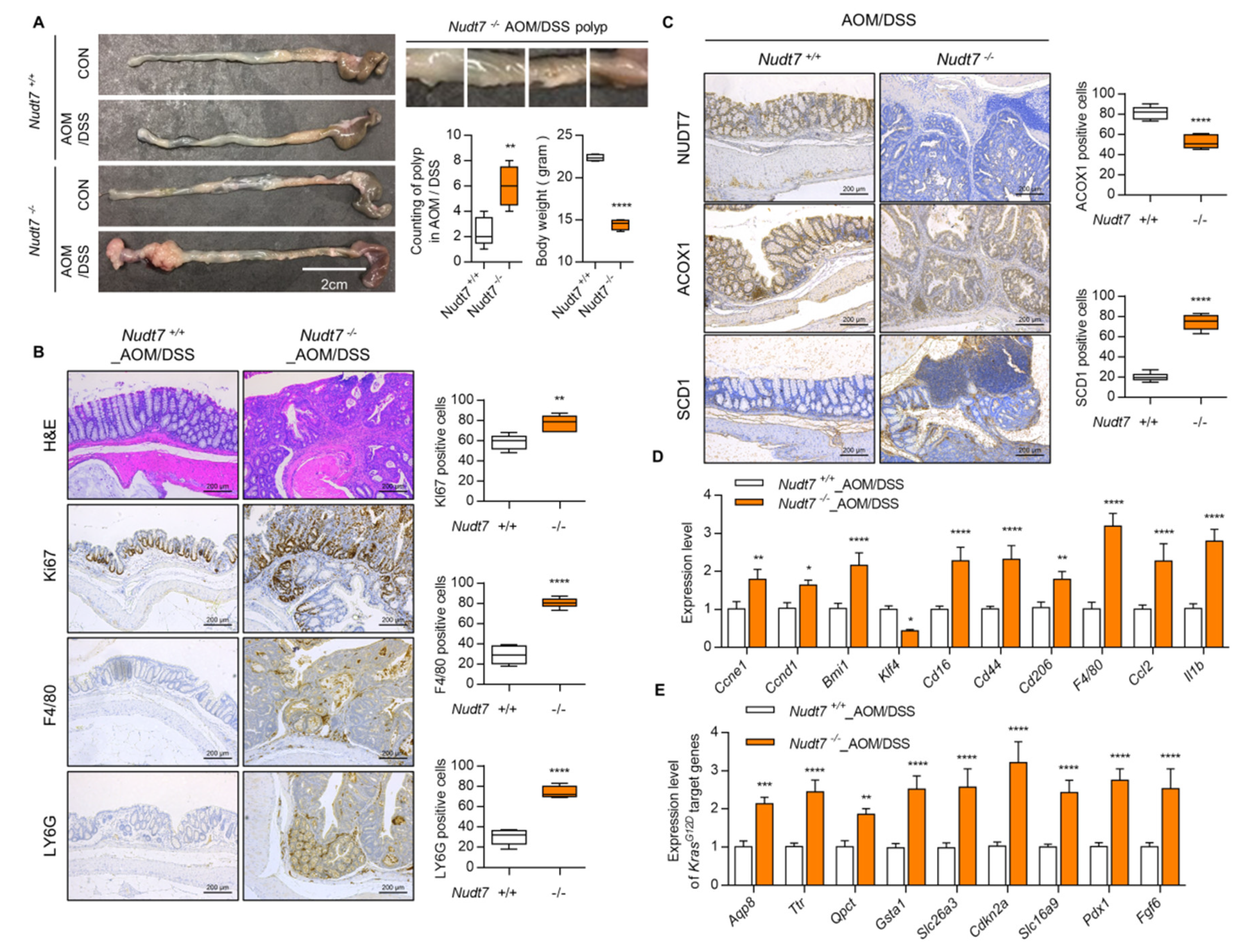
2.2. Dysregulation of Lipid Metabolism by Nudt7 Suppression Is Involved in Development and Progression of KrasG12D CRC
2.3. Increased Palmitic Acid Level by Nudt7 Suppression Is Responsible for CRC Development through Activation of Wnt/β-Catenin Signaling
3. Discussion
4. Materials and Methods
4.1. Clinical Samples
4.2. Ethics Approval
4.3. Animals
4.4. Mouse Xenograft Model
4.5. Cell Culture
4.6. Plasmids and Lentiviral Packaging
4.7. Real-Time PCR
4.8. Quantification of miRNA
4.9. Peroxisomal Gene Profiling
4.10. Microarray Using RNA Isolated from Paraffin Section
4.11. Lipid Accumulation and ROS Staining
4.12. Immunohistochemistry
4.13. Western Blot Analysis
4.14. Preparation of Chitosan Films
4.15. Preparation of Chitosan/Palmitic Acid Composite Films (Chi/PA films)
4.16. Gas Chromatograph/Mass spectrometry (GC/MS) Spectrometry
4.17. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABCA1 | ATP binding cassette subfamily A member 1 | ACC | Acetyl-CoA carboxylase α |
| ACSL1 | Acyl-CoA synthetase long chain fatty acid member 1 | AGPAT | 1-acyl-glycerol-3-phosphate acyltransferase |
| AOM | Azoxymethane | CD36 | Fatty acid translocase |
| CD45 | Protein tyrosine phosphatase receptor type C | CPT1 | Carnitine palmitoyltransferase |
| CRC | Colorectal cancer | CK19 | Cytokeratin 19 |
| DSS | Dextran sulfate | F4/80 | Adhesion G protein-coupled receptor E1 |
| FABP4 | Fatty acid binding protein 4 | FASN | Fatty acid synthase |
| Ki67 | Proliferation marker protein ki-67 | KRAS | Kirsten rat sarcoma viral oncogene homolog |
| LCFA | Long chain fatty acid | LDLR | Low density lipoprotein receptor |
| LY6G | Lymphocyte antigen 6 complex locus G6D | MUFA | Mono-unsaturated fatty acid |
| NUDT7 | Peroxisomal coenzyme A diphosphatase NUDT7 | PPARG | Peroxisome proliferator activated receptor gamma |
| SCD1 | Stearoyl-CoA desaturase-1 | SFA | Saturated fatty acid |
| VLCFA | Very long chain fatty acid | VLDLR | Very low density lipoprotein receptor |
References
- Bhandari, A.; Woodhouse, M.; Gupta, S. Colorectal cancer is a leading cause of cancer incidence and mortality among adults younger than 50 years in the USA: A SEER-based analysis with comparison to other young-onset cancers. J. Investig. Med. 2017, 65, 311–315. [Google Scholar] [CrossRef] [Green Version]
- Esteban-Jurado, C.; Garre, P.; Vila, M.; Lozano, J.J.; Pristoupilova, A.; Beltran, S.; Abuli, A.; Munoz, J.; Balaguer, F.; Ocana, T.; et al. New genes emerging for colorectal cancer predisposition. World J. Gastroenterol. 2014, 20, 1961–1971. [Google Scholar] [CrossRef] [PubMed]
- Gylfe, A.E.; Katainen, R.; Kondelin, J.; Tanskanen, T.; Cajuso, T.; Hanninen, U.; Taipale, J.; Taipale, M.; Renkonen-Sinisalo, L.; Jarvinen, H.; et al. Eleven candidate susceptibility genes for common familial colorectal cancer. PLoS Genet 2013, 9, e1003876. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, E.; De Palma, R.; Orditura, M.; De Vita, F.; Ciardiello, F. Anti-epidermal growth factor receptor monoclonal antibodies in cancer therapy. Clin. Exp. Immunol. 2009, 158, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Kohne, C.H.; Hitre, E.; Zaluski, J.; Chang Chien, C.R.; Makhson, A.; D’Haens, G.; Pinter, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peeters, M.; Price, T.J.; Cervantes, A.; Sobrero, A.F.; Ducreux, M.; Hotko, Y.; Andre, T.; Chan, E.; Lordick, F.; Punt, C.J.; et al. Randomized phase III study of panitumumab with fluorouracil, leucovorin, and irinotecan (FOLFIRI) compared with FOLFIRI alone as second-line treatment in patients with metastatic colorectal cancer. J. Clin. Oncol. 2010, 28, 4706–4713. [Google Scholar] [CrossRef] [PubMed]
- Poehlmann, A.; Kuester, D.; Meyer, F.; Lippert, H.; Roessner, A.; Schneider-Stock, R. K-ras mutation detection in colorectal cancer using the Pyrosequencing technique. Pathol. Res. Pract. 2007, 203, 489–497. [Google Scholar] [CrossRef]
- Raponi, M.; Winkler, H.; Dracopoli, N.C. KRAS mutations predict response to EGFR inhibitors. Curr. Opin. Pharmacol. 2008, 8, 413–418. [Google Scholar] [CrossRef]
- van de Wetering, M.; Sancho, E.; Verweij, C.; de Lau, W.; Oving, I.; Hurlstone, A.; van der Horn, K.; Batlle, E.; Coudreuse, D.; Haramis, A.P.; et al. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell 2002, 111, 241–250. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Kang, Y. Lipid Metabolism Fuels Cancer’s Spread. Cell Metab. 2017, 25, 228–230. [Google Scholar] [CrossRef] [Green Version]
- Maulucci, G.; Cohen, O.; Daniel, B.; Sansone, A.; Petropoulou, P.I.; Filou, S.; Spyridonidis, A.; Pani, G.; De Spirito, M.; Chatgilialoglu, C.; et al. Fatty acid-related modulations of membrane fluidity in cells: detection and implications. Free Radic. Res. 2016, 50, S40–S50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibarguren, M.; Lopez, D.J.; Escriba, P.V. The effect of natural and synthetic fatty acids on membrane structure, microdomain organization, cellular functions and human health. Biochim. Biophys. Acta 2014, 1838, 1518–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igal, R.A. Stearoyl-CoA desaturase-1: A novel key player in the mechanisms of cell proliferation, programmed cell death and transformation to cancer. Carcinogenesis 2010, 31, 1509–1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V., Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padanad, M.S.; Konstantinidou, G.; Venkateswaran, N.; Melegari, M.; Rindhe, S.; Mitsche, M.; Yang, C.; Batten, K.; Huffman, K.E.; Liu, J.; et al. Fatty Acid Oxidation Mediated by Acyl-CoA Synthetase Long Chain 3 Is Required for Mutant KRAS Lung Tumorigenesis. Cell Rep. 2016, 16, 1614–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, R.Z.; Lemonidis, K.M.; Graham, M.J.; Matson, J.E.; Crooke, R.M.; Tribble, D.L.; Wedel, M.K.; Levin, A.A.; Geary, R.S. Cross-species comparison of in vivo PK/PD relationships for second-generation antisense oligonucleotides targeting apolipoprotein B-100. Biochem. Pharmacol. 2009, 77, 910–919. [Google Scholar] [CrossRef]
- Poirier, Y.; Antonenkov, V.D.; Glumoff, T.; Hiltunen, J.K. Peroxisomal beta-oxidation--a metabolic pathway with multiple functions. Biochim. Biophys. Acta 2006, 1763, 1413–1426. [Google Scholar] [CrossRef] [Green Version]
- Reddy, J.K.; Hashimoto, T. Peroxisomal beta-oxidation and peroxisome proliferator-activated receptor alpha: an adaptive metabolic system. Annu. Rev. Nutr. 2001, 21, 193–230. [Google Scholar] [CrossRef]
- Stephen, R.L.; Gustafsson, M.C.; Jarvis, M.; Tatoud, R.; Marshall, B.R.; Knight, D.; Ehrenborg, E.; Harris, A.L.; Wolf, C.R.; Palmer, C.N. Activation of peroxisome proliferator-activated receptor delta stimulates the proliferation of human breast and prostate cancer cell lines. Cancer Res. 2004, 64, 3162–3170. [Google Scholar] [CrossRef] [Green Version]
- Gou, Q.; Gong, X.; Jin, J.; Shi, J.; Hou, Y. Peroxisome proliferator-activated receptors (PPARs) are potential drug targets for cancer therapy. Oncotarget 2017, 8, 60704–60709. [Google Scholar] [CrossRef] [Green Version]
- Youssef, J.; Badr, M. Peroxisome proliferator-activated receptors and cancer: challenges and opportunities. Br. J. Pharmacol. 2011, 164, 68–82. [Google Scholar] [CrossRef] [PubMed]
- Schluter, A.; Fourcade, S.; Domenech-Estevez, E.; Gabaldon, T.; Huerta-Cepas, J.; Berthommier, G.; Ripp, R.; Wanders, R.J.; Poch, O.; Pujol, A. PeroxisomeDB: A database for the peroxisomal proteome, functional genomics and disease. Nucleic Acids Res. 2007, 35, D815–822. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.H.; Ku, J.L.; Yoon, K.A.; Kwon, H.J.; Kim, W.H.; Park, H.S.; Yeo, K.S.; Song, S.Y.; Chung, J.K.; Park, J.G. Establishment and characterization of 12 human colorectal-carcinoma cell lines. Int. J. Cancer 1999, 81, 902–910. [Google Scholar] [CrossRef]
- Ahmed, D.; Eide, P.W.; Eilertsen, I.A.; Danielsen, S.A.; Eknaes, M.; Hektoen, M.; Lind, G.E.; Lothe, R.A. Epigenetic and genetic features of 24 colon cancer cell lines. Oncogenesis 2013, 2, e71. [Google Scholar] [CrossRef]
- Medico, E.; Russo, M.; Picco, G.; Cancelliere, C.; Valtorta, E.; Corti, G.; Buscarino, M.; Isella, C.; Lamba, S.; Martinoglio, B.; et al. The molecular landscape of colorectal cancer cell lines unveils clinically actionable kinase targets. Nat. Commun. 2015, 6, 7002. [Google Scholar] [CrossRef]
- Melone, M.A.B.; Valentino, A.; Margarucci, S.; Galderisi, U.; Giordano, A.; Peluso, G. The carnitine system and cancer metabolic plasticity. Cell Death Dis 2018, 9, 228. [Google Scholar] [CrossRef] [Green Version]
- Valentino, A.; Calarco, A.; Di Salle, A.; Finicelli, M.; Crispi, S.; Calogero, R.A.; Riccardo, F.; Sciarra, A.; Gentilucci, A.; Galderisi, U.; et al. Deregulation of MicroRNAs mediated control of carnitine cycle in prostate cancer: Molecular basis and pathophysiological consequences. Oncogene 2017, 36, 6030–6040. [Google Scholar] [CrossRef]
- Sabates-Bellver, J.; Van der Flier, L.G.; de Palo, M.; Cattaneo, E.; Maake, C.; Rehrauer, H.; Laczko, E.; Kurowski, M.A.; Bujnicki, J.M.; Menigatti, M.; et al. Transcriptome profile of human colorectal adenomas. Mol. Cancer Res. 2007, 5, 1263–1275. [Google Scholar] [CrossRef] [Green Version]
- Boutin, A.T.; Liao, W.T.; Wang, M.; Hwang, S.S.; Karpinets, T.V.; Cheung, H.; Chu, G.C.; Jiang, S.; Hu, J.; Chang, K.; et al. Oncogenic Kras drives invasion and maintains metastases in colorectal cancer. Genes Dev. 2017, 31, 370–382. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Baek, I.J.; Chun, C.H.; Jin, E.J. Dysregulation of the NUDT7-PGAM1 axis is responsible for chondrocyte death during osteoarthritis pathogenesis. Nat. Commun. 2018, 9, 3427. [Google Scholar] [CrossRef]
- Kitajima, S.; Takuma, S.; Morimoto, M. Changes in colonic mucosal permeability in mouse colitis induced with dextran sulfate sodium. Exp. Anim. 1999, 48, 137–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgoudaki, A.M.; Prokopec, K.E.; Boura, V.F.; Hellqvist, E.; Sohn, S.; Ostling, J.; Dahan, R.; Harris, R.A.; Rantalainen, M.; Klevebring, D.; et al. Reprogramming Tumor-Associated Macrophages by Antibody Targeting Inhibits Cancer Progression and Metastasis. Cell Rep. 2016, 15, 2000–2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishigamori, R.; Komiya, M.; Takasu, S.; Mutoh, M.; Imai, T.; Takahashi, M. Osteopontin Deficiency Suppresses Intestinal Tumor Development in Apc-Deficient Min Mice. Int. J. Mol. Sci. 2017, 18, 1058. [Google Scholar] [CrossRef] [PubMed]
- Ducheix, S.; Peres, C.; Hardfeldt, J.; Frau, C.; Mocciaro, G.; Piccinin, E.; Lobaccaro, J.M.; De Santis, S.; Chieppa, M.; Bertrand-Michel, J.; et al. Deletion of Stearoyl-CoA Desaturase-1 From the Intestinal Epithelium Promotes Inflammation and Tumorigenesis, Reversed by Dietary Oleate. Gastroenterology 2018, 155, 1524–1538. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Hinoi, T.; Saito, Y.; Adachi, T.; Miguchi, M.; Niitsu, H.; Sasada, T.; Shimomura, M.; Egi, H.; Oka, S.; et al. Mouse model of proximal colon-specific tumorigenesis driven by microsatellite instability-induced Cre-mediated inactivation of Apc and activation of Kras. J. Gastroenterol. 2016, 51, 447–457. [Google Scholar] [CrossRef]
- Gouw, A.M.; Eberlin, L.S.; Margulis, K.; Sullivan, D.K.; Toal, G.G.; Tong, L.; Zare, R.N.; Felsher, D.W. Oncogene KRAS activates fatty acid synthase, resulting in specific ERK and lipid signatures associated with lung adenocarcinoma. Proc. Natl. Acad. Sci. USA 2017, 114, 4300–4305. [Google Scholar] [CrossRef] [Green Version]
- Barrera, G.; Pizzimenti, S.; Dianzani, M.U. Lipid peroxidation: control of cell proliferation, cell differentiation and cell death. Mol. Aspects. Med. 2008, 29, 1–8. [Google Scholar] [CrossRef]
- Kim, D.; Song, J.; Kang, Y.; Park, S.; Kim, Y.I.; Kwak, S.; Lim, D.; Park, R.; Chun, C.H.; Choe, S.K.; et al. Fis1 depletion in osteoarthritis impairs chondrocyte survival and peroxisomal and lysosomal function. J. Mol. Med. (Berl) 2016, 94, 1373–1384. [Google Scholar] [CrossRef]
- Kohlwein, S.D.; Veenhuis, M.; van der Klei, I.J. Lipid droplets and peroxisomes: key players in cellular lipid homeostasis or a matter of fat--store ’em up or burn ’em down. Genetics 2013, 193, 1–50. [Google Scholar] [CrossRef] [Green Version]
- Lodhi, I.J.; Semenkovich, C.F. Peroxisomes: a nexus for lipid metabolism and cellular signaling. Cell Metab. 2014, 19, 380–392. [Google Scholar] [CrossRef] [Green Version]
- Wanders, R.J.; Jansen, G.A.; Lloyd, M.D. Phytanic acid alpha-oxidation, new insights into an old problem: a review. Biochim. Biophys. Acta 2003, 1631, 119–135. [Google Scholar] [CrossRef]
- Verhoeven, N.M.; Wanders, R.J.; Schor, D.S.; Jansen, G.A.; Jakobs, C. Phytanic acid alpha-oxidation: decarboxylation of 2-hydroxyphytanoyl-CoA to pristanic acid in human liver. J. Lipid. Res. 1997, 38, 2062–2070. [Google Scholar] [PubMed]
- Wierzbicki, A.S.; Lloyd, M.D.; Schofield, C.J.; Feher, M.D.; Gibberd, F.B. Refsum’s disease: a peroxisomal disorder affecting phytanic acid alpha-oxidation. J. Neurochem. 2002, 80, 727–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bessman, M.J.; Frick, D.N.; O’Handley, S.F. The MutT proteins or "Nudix" hydrolases, a family of versatile, widely distributed, "housecleaning" enzymes. J. Biol. Chem. 1996, 271, 25059–25062. [Google Scholar] [CrossRef] [Green Version]
- Dunn, C.A.; O’Handley, S.F.; Frick, D.N.; Bessman, M.J. Studies on the ADP-ribose pyrophosphatase subfamily of the nudix hydrolases and tentative identification of trgB, a gene associated with tellurite resistance. J. Biol. Chem. 1999, 274, 32318–32324. [Google Scholar] [CrossRef] [Green Version]
- Cai, L.; Sutter, B.M.; Li, B.; Tu, B.P. Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Mol. Cell 2011, 42, 426–437. [Google Scholar] [CrossRef] [Green Version]
- Polakis, P. Wnt signaling and cancer. Genes Dev. 2000, 14, 1837–1851. [Google Scholar] [CrossRef] [Green Version]
- Polakis, P. Wnt signaling in cancer. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [Green Version]
- Dimitriadis, A.; Vincan, E.; Mohammed, I.M.; Roczo, N.; Phillips, W.A.; Baindur-Hudson, S. Expression of Wnt genes in human colon cancers. Cancer Lett. 2001, 166, 185–191. [Google Scholar] [CrossRef]
- Holcombe, R.F.; Marsh, J.L.; Waterman, M.L.; Lin, F.; Milovanovic, T.; Truong, T. Expression of Wnt ligands and Frizzled receptors in colonic mucosa and in colon carcinoma. Mol. Pathol. 2002, 55, 220–226. [Google Scholar] [CrossRef]
- de Sousa, E.M.; Vermeulen, L.; Richel, D.; Medema, J.P. Targeting Wnt signaling in colon cancer stem cells. Clin. Cancer Res. 2011, 17, 647–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clements, W.M.; Lowy, A.M.; Groden, J. Adenomatous polyposis coli/beta-catenin interaction and downstream targets: altered gene expression in gastrointestinal tumors. Clin. Colorectal. Cancer 2003, 3, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Tillo, E.; de Barrios, O.; Siles, L.; Cuatrecasas, M.; Castells, A.; Postigo, A. beta-catenin/TCF4 complex induces the epithelial-to-mesenchymal transition (EMT)-activator ZEB1 to regulate tumor invasiveness. Proc. Natl. Acad. Sci. USA 2011, 108, 19204–19209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Abaco, G.M.; Whitehead, R.H.; Burgess, A.W. Synergy between Apc min and an activated ras mutation is sufficient to induce colon carcinomas. Mol. Cell Biol. 1996, 16, 884–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janssen, K.P.; Alberici, P.; Fsihi, H.; Gaspar, C.; Breukel, C.; Franken, P.; Rosty, C.; Abal, M.; El Marjou, F.; Smits, R.; et al. APC and oncogenic KRAS are synergistic in enhancing Wnt signaling in intestinal tumor formation and progression. Gastroenterology 2006, 131, 1096–1109. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.Y.; Yang, J.H.; Kim, N.H.; Lee, K.; Cha, Y.H.; Yun, J.S.; Kang, H.E.; Lee, Y.; Choi, J.; Kim, H.S.; et al. Anti-helminthic niclosamide inhibits Ras-driven oncogenic transformation via activation of GSK-3. Oncotarget 2017, 8, 31856–31863. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Sun, S.; Wang, J.; Fei, F.; Dong, Z.; Ke, A.W.; He, R.; Wang, L.; Zhang, L.; Ji, M.B.; et al. Canonical Wnt Signaling Remodels Lipid Metabolism in Zebrafish Hepatocytes following Ras Oncogenic Insult. Cancer Res. 2018, 78, 5548–5560. [Google Scholar] [CrossRef] [Green Version]
- Veech, R.L. The therapeutic implications of ketone bodies: the effects of ketone bodies in pathological conditions: ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism. Prostaglandins Leukot. Essent. Fatty Acids 2004, 70, 309–319. [Google Scholar] [CrossRef]
- Koo, S.H. Nonalcoholic fatty liver disease: molecular mechanisms for the hepatic steatosis. Clin. Mol. Hepatol. 2013, 19, 210–215. [Google Scholar] [CrossRef]
- Cabral, M.; Martin-Venegas, R.; Moreno, J.J. Role of arachidonic acid metabolites on the control of non-differentiated intestinal epithelial cell growth. Int. J. Biochem. Cell Biol. 2013, 45, 1620–1628. [Google Scholar] [CrossRef]
- Louie, S.M.; Roberts, L.S.; Mulvihill, M.M.; Luo, K.; Nomura, D.K. Cancer cells incorporate and remodel exogenous palmitate into structural and oncogenic signaling lipids. Biochim. Biophys. Acta 2013, 1831, 1566–1572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niso-Santano, M.; Malik, S.A.; Pietrocola, F.; Bravo-San Pedro, J.M.; Marino, G.; Cianfanelli, V.; Ben-Younes, A.; Troncoso, R.; Markaki, M.; Sica, V.; et al. Unsaturated fatty acids induce non-canonical autophagy. EMBO J. 2015, 34, 1025–1041. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.H.; Shui, G.; Zhou, J.; Li, J.J.; Bay, B.H.; Wenk, M.R.; Shen, H.M. Induction of autophagy by palmitic acid via protein kinase C-mediated signaling pathway independent of mTOR (mammalian target of rapamycin). J. Biol. Chem. 2012, 287, 14364–14376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fatima, S.; Hu, X.; Huang, C.; Zhang, W.; Cai, J.; Huang, M.; Gong, R.H.; Chen, M.; Ho, A.H.M.; Su, T.; et al. High-fat diet feeding and palmitic acid increase CRC growth in beta2AR-dependent manner. Cell Death Dis 2019, 10, 711. [Google Scholar] [CrossRef] [Green Version]
- Fujise, T.; Iwakiri, R.; Kakimoto, T.; Shiraishi, R.; Sakata, Y.; Wu, B.; Tsunada, S.; Ootani, A.; Fujimoto, K. Long-term feeding of various fat diets modulates azoxymethane-induced colon carcinogenesis through Wnt/beta-catenin signaling in rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G1150–1156. [Google Scholar] [CrossRef]
- Takada, R.; Satomi, Y.; Kurata, T.; Ueno, N.; Norioka, S.; Kondoh, H.; Takao, T.; Takada, S. Monounsaturated fatty acid modification of Wnt protein: its role in Wnt secretion. Dev. Cell 2006, 11, 791–801. [Google Scholar] [CrossRef] [Green Version]
- Tian, H.; Liu, C.; Zou, X.; Wu, W.; Zhang, C.; Yuan, D. MiRNA-194 Regulates Palmitic Acid-Induced Toll-Like Receptor 4 Inflammatory Responses in THP-1 Cells. Nutrients 2015, 7, 3483–3496. [Google Scholar] [CrossRef] [Green Version]
- Wolfson, B.; Lo, P.K.; Yao, Y.; Li, L.; Wang, H.; Zhou, Q. Impact of miR-140 Deficiency on Non-Alcoholic Fatty Liver Disease. Mol. Nutr. Food Res. 2018, 62, e1800189. [Google Scholar] [CrossRef]
- Yue, Q.; Zhao, C.; Wang, Y.; Zhao, L.; Zhu, Q.; Li, G.; Wu, N.; Jia, D.; Ma, C. Downregulation of growth arrestspecific transcript 5 alleviates palmitic acidinduced myocardial inflammatory injury through the miR26a/HMGB1/NFkappaB axis. Mol. Med. Rep. 2018, 18, 5742–5750. [Google Scholar] [CrossRef] [Green Version]
- Binker-Cosen, M.J.; Richards, D.; Oliver, B.; Gaisano, H.Y.; Binker, M.G.; Cosen-Binker, L.I. Palmitic acid increases invasiveness of pancreatic cancer cells AsPC-1 through TLR4/ROS/NF-kappaB/MMP-9 signaling pathway. Biochem. Biophys. Res. Commun. 2017, 484, 152–158. [Google Scholar] [CrossRef]
- De Robertis, M.; Massi, E.; Poeta, M.L.; Carotti, S.; Morini, S.; Cecchetelli, L.; Signori, E.; Fazio, V.M. The AOM/DSS murine model for the study of colon carcinogenesis: From pathways to diagnosis and therapy studies. J. Carcinog 2011, 10, 9. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Mane, D.R.; Kale, A.D.; Belaldavar, C. Validation of immunoexpression of tenascin-C in oral precancerous and cancerous tissues using ImageJ analysis with novel immunohistochemistry profiler plugin: An immunohistochemical quantitative analysis. J. Oral. Maxillofac. Pathol. 2017, 21, 211–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varghese, F.; Bukhari, A.B.; Malhotra, R.; De, A. IHC Profiler: an open source plugin for the quantitative evaluation and automated scoring of immunohistochemistry images of human tissue samples. PLoS One 2014, 9, e96801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Oh, J.; Kim, Y.I.; Choe, S.K.; Chun, C.H.; Jin, E.J. Suppression of ABCD2 dysregulates lipid metabolism via dysregulation of miR-141:ACSL4 in human osteoarthritis. Cell Biochem. Funct. 2018, 36, 366–376. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, J.; Park, S.; Oh, J.; Kim, D.; Ryu, J.H.; Park, W.C.; Baek, I.-J.; Cheng, X.; Lu, X.; Jin, E.-J. NUDT7 Loss Promotes KrasG12D CRC Development. Cancers 2020, 12, 576. https://doi.org/10.3390/cancers12030576
Song J, Park S, Oh J, Kim D, Ryu JH, Park WC, Baek I-J, Cheng X, Lu X, Jin E-J. NUDT7 Loss Promotes KrasG12D CRC Development. Cancers. 2020; 12(3):576. https://doi.org/10.3390/cancers12030576
Chicago/Turabian StyleSong, Jinsoo, Sujeong Park, Jinjoo Oh, Deokha Kim, Ji Hyun Ryu, Won Cheol Park, In-Jeoung Baek, Xi Cheng, Xin Lu, and Eun-Jung Jin. 2020. "NUDT7 Loss Promotes KrasG12D CRC Development" Cancers 12, no. 3: 576. https://doi.org/10.3390/cancers12030576
APA StyleSong, J., Park, S., Oh, J., Kim, D., Ryu, J. H., Park, W. C., Baek, I.-J., Cheng, X., Lu, X., & Jin, E.-J. (2020). NUDT7 Loss Promotes KrasG12D CRC Development. Cancers, 12(3), 576. https://doi.org/10.3390/cancers12030576