Proteome and Phosphoproteome Changes Associated with Prognosis in Acute Myeloid Leukemia

 , ,
, , 
Abstract
:1. Introduction
2. Results
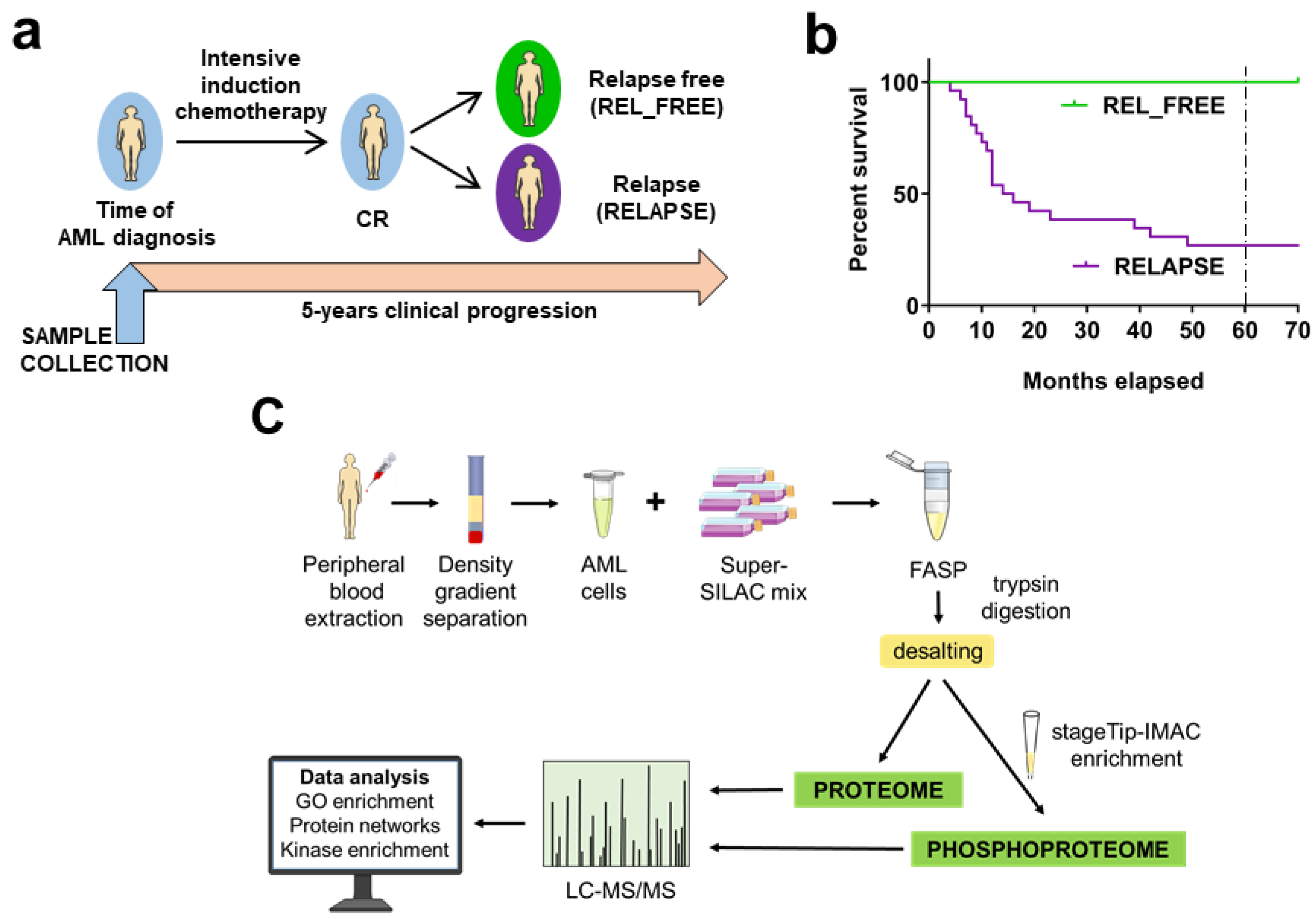
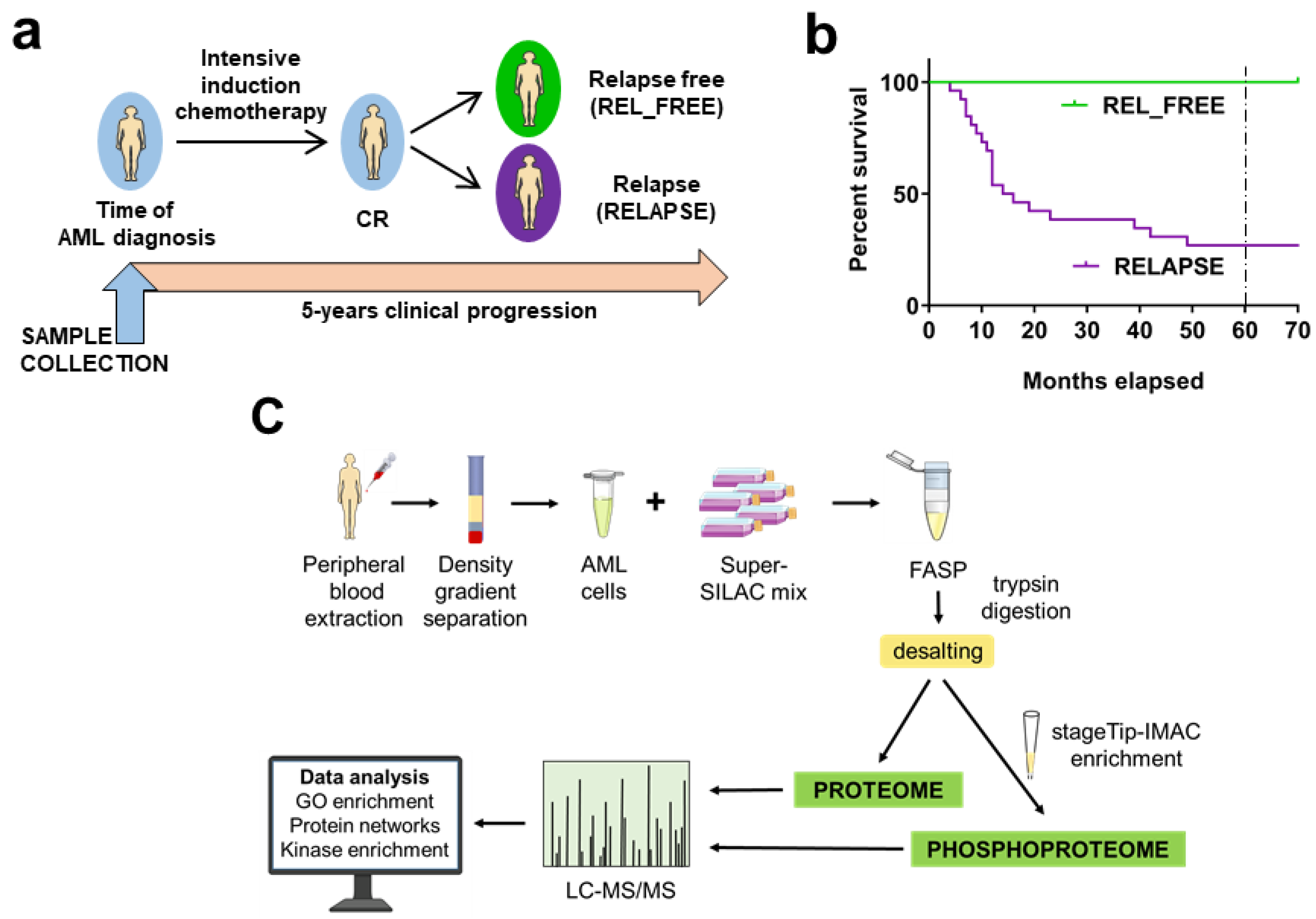
2.1. AML Patients Included in the Study
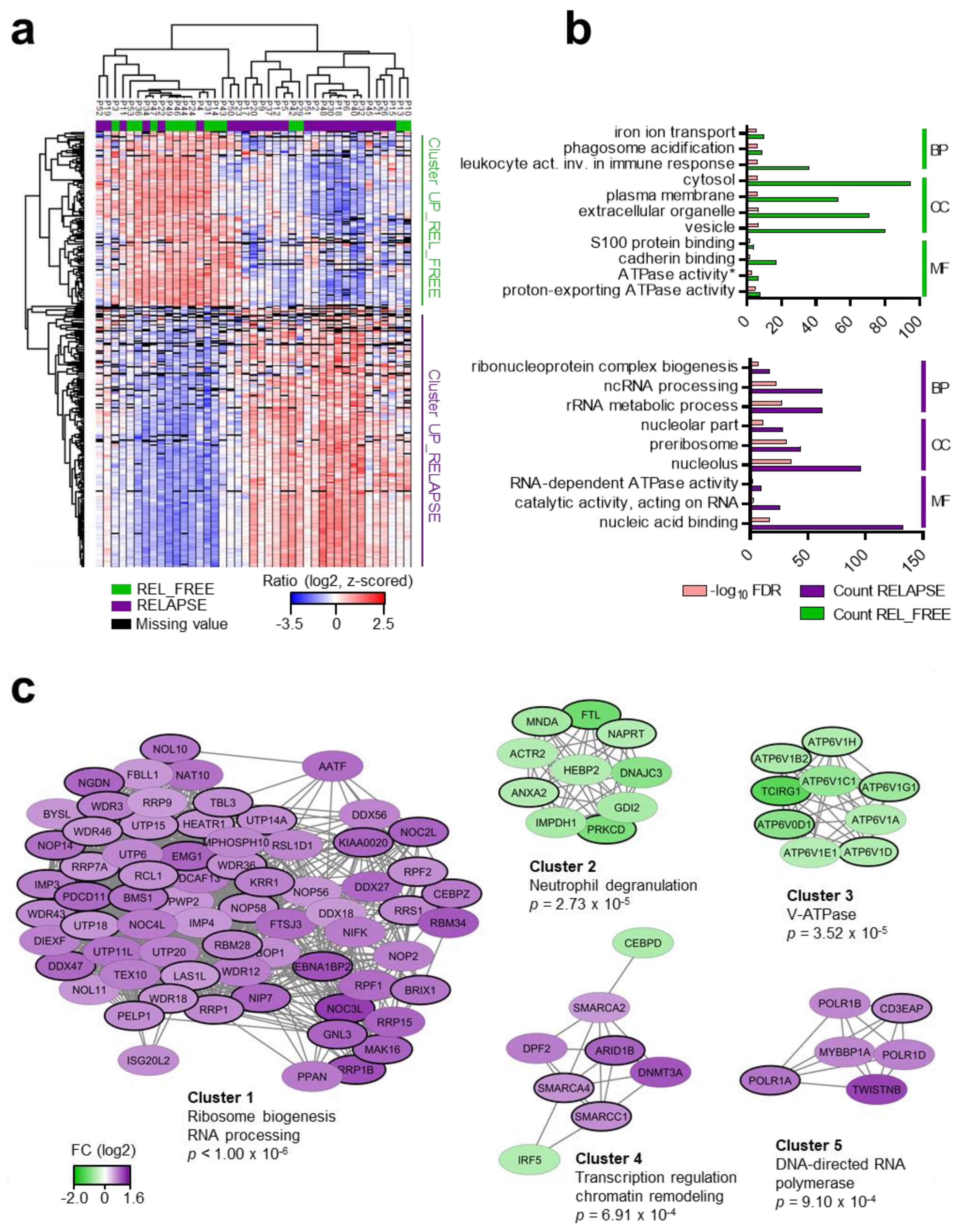
2.2. The Protein Abundances of rRNA Processing Proteins and V-ATPase Subunits Differ Between RELAPSE and REL_FREE Patients
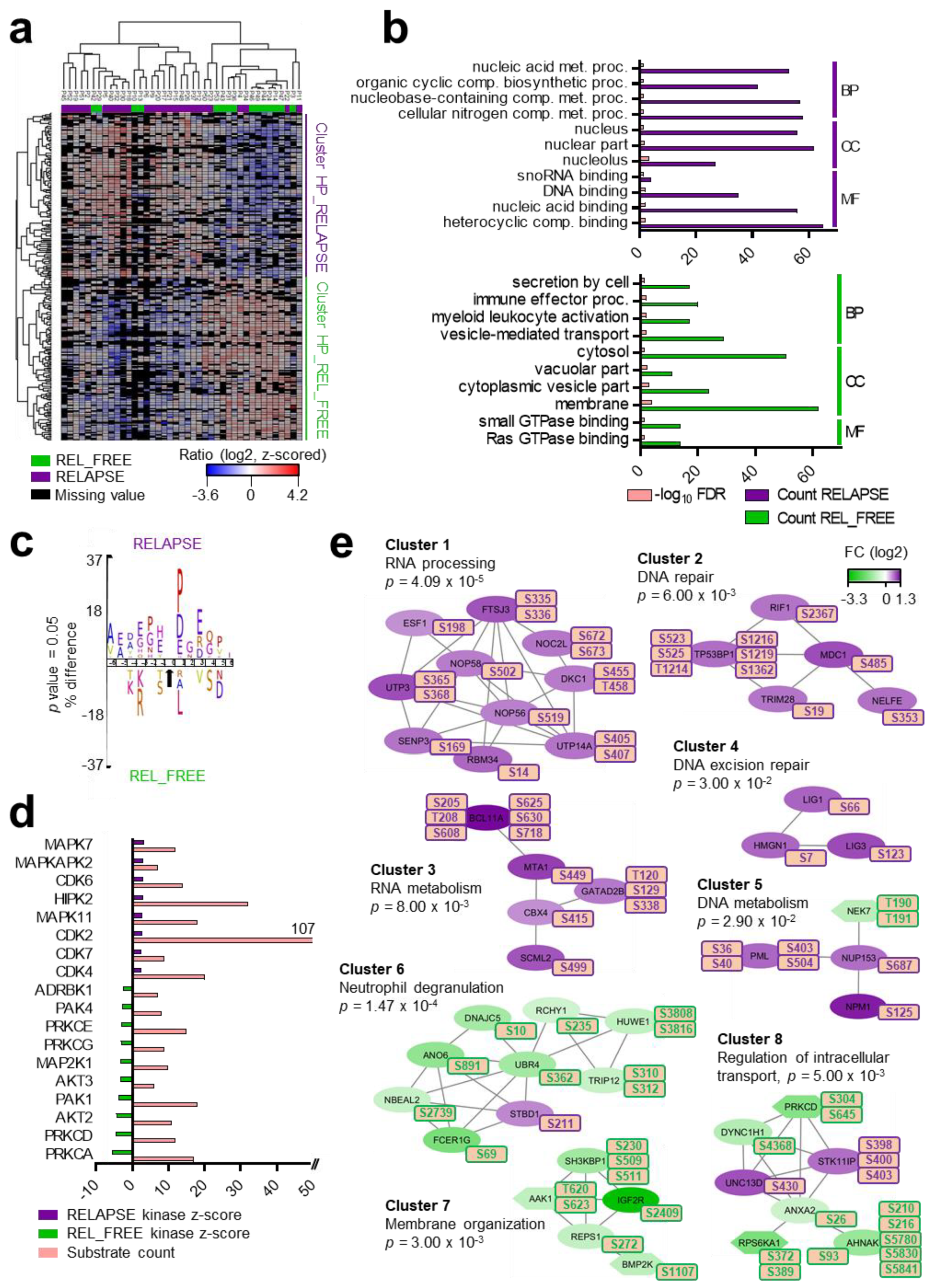
2.3. Differential CDK, CSK2 and PRKCA/D Kinase Activities between RELAPSE and REL_FREE Patients
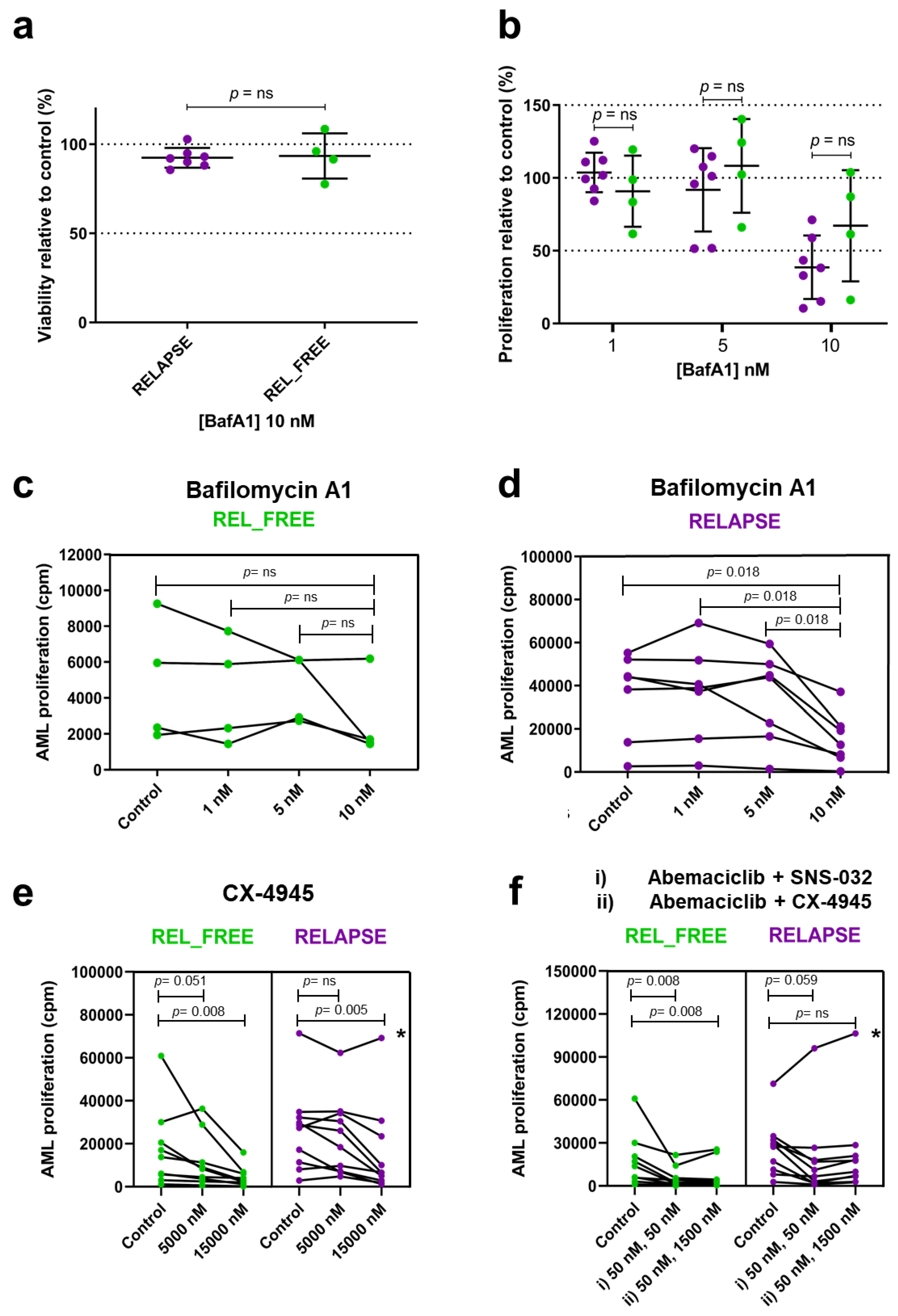
2.4. V-ATPase, CSK2, CDK2/7/9 and CDK4/6 Inhibitors Affect the Proliferation of AML Cells
3. Discussion
4. Materials and Methods
4.1. AML Patients and Sample Collection
4.2. AML Super-SILAC Mix
4.3. Patient Sample Preparation for Proteomic and Phosphoproteomic Analysis
4.4. Nanoflow LC-MS/MS
4.5. Data and Bioinformatics Analysis
4.6. Enrichment Analysis of Transcription Proteins Binding Sites
4.7. Cell Proliferation Assay
4.8. Western Blots
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferrara, F.; Schiffer, C.A. Acute myeloid leukaemia in adults. Lancet 2013, 381, 484–495. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Sanz, M.A.; Fenaux, P.; Tallman, M.S.; Estey, E.H.; Lowenberg, B.; Naoe, T.; Lengfelder, E.; Dohner, H.; Burnett, A.K.; Chen, S.J.; et al. Management of acute promyelocytic leukemia: Updated recommendations from an expert panel of the European LeukemiaNet. Blood 2019, 133, 1630–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Döhner, H.; Estey, E.H.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Burnett, A.K.; Dombret, H.; Fenaux, P.; Grimwade, D.; Larson, R.A.; et al. Diagnosis and management of acute myeloid leukemia in adults: Recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 2010, 115, 453–474. [Google Scholar] [CrossRef] [PubMed]
- Cornelissen, J.J.; Gratwohl, A.; Schlenk, R.F.; Sierra, J.; Bornhäuser, M.; Juliusson, G.; Råcil, Z.; Rowe, J.M.; Russell, N.; Mohty, M.; et al. The European LeukemiaNet AML Working Party consensus statement on allogeneic HSCT for patients with AML in remission: An integrated-risk adapted approach. Nat. Rev. Clin. Oncol. 2012, 9, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Estey, E.H. Acute myeloid leukemia: 2014 update on risk-stratification and management. Am. J. Hematol. 2014, 89, 1063–1081. [Google Scholar] [CrossRef] [PubMed]
- Ossenkoppele, G.J.; Janssen, J.J.; van de Loosdrecht, A.A. Risk factors for relapse after allogeneic transplantation in acute myeloid leukemia. Haematologica 2016, 101, 20–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irish, J.M.; Anensen, N.; Hovland, R.; Skavland, J.; Borresen-Dale, A.L.; Bruserud, O.; Nolan, G.P.; Gjertsen, B.T. Flt3 Y591 duplication and Bc1-2 overexpression are detected in acute myeloid leukemia cells with high levels of phosphorylated wild-type p53. Blood 2007, 109, 2589–2596. [Google Scholar] [CrossRef] [Green Version]
- Nepstad, I.; Hatfield, K.J.; Tvedt, T.H.A.; Reikvam, H.; Bruserud, O. Clonal Heterogeneity Reflected by PI3K-AKT-mTOR Signaling in Human Acute Myeloid Leukemia Cells and Its Association with Adverse Prognosis. Cancers 2018, 10, 332. [Google Scholar] [CrossRef] [Green Version]
- Patel, J.P.; Gönen, M.; Figueroa, M.E.; Fernandez, H.; Sun, Z.; Racevskis, J.; Van Vlierberghe, P.; Dolgalev, I.; Thomas, S.; Aminova, O.; et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N. Engl. J. Med. 2012, 366, 1079–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Bullinger, L.; Dohner, K.; Dohner, H. Genomics of Acute Myeloid Leukemia Diagnosis and Pathways. J. Clin. Oncol. 2017, 35, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [CrossRef] [PubMed] [Green Version]
- Corces, M.R.; Chang, H.Y.; Majeti, R. Preleukemic Hematopoietic Stem Cells in Human Acute Myeloid Leukemia. Front. Oncol. 2017, 7, 263. [Google Scholar] [CrossRef] [Green Version]
- Medeiros, B.C.; Minden, M.D.; Schuh, A.C.; Schimmer, A.D.; Yee, K.; Lipton, J.H.; Messner, H.A.; Gupta, V.; Chun, K.; Xu, W.; et al. Characteristics and outcomes of acute myelogenous leukemia patients with very late relapse (>5 years). Leuk. Lymphoma 2007, 48, 65–71. [Google Scholar] [CrossRef]
- Pedersen-Bjergaard, J.; Andersen, M.T.; Andersen, M.K. Genetic pathways in the pathogenesis of therapy-related myelodysplasia and acute myeloid leukemia. Hematol. Am. Soc. Hematol. Educ. Progr. 2007. [Google Scholar] [CrossRef] [PubMed]
- Cocciardi, S.; Dolnik, A.; Kapp-Schwoerer, S.; Rucker, F.G.; Lux, S.; Blatte, T.J.; Skambraks, S.; Kronke, J.; Heidel, F.H.; Schnoder, T.M.; et al. Clonal evolution patterns in acute myeloid leukemia with NPM1 mutation. Nat. Commun. 2019, 10, 2031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollein, A.; Meggendorfer, M.; Dicker, F.; Jeromin, S.; Nadarajah, N.; Kern, W.; Haferlach, C.; Haferlach, T. NPM1 mutated AML can relapse with wild-type NPM1: Persistent clonal hematopoiesis can drive relapse. Blood Adv. 2018, 2, 3118–3125. [Google Scholar] [CrossRef]
- Greif, P.A.; Hartmann, L.; Vosberg, S.; Stief, S.M.; Mattes, R.; Hellmann, I.; Metzeler, K.H.; Herold, T.; Bamopoulos, S.A.; Kerbs, P.; et al. Evolution of Cytogenetically Normal Acute Myeloid Leukemia During Therapy and Relapse: An Exome Sequencing Study of 50 Patients. Clin. Cancer Res. 2018, 24, 1716–1726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vosberg, S.; Greif, P.A. Clonal evolution of acute myeloid leukemia from diagnosis to relapse. Genes Chromosomes Cancer 2019, 58, 839–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yilmaz, M.; Wang, F.; Loghavi, S.; Bueso-Ramos, C.; Gumbs, C.; Little, L.; Song, X.Z.; Zhang, J.H.; Kadia, T.; Borthakur, G.; et al. Late relapse in acute myeloid leukemia (AML): Clonal evolution or therapy-related leukemia? Blood Cancer J. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Aasebø, E.; Forthun, R.B.; Berven, F.; Selheim, F.; Hernandez-Valladares, M. Global Cell Proteome Profiling, Phospho-signaling and Quantitative Proteomics for Identification of New Biomarkers in Acute Myeloid Leukemia Patients. Curr. Pharm. Biotechnol. 2016, 17, 52–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foss, E.J.; Radulovic, D.; Stirewalt, D.L.; Radich, J.; Sala-Torra, O.; Pogosova-Agadjanyan, E.L.; Hengel, S.M.; Loeb, K.R.; Deeg, H.J.; Meshinchi, S.; et al. Proteomic classification of acute leukemias by alignment-based quantitation of LC-MS/MS data sets. J. Proteome Res. 2012, 11, 5005–5010. [Google Scholar] [CrossRef] [Green Version]
- Forthun, R.B.; Aasebø, E.; Rasinger, J.D.; Bedringaas, S.L.; Berven, F.; Selheim, F.; Bruserud, Ø.; Gjertsen, B.T. Phosphoprotein DIGE profiles reflect blast differentiation, cytogenetic risk stratification, FLT3/NPM1 mutations and therapy response in acute myeloid leukaemia. J. Proteom. 2018, 173, 32–41. [Google Scholar] [CrossRef]
- Wojtuszkiewicz, A.; Schuurhuis, G.J.; Kessler, F.L.; Piersma, S.R.; Knol, J.C.; Pham, T.V.; Jansen, G.; Musters, R.J.; van Meerloo, J.; Assaraf, Y.G.; et al. Exosomes Secreted by Apoptosis-Resistant Acute Myeloid Leukemia (AML) Blasts Harbor Regulatory Network Proteins Potentially Involved in Antagonism of Apoptosis. Mol. Cell. Proteom. 2016, 15, 1281–1298. [Google Scholar] [CrossRef] [Green Version]
- Schaab, C.; Oppermann, F.S.; Klammer, M.; Pfeifer, H.; Tebbe, A.; Oellerich, T.; Krauter, J.; Levis, M.; Perl, A.E.; Daub, H.; et al. Global phosphoproteome analysis of human bone marrow reveals predictive phosphorylation markers for the treatment of acute myeloid leukemia with quizartinib. Leukemia 2014, 28, 716–719. [Google Scholar] [CrossRef] [Green Version]
- Tong, J.; Helmy, M.; Cavalli, F.M.; Jin, L.; St-Germain, J.; Karisch, R.; Taylor, P.; Minden, M.D.; Taylor, M.D.; Neel, B.G.; et al. Integrated analysis of proteome, phosphotyrosine-proteome, tyrosine-kinome, and tyrosine-phosphatome in acute myeloid leukemia. Proteomics 2017, 17. [Google Scholar] [CrossRef]
- Casado, P.; Wilkes, E.H.; Miraki-Moud, F.; Hadi, M.M.; Rio-Machin, A.; Rajeeve, V.; Pike, R.; Iqbal, S.; Marfa, S.; Lea, N.; et al. Proteomic and genomic integration identifies kinase and differentiation determinants of kinase inhibitor sensitivity in leukemia cells. Leukemia 2018, 32, 1818–1822. [Google Scholar] [CrossRef]
- Aasebø, E.; Vaudel, M.; Mjaavatten, O.; Gausdal, G.; Van der Burgh, A.; Gjertsen, B.T.; Døskeland, S.O.; Bruserud, Ø.; Berven, F.S.; Selheim, F. Performance of super-SILAC based quantitative proteomics for comparison of different acute myeloid leukemia (AML) cell lines. Proteomics 2014, 14, 1971–1976. [Google Scholar] [CrossRef]
- Aasebø, E.; Mjaavatten, O.; Vaudel, M.; Farag, Y.; Selheim, F.; Berven, F.; Bruserud, Ø.; Hernandez-Valladares, M. Freezing effects on the acute myeloid leukemia cell proteome and phosphoproteome revealed using optimal quantitative workflows. J. Proteom. 2016, 145, 214–225. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Valladares, M.; Aasebø, E.; Mjaavatten, O.; Vaudel, M.; Bruserud, Ø.; Berven, F.; Selheim, F. Reliable FASP-based procedures for optimal quantitative proteomic and phosphoproteomic analysis on samples from acute myeloid leukemia patients. Biol. Proced. Online 2016, 18, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez-Valladares, M.; Aasebo, E.; Selheim, F.; Berven, F.S.; Bruserud, O. Selecting Sample Preparation Workflows for Mass Spectrometry-Based Proteomic and Phosphoproteomic Analysis of Patient Samples with Acute Myeloid Leukemia. Proteomes 2016, 4, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reikvam, H.; Aasebo, E.; Brenner, A.K.; Bartaula-Brevik, S.; Gronningsaeter, I.S.; Forthun, R.B.; Hovland, R.; Bruserud, O. High Constitutive Cytokine Release by Primary Human Acute Myeloid Leukemia Cells Is Associated with a Specific Intercellular Communication Phenotype. J. Clin. Med. 2019, 8, 970. [Google Scholar] [CrossRef] [Green Version]
- Tsykunova, G.; Reikvam, H.; Hovland, R.; Bruserud, O. The surface molecule signature of primary human acute myeloid leukemia (AML) cells is highly associated with NPM1 mutation status. Leukemia 2012, 26, 557–559. [Google Scholar] [CrossRef] [Green Version]
- Stransky, L.; Cotter, K.; Forgac, M. The Function of V-Atpases in Cancer. Physiol. Rev. 2016, 96, 1071–1091. [Google Scholar] [CrossRef]
- Bammert, L.; Jonas, S.; Ungricht, R.; Kutay, U. Human AATF/Che-1 forms a nucleolar protein complex with NGDN and NOL10 required for 40S ribosomal subunit synthesis. Nucleic Acids Res. 2016, 44, 9803–9820. [Google Scholar] [CrossRef] [Green Version]
- Folgiero, V.; Sorino, C.; Locatelli, F.; Fanciulli, M. A new baby in the c-Myc-directed transcriptional machinery: Che-1/AATF. Cell Cycle 2018, 17, 1286–1290. [Google Scholar] [CrossRef] [Green Version]
- Kaul, D.; Mehrotra, A. Functional characterization of AATF transcriptome in human leukemic cells. Mol. Cell. Biochem. 2007, 297, 215–220. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Breems, D.A.; Van Putten, W.L.; Huijgens, P.C.; Ossenkoppele, G.J.; Verhoef, G.E.; Verdonck, L.F.; Vellenga, E.; De Greef, G.E.; Jacky, E.; Van der Lelie, J.; et al. Prognostic index for adult patients with acute myeloid leukemia in first relapse. J. Clin. Oncol. 2005, 23, 1969–1978. [Google Scholar] [CrossRef] [PubMed]
- Short, N.J.; Rytting, M.E.; Cortes, J.E. Acute myeloid leukaemia. Lancet 2018, 392, 593–606. [Google Scholar] [CrossRef]
- Colaert, N.; Helsens, K.; Martens, L.; Vandekerckhove, J.; Gevaert, K. Improved visualization of protein consensus sequences by iceLogo. Nat. Methods 2009, 6, 786–787. [Google Scholar] [CrossRef]
- Wiredja, D.D.; Koyuturk, M.; Chance, M.R. The KSEA App: A web-based tool for kinase activity inference from quantitative phosphoproteomics. Bioinformatics 2017, 33, 3489–3491. [Google Scholar] [CrossRef] [PubMed]
- Casado, P.; Rodriguez-Prados, J.C.; Cosulich, S.C.; Guichard, S.; Vanhaesebroeck, B.; Joel, S.; Cutillas, P.R. Kinase-substrate enrichment analysis provides insights into the heterogeneity of signaling pathway activation in leukemia cells. Sci. Signal. 2013, 6, rs6. [Google Scholar] [CrossRef] [PubMed]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [Green Version]
- Negi, S.S.; Olson, M.O. Effects of interphase and mitotic phosphorylation on the mobility and location of nucleolar protein B23. J. Cell Sci. 2006, 119, 3676–3685. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Jiang, X.; Wang, Y. Identification of novel in vivo phosphorylation sites in high mobility group N1 protein from the MCF-7 human breast cancer cells. Biochemistry 2004, 43, 6322–6329. [Google Scholar] [CrossRef]
- Beausoleil, S.A.; Jedrychowski, M.; Schwartz, D.; Elias, J.E.; Villen, J.; Li, J.; Cohn, M.A.; Cantley, L.C.; Gygi, S.P. Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc. Natl. Acad. Sci. USA 2004, 101, 12130–12135. [Google Scholar] [CrossRef] [Green Version]
- Misra, R.N.; Xiao, H.Y.; Kim, K.S.; Lu, S.F.; Han, W.C.; Barbosa, S.A.; Hunt, J.T.; Rawlins, D.B.; Shan, W.F.; Ahmed, S.Z.; et al. N-(Cycloalkylamino)acyl-2-aminothiazole inhibitors of cyclin-dependent kinase-2. N-[5-[[[5-(1,1-dimethylethyl)-2-oxazolyl]methyl]thio]-2-thiazolyl]-4-piperidinecarboxamide (BMS-387032), a highly efficacious and selective antitumor agent. J. Med. Chem. 2004, 47, 1719–1728. [Google Scholar] [CrossRef]
- Lemke, J.; von Karstedt, S.; Abd El Hay, M.; Conti, A.; Arce, F.; Montinaro, A.; Papenfuss, K.; El-Bahrawy, M.A.; Walczak, H. Selective CDK9 inhibition overcomes TRAIL resistance by concomitant suppression of cFlip and Mcl-1. Cell Death Differ. 2014, 21, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Tang, H.; Wu, S.; Chen, J.; Liu, J.; Liao, C. The cyclin-dependent kinase inhibitor SNS-032 induces apoptosis in breast cancer cells via depletion of Mcl-1 and X-linked inhibitor of apoptosis protein and displays antitumor activity in vivo. Int. J. Oncol. 2014, 45, 804–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, S.; Morris, E.J.; Hruza, A.; Mansueto, M.S.; Schroeder, G.K.; Arbanas, J.; McMasters, D.; Restaino, C.R.; Dayananth, P.; Black, S.; et al. Dissecting Therapeutic Resistance to ERK Inhibition. Mol. Cancer Ther. 2016, 15, 548–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gjertsen, B.T.; Øyan, A.M.; Marzolf, B.; Hovland, R.; Gausdal, G.; Døskeland, S.O.; Dimitrov, K.; Golden, A.; Kalland, K.H.; Hood, L.; et al. Analysis of acute myelogenous leukemia: Preparation of samples for genomic and proteomic analyses. J. Hematother. Stem Cell Res. 2002, 11, 469–481. [Google Scholar] [CrossRef]
- Bruserud, O.; Tjonnfjord, G.; Gjertsen, B.T.; Foss, B.; Ernst, P. New strategies in the treatment of acute myelogenous leukemia: Mobilization and transplantation of autologous peripheral blood stem cells in adult patients. Stem Cells 2001, 18, 341–351. [Google Scholar]
- Bruserud, Ø.; Hovland, R.; Wergeland, L.; Huang, T.S.; Gjertsen, B.T. Flt3-mediated signaling in human acute myelogenous leukemia (AML) blasts: A functional characterization of Flt3-ligand effects in AML cell populations with and without genetic Flt3 abnormalities. Haematologica 2003, 88, 416–428. [Google Scholar]
- Hu, C.W.; Qiu, Y.; Ligeralde, A.; Raybon, A.Y.; Yoo, S.Y.; Coombes, K.R.; Qutub, A.A.; Kornblau, S.M. A quantitative analysis of heterogeneities and hallmarks in acute myelogenous leukaemia. Nat. Biomed. Eng. 2019, 3, 889–901. [Google Scholar] [CrossRef]
- Wheatley, K.; Burnett, A.K.; Goldstone, A.H.; Gray, R.G.; Hann, I.M.; Harrison, C.J.; Rees, J.K.H.; Stevens, R.F.; Walker, H. A simple, robust, validated and highly predictive index for the determination of risk-directed therapy in acute myeloid leukaemia derived from the MRC AML 10 trial. Br. J. Haematol. 1999, 107, 69–79. [Google Scholar] [CrossRef]
- de Jonge, H.J.M.; Valk, P.J.M.; de Bont, E.S.J.M.; Schuringa, J.J.; Ossenkoppele, G.; Vellenga, E.; Huls, G. Prognostic impact of white blood cell count in intermediate risk acute myeloid leukemia: Relevance of mutated NPM1 and FLT3-ITD. Haematologica 2011, 96, 1310–1317. [Google Scholar] [CrossRef]
- Lin, P.; Chen, L.; Luthra, R.; Konoplev, S.N.; Wang, X.M.; Medeiros, L.J. Acute myeloid leukemia harboring t(8;21)(q22;q22): A heterogeneous disease with poor outcome in a subset of patients unrelated to secondary cytogenetic aberrations. Mod. Pathol. 2008, 21, 1029–1036. [Google Scholar] [CrossRef] [Green Version]
- Feng, S.L.; Zhou, L.; Zhang, X.H.; Tang, B.L.; Zhu, X.Y.; Liu, H.L.; Sun, Z.M.; Zheng, C.C. Impact Of ELN Risk Stratification, Induction Chemotherapy Regimens And Hematopoietic Stem Cell Transplantation On Outcomes In Hyperleukocytic Acute Myeloid Leukemia With Initial White Blood Cell Count More Than 100 x 10(9)/L. Cancer Manag. Res. 2019, 11, 9495–9503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- How, J.; Sykes, J.; Gupta, V.; Yee, K.W.; Schimmer, A.D.; Schuh, A.C.; Minden, M.D.; Kamel-Reid, S.; Brandwein, J.M. Influence of FLT3-internal tandem duplication allele burden and white blood cell count on the outcome in patients with intermediate-risk karyotype acute myeloid leukemia. Cancer 2012, 118, 6110–6117. [Google Scholar] [CrossRef] [PubMed]
- Reuss-Borst, M.A.; Klein, G.; Waller, H.D.; Muller, C.A. Differential expression of adhesion molecules in acute leukemia. Leukemia 1995, 9, 869–874. [Google Scholar] [PubMed]
- Brenner, A.K.; Aasebo, E.; Hernandez-Valladares, M.; Selheim, F.; Berven, F.; Gronningsaeter, I.S.; Bartaula-Brevik, S.; Bruserud, O. The Capacity of Long-Term In Vitro Proliferation of Acute Myeloid Leukemia Cells Supported Only by Exogenous Cytokines Is Associated with a Patient Subset with Adverse Outcome. Cancers 2019, 11, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nepstad, I.; Reikvam, H.; Brenner, A.K.; Bruserud, O.; Hatfield, K.J. Resistance to the Antiproliferative In Vitro Effect of PI3K-Akt-mTOR Inhibition in Primary Human Acute Myeloid Leukemia Cells Is Associated with Altered Cell Metabolism. Int. J. Mol. Sci. 2018, 19, 382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenner, A.K.; Tvedt, T.H.; Nepstad, I.; Rye, K.P.; Hagen, K.M.; Reikvam, H.; Bruserud, O. Patients with acute myeloid leukemia can be subclassified based on the constitutive cytokine release of the leukemic cells; the possible clinical relevance and the importance of cellular iron metabolism. Expert Opin. Ther. Targets 2017, 21, 357–369. [Google Scholar] [CrossRef]
- Bullinger, L.; Dohner, K.; Bair, E.; Frohling, S.; Schlenk, R.F.; Tibshirani, R.; Dohner, H.; Pollack, J.R. Use of gene-expression profiling to identify prognostic subclasses in adult acute myeloid leukemia. N. Engl. J. Med. 2004, 350, 1605–1616. [Google Scholar] [CrossRef] [Green Version]
- Heuser, M.; Wingen, L.U.; Steinemann, D.; Cario, G.; von Neuhoff, N.; Tauscher, M.; Bullinger, L.; Krauter, J.; Heil, G.; Dohner, H.; et al. Gene-expression profiles and their association with drug resistance in adult acute myeloid leukemia. Haematologica 2005, 90, 1484–1492. [Google Scholar]
- Eppert, K.; Takenaka, K.; Lechman, E.R.; Waldron, L.; Nilsson, B.; van Galen, P.; Metzeler, K.H.; Poeppl, A.; Ling, V.; Beyene, J.; et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat. Med. 2011, 17, 1086–1093. [Google Scholar] [CrossRef]
- Bullinger, L.; Ehrich, M.; Dohner, K.; Schlenk, R.F.; Dohner, H.; Nelson, M.R.; van den Boom, D. Quantitative DNA methylation predicts survival in adult acute myeloid leukemia. Blood 2010, 115, 636–642. [Google Scholar] [CrossRef]
- Li, S.; Garrett-Bakelman, F.E.; Chung, S.S.; Sanders, M.A.; Hricik, T.; Rapaport, F.; Patel, J.; Dillon, R.; Vijay, P.; Brown, A.L.; et al. Distinct evolution and dynamics of epigenetic and genetic heterogeneity in acute myeloid leukemia. Nat. Med. 2016, 22, 792–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irish, J.M.; Hovland, R.; Krutzik, P.O.; Perez, O.D.; Bruserud, O.; Gjertsen, B.T.; Nolan, G.P. Single cell profiling of potentiated phospho-protein networks in cancer cells. Cell 2004, 118, 217–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruserud, O.; Aasebo, E.; Hernandez-Valladares, M.; Tsykunova, G.; Reikvam, H. Therapeutic targeting of leukemic stem cells in acute myeloid leukemia—The biological background for possible strategies. Expert Opin. Drug Dis. 2017, 12, 1053–1065. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, J.; Thomas, G.; Volarevic, S. Ribosome biogenesis in cancer: New players and therapeutic avenues. Nat. Rev. Cancer 2018, 18, 51–63. [Google Scholar] [CrossRef]
- Wang, E.; Lu, S.X.; Pastore, A.; Chen, X.; Imig, J.; Chun-Wei Lee, S.; Hockemeyer, K.; Ghebrechristos, Y.E.; Yoshimi, A.; Inoue, D.; et al. Targeting an RNA-Binding Protein Network in Acute Myeloid Leukemia. Cancer Cell 2019, 35, 369–384. [Google Scholar] [CrossRef] [Green Version]
- Delgado, M.D.; Albajar, M.; Gomez-Casares, M.T.; Batlle, A.; Leon, J. MYC oncogene in myeloid neoplasias. Clin. Transl. Oncol. 2013, 15, 87–94. [Google Scholar] [CrossRef]
- Delgado, M.D.; Leon, J. Myc roles in hematopoiesis and leukemia. Genes Cancer 2010, 1, 605–616. [Google Scholar] [CrossRef] [Green Version]
- van Riggelen, J.; Yetil, A.; Felsher, D.W. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat. Rev. Cancer 2010, 10, 301–309. [Google Scholar] [CrossRef]
- Grandori, C.; Gomez-Roman, N.; Felton-Edkins, Z.A.; Ngouenet, C.; Galloway, D.A.; Eisenman, R.N.; White, R.J. c-Myc binds to human ribosomal DNA and stimulates transcription of rRNA genes by RNA polymerase I. Nat. Cell Biol. 2005, 7, 311–318. [Google Scholar] [CrossRef]
- Herter, E.K.; Stauch, M.; Gallant, M.; Wolf, E.; Raabe, T.; Gallant, P. snoRNAs are a novel class of biologically relevant Myc targets. BMC Biol. 2015, 13, 25. [Google Scholar] [CrossRef] [Green Version]
- Derenzini, E.; Rossi, A.; Trere, D. Treating hematological malignancies with drugs inhibiting ribosome biogenesis: When and why. J. Hematol. Oncol. 2018, 11, 75. [Google Scholar] [CrossRef] [PubMed]
- Bywater, M.J.; Poortinga, G.; Sanij, E.; Hein, N.; Peck, A.; Cullinane, C.; Wall, M.; Cluse, L.; Drygin, D.; Anderes, K.; et al. Inhibition of RNA polymerase I as a therapeutic strategy to promote cancer-specific activation of p53. Cancer Cell 2012, 22, 51–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stransky, L.A.; Forgac, M. Amino Acid Availability Modulates Vacuolar H+-ATPase Assembly. J. Biol. Chem. 2015, 290, 27360–27369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoncu, R.; Bar-Peled, L.; Efeyan, A.; Wang, S.; Sancak, Y.; Sabatini, D.M. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 2011, 334, 678–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobia, F.; Duchi, S.; Deflorian, G.; Vaccari, T. Pharmacologic inhibition of vacuolar H+ ATPase reduces physiologic and oncogenic Notch signaling. Mol. Oncol. 2014, 8, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Bernhard, S.M.; Seidel, K.; Schmitz, J.; Klare, S.; Kirsch, S.; Schrezenmeier, E.; Zaade, D.; Meyborg, H.; Goldin-Lang, P.; Stawowy, P.; et al. The (pro)renin receptor ((P)RR) can act as a repressor of Wnt signalling. Biochem. Pharmacol. 2012, 84, 1643–1650. [Google Scholar] [CrossRef]
- Rothe, K.; Porter, V.; Jiang, X. Current Outlook on Autophagy in Human Leukemia: Foe in Cancer Stem Cells and Drug Resistance, Friend in New Therapeutic Interventions. Int. J. Mol. Sci. 2019, 20, 461. [Google Scholar] [CrossRef] [Green Version]
- Zare-Abdollahi, D.; Safari, S.; Movafagh, A.; Ghadiani, M.; Tabarraee, M.; Riazi-Isfahani, S.; Gorji, S.; Keyvan, L.; Gachkar, L. Expression analysis of BECN1 in acute myeloid leukemia: Association with distinct cytogenetic and molecular abnormalities. Int. J. Lab. Hematol. 2016, 38, 125–132. [Google Scholar] [CrossRef]
- Radwan, S.M.; Hamdy, N.M.; Hegab, H.M.; El-Mesallamy, H.O. Beclin-1 and hypoxia-inducible factor-1alpha genes expression: Potential biomarkers in acute leukemia patients. Cancer Biomark. 2016, 16, 619–626. [Google Scholar] [CrossRef]
- Aasebø, E.; Bartaula-Brevik, S.; Hernandez-Valladares, M.; Bruserud, Ø. Vacuolar ATPase as a possible therapeutic target in human acute myeloid leukemia. Expert Rev. Hematol. 2018, 11, 13–24. [Google Scholar] [CrossRef]
- Watson, A.S.; Riffelmacher, T.; Stranks, A.; Williams, O.; De Boer, J.; Cain, K.; MacFarlane, M.; McGouran, J.; Kessler, B.; Khandwala, S.; et al. Autophagy limits proliferation and glycolytic metabolism in acute myeloid leukemia. Cell Death Discov. 2015, 1. [Google Scholar] [CrossRef] [PubMed]
- Rothe, K.; Lin, H.; Lin, K.B.; Leung, A.; Wang, H.M.; Malekesmaeili, M.; Brinkman, R.R.; Forrest, D.L.; Gorski, S.M.; Jiang, X. The core autophagy protein ATG4B is a potential biomarker and therapeutic target in CML stem/progenitor cells. Blood 2014, 123, 3622–3634. [Google Scholar] [CrossRef]
- Piya, S.; Kornblau, S.M.; Ruvolo, V.R.; Mu, H.; Ruvolo, P.P.; McQueen, T.; Davis, R.E.; Hail, N., Jr.; Kantarjian, H.; Andreeff, M.; et al. Atg7 suppression enhances chemotherapeutic agent sensitivity and overcomes stroma-mediated chemoresistance in acute myeloid leukemia. Blood 2016, 128, 1260–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumitomo, Y.; Koya, J.; Nakazaki, K.; Kataoka, K.; Tsuruta-Kishino, T.; Morita, K.; Sato, T.; Kurokawa, M. Cytoprotective autophagy maintains leukemia-initiating cells in murine myeloid leukemia. Blood 2016, 128, 1614–1624. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Jin, J.; Pan, J.; Yao, R.; Li, X.; Huang, X.; Ma, Z.; Huang, S.; Yan, X.; Jin, J.; et al. The change of nuclear LC3 distribution in acute myeloid leukemia cells. Exp. Cell Res. 2018, 369, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Buontempo, F.; McCubrey, J.A.; Orsini, E.; Ruzzene, M.; Cappellini, A.; Lonetti, A.; Evangelisti, C.; Chiarini, F.; Evangelisti, C.; Barata, J.T.; et al. Therapeutic targeting of CK2 in acute and chronic leukemias. Leukemia 2018, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.J.; Anders, L. Signaling through cyclin D-dependent kinases. Oncogene 2014, 33, 1890–1903. [Google Scholar] [CrossRef] [Green Version]
- Saiki, Y.; Yamazaki, Y.; Yoshida, M.; Katoh, O.; Nakamura, T. Human EVI9, a homologue of the mouse myeloid leukemia gene, is expressed in the hematopoietic progenitors and down-regulated during myeloid differentiation of HL60 cells. Genomics 2000, 70, 387–391. [Google Scholar] [CrossRef]
- Yin, B.; Delwel, R.; Valk, P.J.; Wallace, M.R.; Loh, M.L.; Shannon, K.M.; Largaespada, D.A. A retroviral mutagenesis screen reveals strong cooperation between Bcl11a overexpression and loss of the Nf1 tumor suppressor gene. Blood 2009, 113, 1075–1085. [Google Scholar] [CrossRef] [Green Version]
- Jowsey, P.; Morrice, N.A.; Hastie, C.J.; McLauchlan, H.; Toth, R.; Rouse, J. Characterisation of the sites of DNA damage-induced 53BP1 phosphorylation catalysed by ATM and ATR. DNA Repair 2007, 6, 1536–1544. [Google Scholar] [CrossRef]
- Drane, P.; Brault, M.E.; Cui, G.; Meghani, K.; Chaubey, S.; Detappe, A.; Parnandi, N.; He, Y.; Zheng, X.F.; Botuyan, M.V.; et al. TIRR regulates 53BP1 by masking its histone methyl-lysine binding function. Nature 2017, 543, 211–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Kim, S.; Rhee, K. NEK7 is essential for centriole duplication and centrosomal accumulation of pericentriolar material proteins in interphase cells. J. Cell Sci. 2011, 124, 3760–3770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salem, H.; Rachmin, I.; Yissachar, N.; Cohen, S.; Amiel, A.; Haffner, R.; Lavi, L.; Motro, B. Nek7 kinase targeting leads to early mortality, cytokinesis disturbance and polyploidy. Oncogene 2010, 29, 4046–4057. [Google Scholar] [CrossRef] [PubMed]
- Belham, C.; Comb, M.J.; Avruch, J. Identification of the NIMA family kinases NEK6/7 as regulators of the p70 ribosomal S6 kinase. Curr. Biol. 2001, 11, 1155–1167. [Google Scholar] [CrossRef] [Green Version]
- Dalby, K.N.; Morrice, N.; Caudwell, F.B.; Avruch, J.; Cohen, P. Identification of regulatory phosphorylation sites in mitogen-activated protein kinase (MAPK)-activated protein kinase-1a/p90rsk that are inducible by MAPK. J. Biol. Chem. 1998, 273, 1496–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulzmaier, F.J.; Ramos, J.W. RSK isoforms in cancer cell invasion and metastasis. Cancer Res. 2013, 73, 6099–6105. [Google Scholar] [CrossRef] [Green Version]
- Hatfield, K.J.; Hovland, R.; Øyan, A.M.; Kalland, K.H.; Ryningen, A.; Gjertsen, B.T.; Bruserud, Ø. Release of angiopoietin-1 by primary human acute myelogenous leukemia cells is associated with mutations of nucleophosmin, increased by bone marrow stromal cells and possibly antagonized by high systemic angiopoietin-2 levels. Leukemia 2008, 22, 287–293. [Google Scholar] [CrossRef]
- Reikvam, H.; Hovland, R.; Forthun, R.B.; Erdal, S.; Gjertsen, B.T.; Fredly, H.; Bruserud, O. Disease-stabilizing treatment based on all-trans retinoic acid and valproic acid in acute myeloid leukemia—Identification of responders by gene expression profiling of pretreatment leukemic cells. BMC Cancer 2017, 17, 630. [Google Scholar] [CrossRef] [Green Version]
- Geiger, T.; Cox, J.; Ostasiewicz, P.; Wisniewski, J.R.; Mann, M. Super-SILAC mix for quantitative proteomics of human tumor tissue. Nat. Methods 2010, 7, 383–385. [Google Scholar] [CrossRef]
- Wisniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef]
- Kulak, N.A.; Pichler, G.; Paron, I.; Nagaraj, N.; Mann, M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 2014, 11, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Aasebø, E.; Hernandez-Valladares, M.; Selheim, F.; Berven, F.S.; Brenner, A.K.; Bruserud, Ø. Proteomic Profiling of Primary Human Acute Myeloid Leukemia Cells Does Not Reflect Their Constitutive Release of Soluble Mediators. Proteomes 2018, 7, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Matic, I.; Hilger, M.; Nagaraj, N.; Selbach, M.; Olsen, J.V.; Mann, M. A practical guide to the MaxQuant computational platform for SILAC-based quantitative proteomics. Nat. Protoc. 2009, 4, 698–705. [Google Scholar] [CrossRef]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef]
- Vizcaino, J.A.; Deutsch, E.W.; Wang, R.; Csordas, A.; Reisinger, F.; Rios, D.; Dianes, J.A.; Sun, Z.; Farrah, T.; Bandeira, N.; et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 2014, 32, 223–226. [Google Scholar] [CrossRef]
- Vizcaino, J.A.; Csordas, A.; del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, D447–D456. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Arntzen, M.Ø.; Koehler, C.J.; Barsnes, H.; Berven, F.S.; Treumann, A.; Thiede, B. IsobariQ: Software for isobaric quantitative proteomics using IPTL, iTRAQ, and TMT. J. Proteome Res. 2011, 10, 913–920. [Google Scholar] [CrossRef]
- Scholz, C.; Lyon, D.; Refsgaard, J.C.; Jensen, L.J.; Choudhary, C.; Weinert, B.T. Avoiding abundance bias in the functional annotation of post-translationally modified proteins. Nat. Methods 2015, 12, 1003–1004. [Google Scholar] [CrossRef]
- Hulsen, T.; de Vlieg, J.; Alkema, W. BioVenn—A web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genom. 2008, 9, 488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linding, R.; Jensen, L.J.; Ostheimer, G.J.; van Vugt, M.A.; Jorgensen, C.; Miron, I.M.; Diella, F.; Colwill, K.; Taylor, L.; Elder, K.; et al. Systematic discovery of in vivo phosphorylation networks. Cell 2007, 129, 1415–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Nepusz, T.; Yu, H.; Paccanaro, A. Detecting overlapping protein complexes in protein-protein interaction networks. Nat. Methods 2012, 9, 471–472. [Google Scholar] [CrossRef] [PubMed]
- Andersson, L.C.; Nilsson, K.; Gahmberg, C.G. K562—Human Erythroleukemic Cell-Line. Int. J. Cancer 1979, 23, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Fajtova, M.; Babusikova, O. Immunophenotype characterization of hematopoietic stem cells, progenitor cells restricted to myeloid lineage and their leukemia counterparts. Neoplasma 2010, 57, 392–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korfanty, J.; Stokowy, T.; Chadalski, M.; Toma-Jonik, A.; Vydra, N.; Widlak, P.; Wojtas, B.; Gielniewski, B.; Widlak, W. SPEN protein expression and interactions with chromatin in mouse testicular cells. Reproduction 2018, 156, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Janus, P.; Szoltysek, K.; Zajac, G.; Stokowy, T.; Walaszczyk, A.; Widlak, W.; Wojtas, B.; Gielniewski, B.; Iwanaszko, M.; Braun, R.; et al. Pro-inflammatory cytokine and high doses of ionizing radiation have similar effects on the expression of NF-kappaB-dependent genes. Cell. Signal. 2018, 46, 23–31. [Google Scholar] [CrossRef]
- Janus, P.; Stokowy, T.; Jaksik, R.; Szoltysek, K.; Handschuh, L.; Podkowinski, J.; Widlak, W.; Kimmel, M.; Widlak, P. Cross talk between cytokine and hyperthermia-induced pathways: Identification of different subsets of NF-kappaB-dependent genes regulated by TNFalpha and heat shock. Mol. Genet. Genom. 2015, 290, 1979–1990. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Sun, H.; Ma, J.; Zang, C.; Wang, C.; Wang, J.; Tang, Q.; Meyer, C.A.; Zhang, Y.; Liu, X.S. Target analysis by integration of transcriptome and ChIP-seq data with BETA. Nat. Protoc. 2013, 8, 2502–2515. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | REL_FREE | RELAPSE |
|---|---|---|
| Age average (range) in years | 49.5 (36–65) | 50.5 (18–68) |
| Number of patients | 15 | 26 |
| FAB classification | ||
| M0-M1 | 1 | 11 |
| M2 | 0 | 2 |
| M4-M5 | 14 | 12 |
| uncertain | 0 | 1 |
| FLT3 | ||
| WT | 14 | 14 |
| ITD | 1 | 8 |
| ND | 0 | 4 |
| NPM1 | ||
| WT | 6 | 16 |
| Ins | 8 | 7 |
| ND | 1 | 3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aasebø, E.; Berven, F.S.; Bartaula-Brevik, S.; Stokowy, T.; Hovland, R.; Vaudel, M.; Døskeland, S.O.; McCormack, E.; Batth, T.S.; Olsen, J.V.; et al. Proteome and Phosphoproteome Changes Associated with Prognosis in Acute Myeloid Leukemia. Cancers 2020, 12, 709. https://doi.org/10.3390/cancers12030709
Aasebø E, Berven FS, Bartaula-Brevik S, Stokowy T, Hovland R, Vaudel M, Døskeland SO, McCormack E, Batth TS, Olsen JV, et al. Proteome and Phosphoproteome Changes Associated with Prognosis in Acute Myeloid Leukemia. Cancers. 2020; 12(3):709. https://doi.org/10.3390/cancers12030709
Chicago/Turabian StyleAasebø, Elise, Frode S. Berven, Sushma Bartaula-Brevik, Tomasz Stokowy, Randi Hovland, Marc Vaudel, Stein Ove Døskeland, Emmet McCormack, Tanveer S. Batth, Jesper V. Olsen, and et al. 2020. "Proteome and Phosphoproteome Changes Associated with Prognosis in Acute Myeloid Leukemia" Cancers 12, no. 3: 709. https://doi.org/10.3390/cancers12030709
APA StyleAasebø, E., Berven, F. S., Bartaula-Brevik, S., Stokowy, T., Hovland, R., Vaudel, M., Døskeland, S. O., McCormack, E., Batth, T. S., Olsen, J. V., Bruserud, Ø., Selheim, F., & Hernandez-Valladares, M. (2020). Proteome and Phosphoproteome Changes Associated with Prognosis in Acute Myeloid Leukemia. Cancers, 12(3), 709. https://doi.org/10.3390/cancers12030709