Karyotypic Flexibility of the Complex Cancer Genome and the Role of Polyploidization in Maintenance of Structural Integrity of Cancer Chromosomes


 , and
, and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
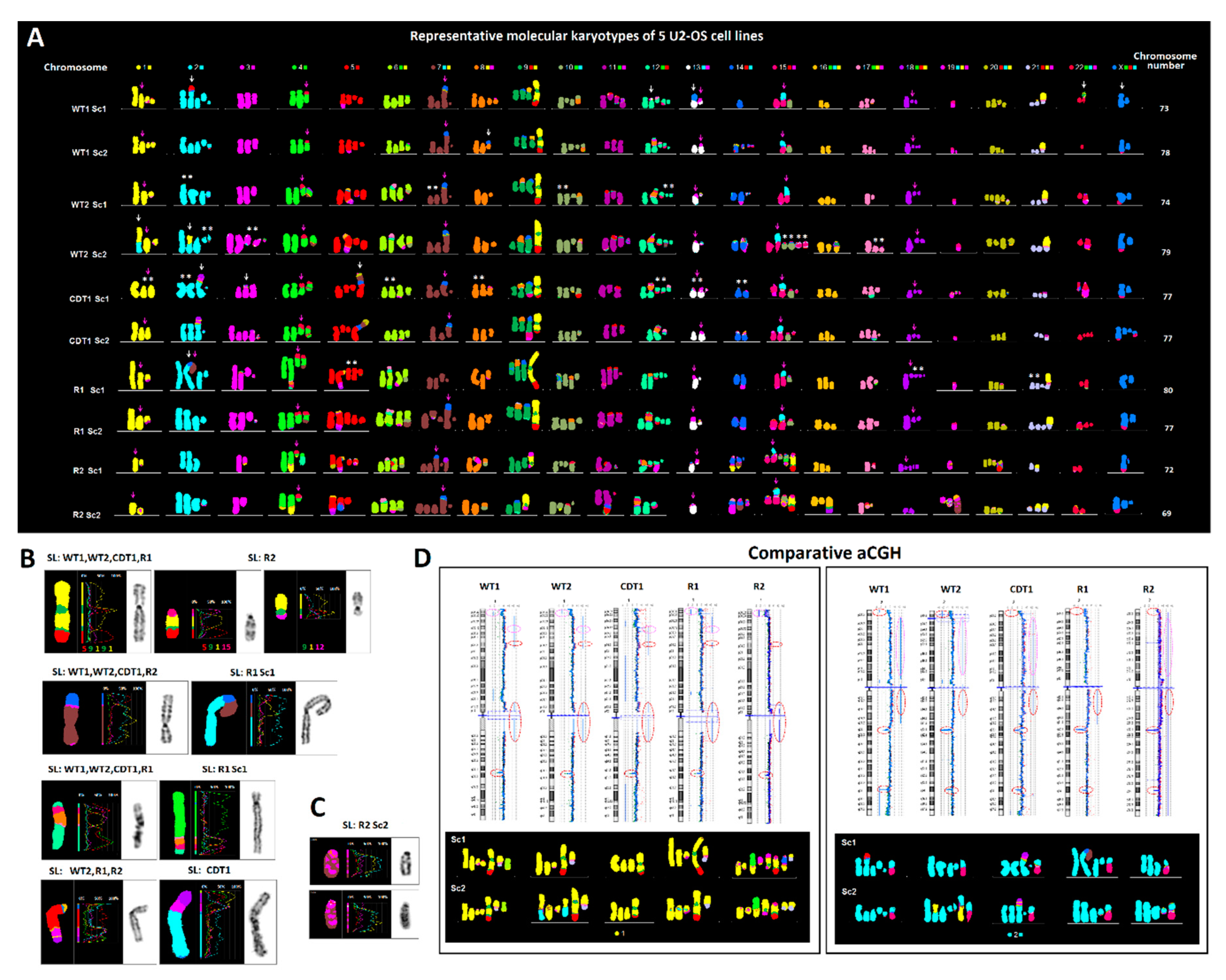
2.1. Clonal Evolution of the ALT Cancer Genome Is Characterized by Narrow Karyotypic Flexibility and the Tendency to Maintain Cancer-Genome Dosage Stability
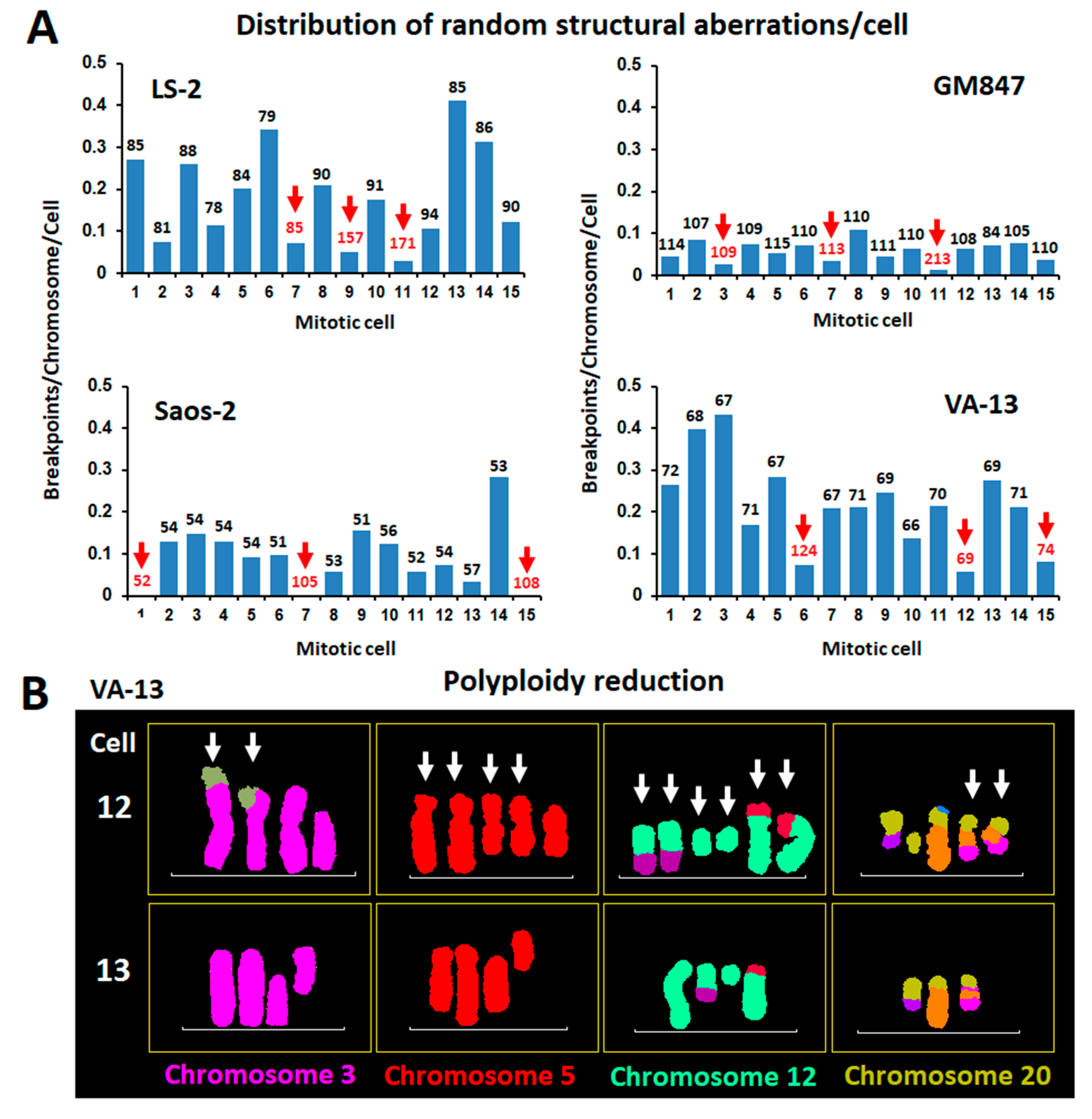
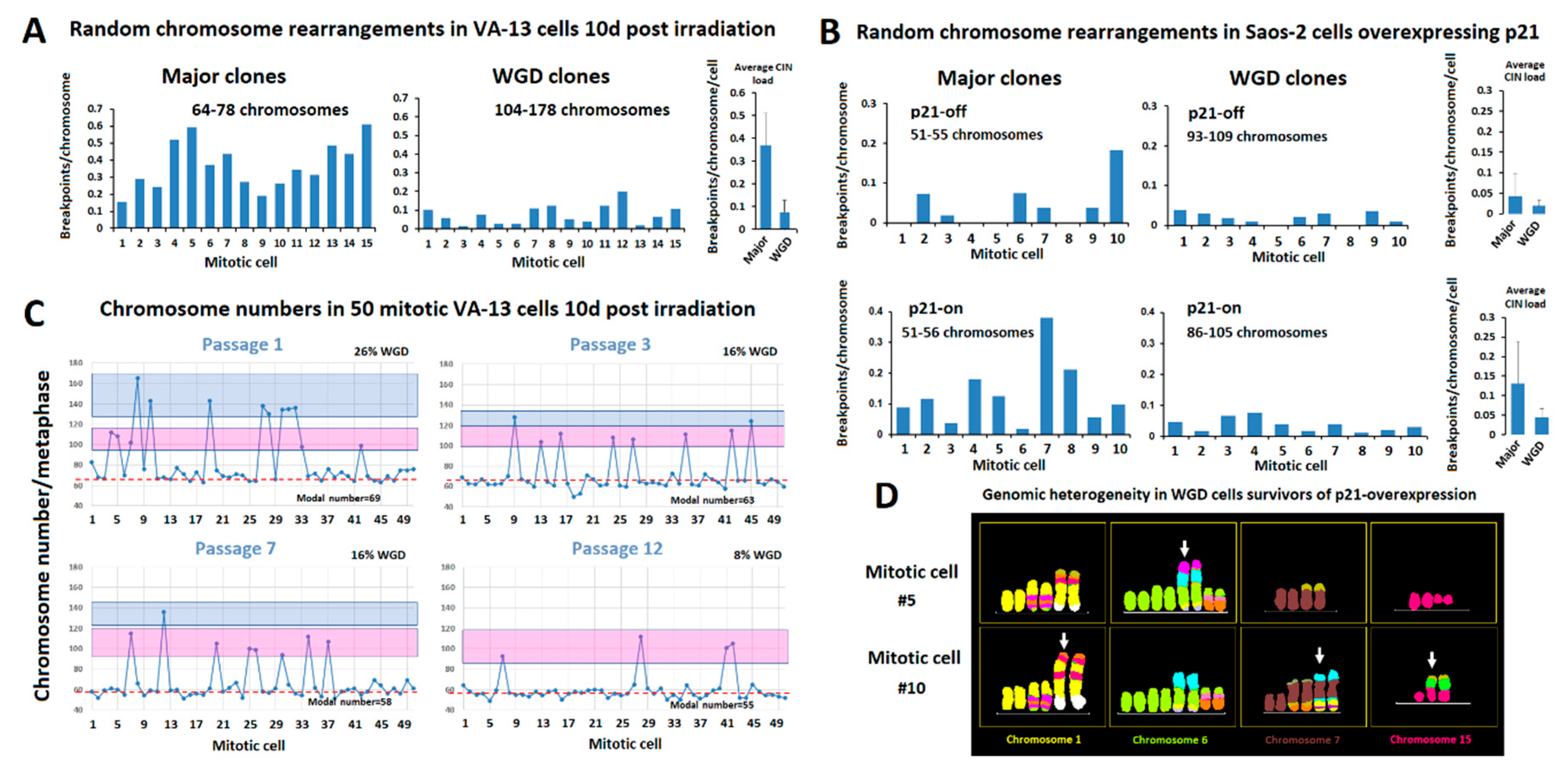
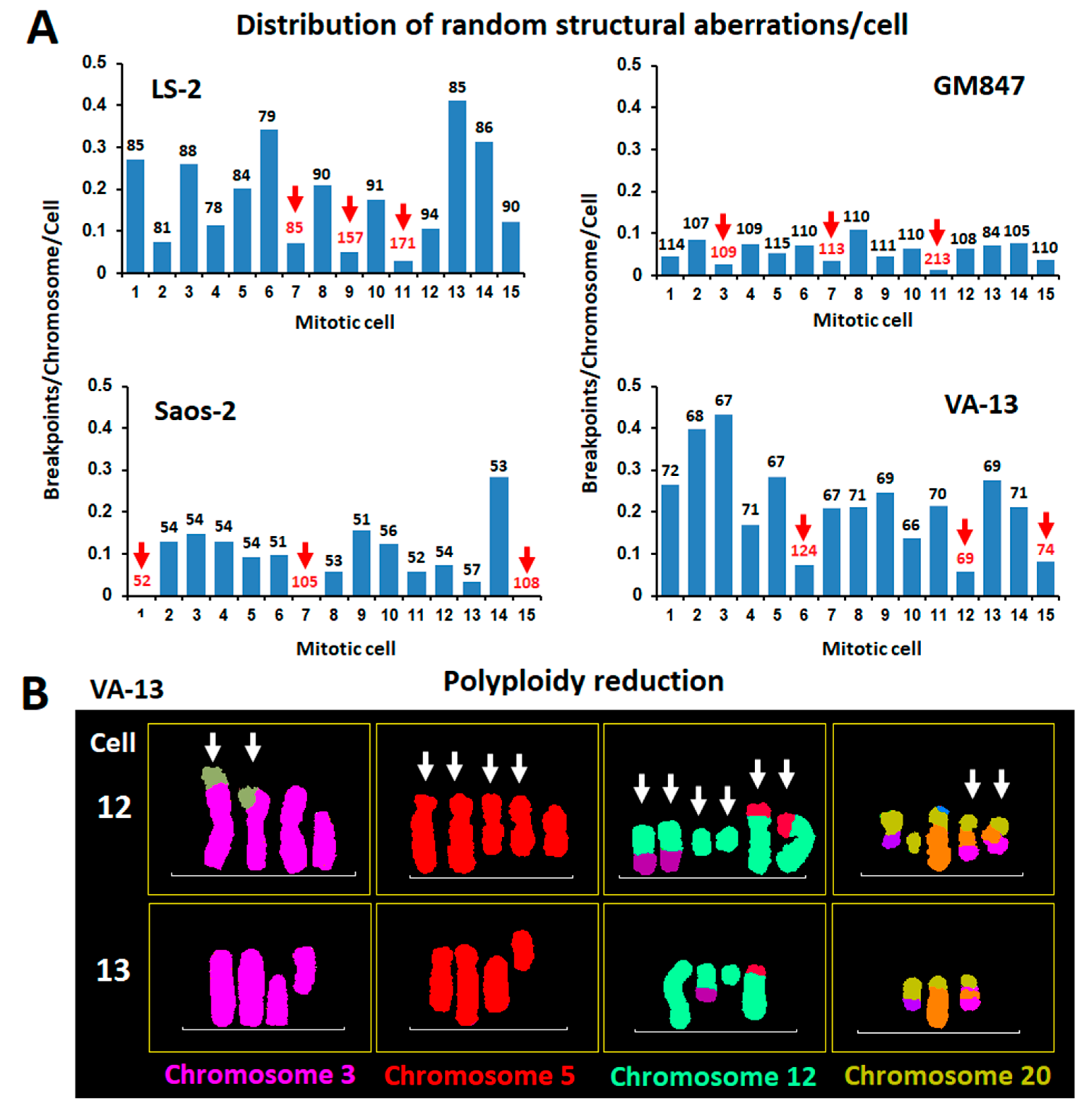
2.2. Distribution of Random Structural Chromosome Anomalies between Co-Dividing ALT Cells and a Putative Role of Polyploidy in the Protection of Cancer Genome Integrity
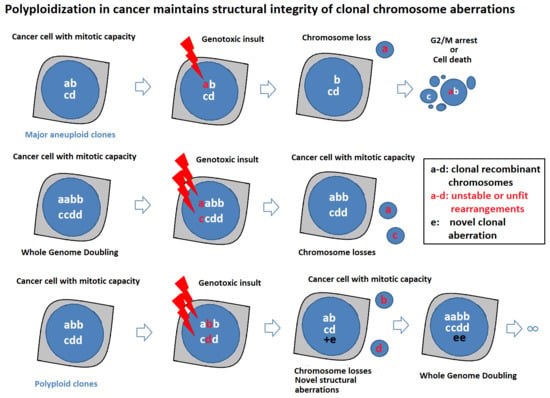
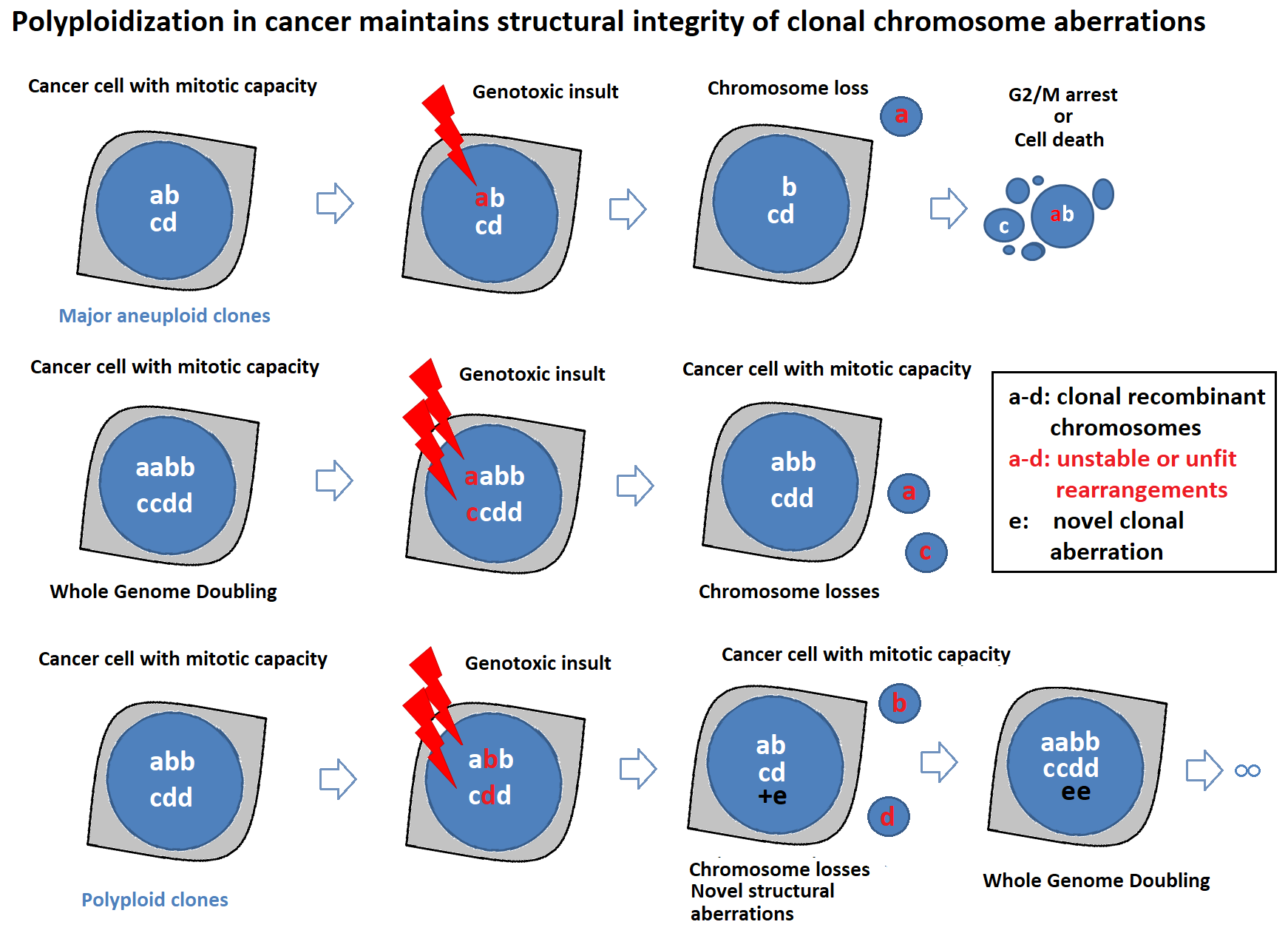
2.3. Polyploidization Protects the Abnormal Cancer Genome from Extreme Structural CIN and Promotes Intratumor Genomic Heterogeneity
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture Conditions
4.2. Cytogenetics
4.3. Comparative Genomic Hybridization
4.4. γ Irradiation
4.5. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nowell, P.C. The clonal evolution of tumor cell populations. Science (80) 1976, 194, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.J.; Cleveland, D.W. Boveri revisited: Chromosomal instability, aneuploidy and tumorigenesis. Nat. Rev. Mol. Cell Biol. 2009, 10, 478–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shay, J.W.; Wright, W.E. Senescence and immortalization: Role of telomeres and telomerase. Carcinogenesis 2005, 26, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An oncogene-induced DNA damage model for cancer development. Science (80) 2008, 319, 1352–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajagopalan, H.; Jallepalli, P.V.; Rago, C.; Velculescu, V.E.; Kinzler, K.W.; Vogelstein, B.; Lengauer, C. Inactivation of hCDC4 can cause chromosomal instability. Nature 2004, 428, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Bakhoum, S.F.; Cantley, L.C. The Multifaceted Role of Chromosomal Instability in Cancer and Its Microenvironment. Cell 2018, 174, 1347–1360. [Google Scholar] [CrossRef] [Green Version]
- Turajlic, S.; Sottoriva, A.; Graham, T.; Swanton, C. Resolving genetic heterogeneity in cancer. Nat. Rev. Genet. 2019, 20, 404–416. [Google Scholar] [CrossRef]
- Gorgoulis, V.G.; Vassiliou, L.V.F.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; DiTullio, R.A.; Kastrinakis, N.G.; Levy, B.; et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005, 434, 907–913. [Google Scholar] [CrossRef]
- Vogelstein, B.; Kinzler, K.W. Cancer genes and the pathways they control. Nat. Med. 2004, 10, 789–799. [Google Scholar] [CrossRef]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instabilities in human cancers. Nature 1998, 396, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Roschke, A.V.; Kirsch, I.R. Targeting karyotypic complexity and chromosomal instability of cancer cells. Curr. Drug Targets 2010, 11, 1341–1350. [Google Scholar] [CrossRef] [PubMed]
- Fabarius, A.; Hehlmann, R.; Duesberg, P.H. Instability of chromosome structure in cancer cells increases exponentially with degrees of aneuploidy. Cancer Genet. Cytogenet. 2003, 143, 59–72. [Google Scholar] [CrossRef]
- Mitelman, F.; Johansson, B.; Mertens, F. The impact of translocations and gene fusions on cancer causation. Nat. Rev. Cancer 2007, 7, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Davoli, T.; Denchi, E.L.; de Lange, T. Persistent Telomere Damage Induces Bypass of Mitosis and Tetraploidy. Cell 2010, 141, 81–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davoli, T.; de Lange, T. The Causes and Consequences of Polyploidy in Normal Development and Cancer. Annu. Rev. Cell Dev. Biol. 2011, 27, 585–610. [Google Scholar] [CrossRef] [Green Version]
- Davoli, T.; de Lange, T. Telomere-Driven Tetraploidization Occurs in Human Cells Undergoing Crisis and Promotes Transformation of Mouse Cells. Cancer Cell 2012, 21, 765–776. [Google Scholar] [CrossRef] [Green Version]
- Christodoulidou, A.; Raftopoulou, C.; Chiourea, M.; Papaioannou, G.K.; Hoshiyama, H.; Wright, W.E.; Shay, J.W.; Gagos, S. The roles of telomerase in the generation of polyploidy during neoplastic cell growth. Neoplasia (United States) 2013, 15, 156–168. [Google Scholar] [CrossRef] [Green Version]
- Storchova, Z.; Pellman, D. From polyploidy to aneuploidy, genome instability and cancer. Nat. Rev. Mol. Cell Biol. 2004, 5, 45–54. [Google Scholar] [CrossRef]
- Bardi, G.; Sukhikh, T.; Pandis, N.; Fenger, C.; Kronborg, O.; Heim, S. Karyotypic characterization of colorectal adenocarcinomas. Genes, Chromosom. Cancer 1995, 12, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Dewhurst, S.M.; McGranahan, N.; Burrell, R.A.; Rowan, A.J.; Grönroos, E.; Endesfelder, D.; Joshi, T.; Mouradov, D.; Gibbs, P.; Ward, R.L.; et al. Tolerance of whole- genome doubling propagates chromosomal instability and accelerates cancer genome evolution. Cancer Discov. 2014, 4, 175–185. [Google Scholar] [CrossRef] [Green Version]
- Kuznetsova, A.Y.; Seget, K.; Moeller, G.K.; de Pagter, M.S.; de Roos, J.A.D.M.; Dürrbaum, M.; Kuffer, C.; Müller, S.; Zaman, G.J.R.; Kloosterman, W.P.; et al. Chromosomal instability, tolerance of mitotic errors and multidrug resistance are promoted by tetraploidization in human cells. Cell Cycle 2015, 14, 2810–2820. [Google Scholar] [CrossRef] [PubMed]
- Coward, J.; Harding, A. Size does matter: Why polyploid tumor cells are critical drug targets in the war on cancer. Front. Oncol. 2014, 4, 123. [Google Scholar] [CrossRef] [PubMed]
- Mosieniak, G.; Sliwinska, M.A.; Alster, O.; Strzeszewska, A.; Sunderland, P.; Piechota, M.; Was, H.; Sikora, E. Polyploidy Formation in Doxorubicin-Treated Cancer Cells Can Favor Escape from Senescence. Neoplasia 2015, 17, 882–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogden, A.; Rida, P.C.G.; Knudsen, B.S.; Kucuk, O.; Aneja, R. Docetaxel-induced polyploidization may underlie chemoresistance and disease relapse. Cancer Lett. 2015, 367, 89–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castedo, M.; Coquelle, A.; Vitale, I.; Vivet, S.; Mouhamad, S.; Viaud, S.; Zitvogel, L.; Kroemer, G. Selective resistance of tetraploid cancer cells against DNA damage-induced apoptosis. In Annals of the New York Academy of Sciences; Blackwell Publishing Inc.: Malden, MA, USA, 2006; Volume 1090, pp. 35–49. [Google Scholar]
- Zhang, D.; Wang, Y.; Zhang, S. Asymmetric cell division in polyploid giant cancer cells and low eukaryotic cells. Biomed. Res. Int. 2014, 2014, 432652. [Google Scholar] [CrossRef] [Green Version]
- Erenpreisa, J.; Cragg, M.S. MOS, aneuploidy and the ploidy cycle of cancer cells. Oncogene 2010, 29, 5447–5451. [Google Scholar] [CrossRef] [Green Version]
- Erenpreisa, J.; Cragg, M.S. Three steps to the immortality of cancer cells: Senescence, polyploidy and self-renewal. Cancer Cell Int. 2013, 13, 92. [Google Scholar] [CrossRef] [Green Version]
- Salmina, K.; Huna, A.; Kalejs, M.; Pjanova, D.; Scherthan, H.; Cragg, M.S.; Erenpreisa, J. The cancer aneuploidy paradox: In the light of evolution. Genes (Basel) 2019, 10, 83. [Google Scholar] [CrossRef] [Green Version]
- De Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabatier, L.; Ricoul, M.; Pottier, G.; Murnane, J.P. The loss of a single telomere can result in instability of multiple chromosomes in a human tumor cell line. Mol. Cancer Res. 2005, 3, 139–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewénius, Y.; Gorunova, L.; Jonson, T.; Larsson, N.; Höglund, M.; Mandahl, N.; Mertens, F.; Mitelman, F.; Gisselsson, D. Structural and numerical chromosome changes in colon cancer develop through telomere-mediated anaphase bridges, not through mitotic multipolarity. Proc. Natl. Acad. Sci. USA 2005, 102, 5541–5546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gagos, S.; Chiourea, M.; Christodoulidou, A.; Apostolou, E.; Raftopoulou, C.; Deustch, S.; Jefford, C.E.; Irminger-Finger, I.; Shay, J.W.; Antonarakis, S.E. Pericentromeric instability and spontaneous emergence of human neoacrocentric and minute chromosomes in the alternative pathway of telomere lengthening. Cancer Res. 2008, 68, 8146–8155. [Google Scholar] [CrossRef] [Green Version]
- Sakellariou, D.; Chiourea, M.; Raftopoulou, C.; Gagos, S. Alternative lengthening of telomeres: Recurrent cytogenetic aberrations and chromosome stability under extreme telomere dysfunction. Neoplasia (United States) 2013, 15, 1301–1313. [Google Scholar] [CrossRef] [Green Version]
- Kirsch-Volders, M.; Elhajouji, A.; Cundari, E.; Van Hummelen, P. The in vitro micronucleus test: a multi-endpoint assay to detect simultaneously mitotic delay, apoptosis, chromosome breakage, chromosome loss and non-disjunction. Mutat. Res. 1997, 392, 19–30. [Google Scholar] [CrossRef]
- Ye, C.J.; Sharpe, Z.; Alemara, S.; Mackenzie, S.; Liu, G.; Abdallah, B.; Horne, S.; Regan, S.; Heng, H.H. Micronuclei and genome chaos: Changing the system inheritance. Genes (Basel) 2019, 10, 366. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.C.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific association of human telomerase activity with immortal cells and cancer. Science (80) 1994, 266, 2011–2015. [Google Scholar] [CrossRef]
- Pickett, H.A.; Reddel, R.R. Molecular mechanisms of activity and derepression of alternative lengthening of telomeres. Nat. Struct. Mol. Biol. 2015, 22, 875–880. [Google Scholar] [CrossRef]
- Roumelioti, F.; Sotiriou, S.K.; Katsini, V.; Chiourea, M.; Halazonetis, T.D.; Gagos, S. Alternative lengthening of human telomeres is a conservative DNA replication process with features of break-induced replication. EMBO Rep. 2016, 17, 1731–1737. [Google Scholar] [CrossRef]
- Loeb, L.A. Mutator Phenotype May Be Required for Multistage Carcinogenesis. Cancer Res. 1991, 51, 3075–3079. [Google Scholar] [PubMed]
- Ponten, J.; Saksela, E. Two established in vitro cell lines from human mesenchymal tumours. Int. J. Cancer 1967, 2, 434–447. [Google Scholar] [CrossRef] [PubMed]
- Lourda, M.; Trougakos, I.P.; Gonos, E.S. Development of resistance to chemotherapeutic drugs in human osteosarcoma cell lines largely depends on up-regulation of Clusterin/Apolipoprotein. J. Int. J. Cancer 2007, 120, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Liontos, M.; Koutsami, M.; Sideridou, M.; Evangelou, K.; Kletsas, D.; Levy, B.; Kotsinas, A.; Nahum, O.; Zoumpourlis, V.; Kouloukoussa, M.; et al. Deregulated overexpression of hCdt1 and hCdc6 promotes malignant behavior. Cancer Res. 2007, 67, 10899–10909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holland, A.J.; Cleveland, D.W. Chromoanagenesis and cancer: Mechanisms and consequences of localized, complex chromosomal rearrangements. Nat. Med. 2012, 18, 1630–1638. [Google Scholar] [CrossRef] [Green Version]
- Londoño-Vallejo, J.A.; Der-Sarkissian, H.; Cazes, L.; Bacchetti, S.; Reddel, R.R. Alternative Lengthening of Telomeres Is Characterized by High Rates of Telomeric Exchange. Cancer Res. 2004, 64, 2324–2327. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, M.A.; Johnson, J.E.; Pascarelli, K.; Beeharry, N.; Chiourea, M.; Gagos, S.; Lev, D.; Von Mehren, M.; Kipling, D.; Broccoli, D. Doxorubicin resistance in a novel in vitro model of human pleomorphic liposarcoma associated with alternative lengthening of telomeres. Mol. Cancer Ther. 2010, 9, 682–692. [Google Scholar] [CrossRef] [Green Version]
- Galanos, P.; Vougas, K.; Walter, D.; Polyzos, A.; Maya-Mendoza, A.; Haagensen, E.J.; Kokkalis, A.; Roumelioti, F.M.; Gagos, S.; Tzetis, M.; et al. Chronic p53-independent p21 expression causes genomic instability by deregulating replication licensing. Nat. Cell Biol. 2016, 18, 777–789. [Google Scholar] [CrossRef]
- Blasco, M.A. Telomeres and human disease: Ageing, cancer and beyond. Nat. Rev. Genet. 2005, 6, 611–622. [Google Scholar] [CrossRef]
- Crouch, J.D.; Brosh, R.M. Mechanistic and biological considerations of oxidatively damaged DNA for helicase-dependent pathways of nucleic acid metabolism. Free Radic. Biol. Med. 2017, 107, 245–257. [Google Scholar] [CrossRef]
- Venkatesan, R.N.; Loeb, L.A. The multiplicity of mutations in human cancers. Adv. Exp. Med. Biol. 2005, 570, 3–17. [Google Scholar] [PubMed]
- Shibata, M.; Shen, M.M. The roots of cancer: Stem cells and the basis for tumor heterogeneity. BioEssays 2013, 35, 253–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson-Rees, W.A.; Flandermeyer, R.R.; Hawthorne, P.K. Banded marker chromosomes as indicators of intraspecies cellular contamination. Science (80) 1974, 184, 1093–1096. [Google Scholar] [CrossRef] [PubMed]
- Lavappa, K.S. Survey of ATCC stocks of human cell lines for hela contamination. In Vitro 1978, 14, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Vcelar, S.; Melcher, M.; Auer, N.; Hrdina, A.; Puklowski, A.; Leisch, F.; Jadhav, V.; Wenger, T.; Baumann, M.; Borth, N. Changes in Chromosome Counts and Patterns in CHO Cell Lines upon Generation of Recombinant Cell Lines and Subcloning. Biotechnol. J. 2018, 13, 1700495. [Google Scholar] [CrossRef] [PubMed]
- Roschke, A.V.; Stover, K.; Tonon, G.; Schäffer, A.A.; Kirsch, I.R. Stable Karyotypes in Epithelial Cancer Cell Lines Despite High Rates of Ongoing Structural and Numerical Chromosomal Instability. Neoplasia 2002, 4, 19–31. [Google Scholar] [CrossRef] [Green Version]
- Roschke, A.V.; Tonon, G.; Gehlhaus, K.S.; McTyre, N.; Bussey, K.J.; Lababidi, S.; Scudiero, D.A.; Weinstein, J.N.; Kirsch, I.R. Karyotypic Complexity of the NCI-60 Drug-Screening Panel. Cancer Res. 2003, 63, 8634–8647. [Google Scholar]
- Padilla-Nash, H.M.; Heselmeyer-Haddad, K.; Wangsa, D.; Zhang, H.; Ghadimi, B.M.; Macville, M.; Augustus, M.; Schröck, E.; Hilgenfeld, E.; Ried, T. Jumping translocations are common in solid tumor cell lines and result in recurrent fusions of whole chromosome arms. Genes Chromosom. Cancer 2001, 30, 349–363. [Google Scholar] [CrossRef]
- Manola, K.N.; Georgakakos, V.N.; Stavropoulou, C.; Spyridonidis, A.; Angelopoulou, M.K.; Vlachadami, I.; Katsigiannis, A.; Roussou, P.; Pantelias, G.E.; Sambani, C. Jumping translocations in hematological malignancies: a cytogenetic study of five cases. Cancer Genet. Cytogenet. 2008, 187, 85–94. [Google Scholar] [CrossRef]
- Jackson-Cook, C.; Zou, Y.; Turner, K.; Astbury, C.; Ware, J. A novel tumorigenic human prostate epithelial cell line (M2205): molecular cytogenetic characterization demonstrates C-MYC amplification and jumping translocations. Cancer Genet. Cytogenet. 2003, 141, 56–64. [Google Scholar] [CrossRef]
- Vukovic, B.; Beheshti, B.; Park, P.; Lim, G.; Bayani, J.; Zielenska, M.; Squire, J.A. Correlating breakage-fusion-bridge events with the overall chromosomal instability and in vitro karyotype evolution in prostate cancer. Cytogenet. Genome Res. 2007, 116, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Z.; Leibowitz, M.L.; Pellman, D. Chromothripsis and beyond: Rapid genome evolution from complex chromosomal rearrangements. Genes Dev. 2013, 27, 2513–2530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maciejowski, J.; Li, Y.; Bosco, N.; Campbell, P.J.; De Lange, T. Chromothripsis and Kataegis Induced by Telomere Crisis. Cell 2015, 163, 1641–1654. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Dai, H.; Zhou, M.; Li, X.; Liu, C.; Guo, Z.; Wu, X.; Wu, J.; Wang, C.; Zhong, J.; et al. Polyploid cells rewire DNA damage response networks to overcome replication stress-induced barriers for tumour progression. Nat. Commun. 2012, 3, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Storchova, Z.; Kuffer, C. The consequences of tetraploidy and aneuploidy. J. Cell Sci. 2008, 121, 3859–3866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahanban-Esfahlan, R.; Seidi, K.; Manjili, M.H.; Jahanban-Esfahlan, A.; Javaheri, T.; Zare, P. Tumor Cell Dormancy: Threat or Opportunity in the Fight against Cancer. Cancers (Basel) 2019, 11, 1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.J.X.; Endesfelder, D.; Rowan, A.J.; Walther, A.; Birkbak, N.J.; Futreal, P.A.; Downward, J.; Szallasi, Z.; Tomlinson, I.P.M.; Howell, M.; et al. Chromosomal instability confers intrinsic multidrug resistance. Cancer Res. 2011, 71, 1858–1870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeung, Y.T.; Yin, S.; Lu, B.; Fan, S.; Yang, R.; Bai, R.; Zhang, C.; Bode, A.M.; Liu, K.; Dong, Z. Losmapimod Overcomes Gefitinib Resistance in Non-small Cell Lung Cancer by Preventing Tetraploidization. EBioMedicine 2018, 28, 51–61. [Google Scholar] [CrossRef] [Green Version]
- Speicher, M.R.; Carter, N.P. The new cytogenetics: Blurring the boundaries with molecular biology. Nat. Rev. Genet. 2005, 6, 782–792. [Google Scholar] [CrossRef]
- Li, Y.; Laterra, J. Cancer stem cells: Distinct entities or dynamically regulated phenotypes? Cancer Res. 2012, 72, 576–580. [Google Scholar] [CrossRef] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raftopoulou, C.; Roumelioti, F.-M.; Dragona, E.; Gimelli, S.; Sloan-Béna, F.; Gorgoulis, V.; Antonarakis, S.E.; Gagos, S. Karyotypic Flexibility of the Complex Cancer Genome and the Role of Polyploidization in Maintenance of Structural Integrity of Cancer Chromosomes. Cancers 2020, 12, 591. https://doi.org/10.3390/cancers12030591
Raftopoulou C, Roumelioti F-M, Dragona E, Gimelli S, Sloan-Béna F, Gorgoulis V, Antonarakis SE, Gagos S. Karyotypic Flexibility of the Complex Cancer Genome and the Role of Polyploidization in Maintenance of Structural Integrity of Cancer Chromosomes. Cancers. 2020; 12(3):591. https://doi.org/10.3390/cancers12030591
Chicago/Turabian StyleRaftopoulou, Christina, Fani-Marlen Roumelioti, Eleni Dragona, Stefanie Gimelli, Frédérique Sloan-Béna, Vasilis Gorgoulis, Stylianos E. Antonarakis, and Sarantis Gagos. 2020. "Karyotypic Flexibility of the Complex Cancer Genome and the Role of Polyploidization in Maintenance of Structural Integrity of Cancer Chromosomes" Cancers 12, no. 3: 591. https://doi.org/10.3390/cancers12030591
APA StyleRaftopoulou, C., Roumelioti, F.-M., Dragona, E., Gimelli, S., Sloan-Béna, F., Gorgoulis, V., Antonarakis, S. E., & Gagos, S. (2020). Karyotypic Flexibility of the Complex Cancer Genome and the Role of Polyploidization in Maintenance of Structural Integrity of Cancer Chromosomes. Cancers, 12(3), 591. https://doi.org/10.3390/cancers12030591