Recognition Dynamics of Cancer Mutations on the ERp57-Tapasin Interface

 , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
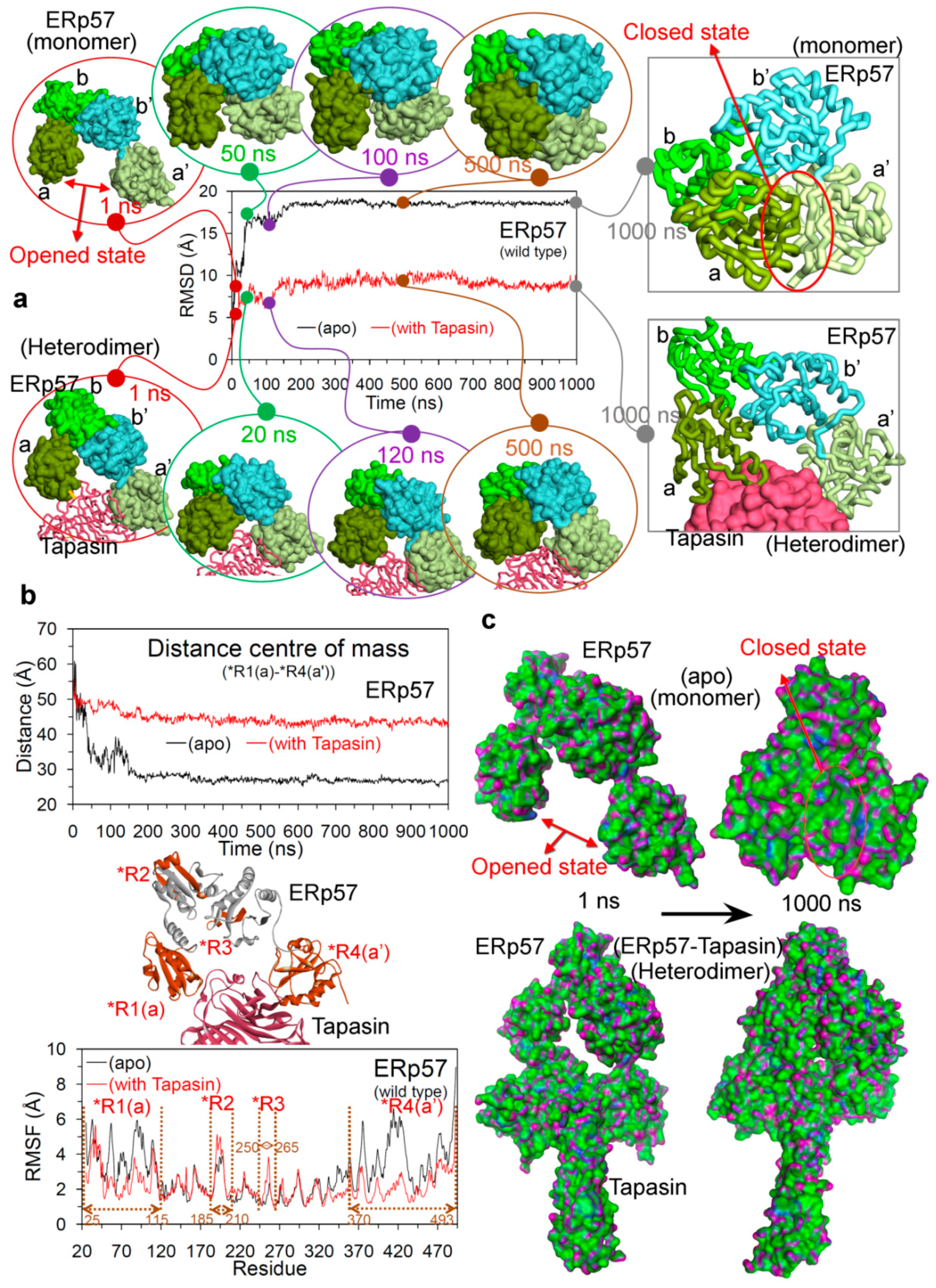
2.1. Open and Closed Conformation of ERp57, and ERp57-Tapasin Wild-Type Systems
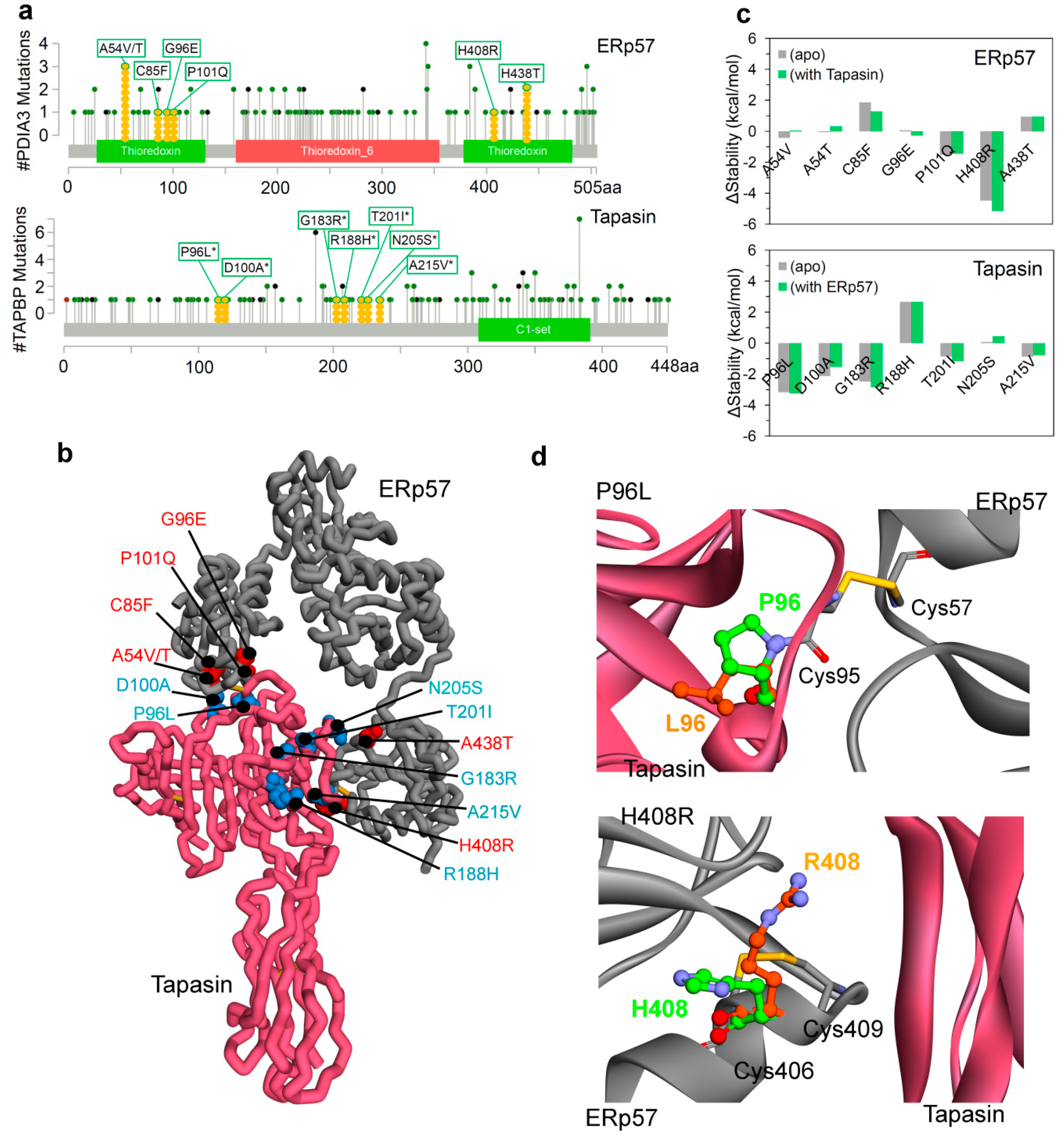
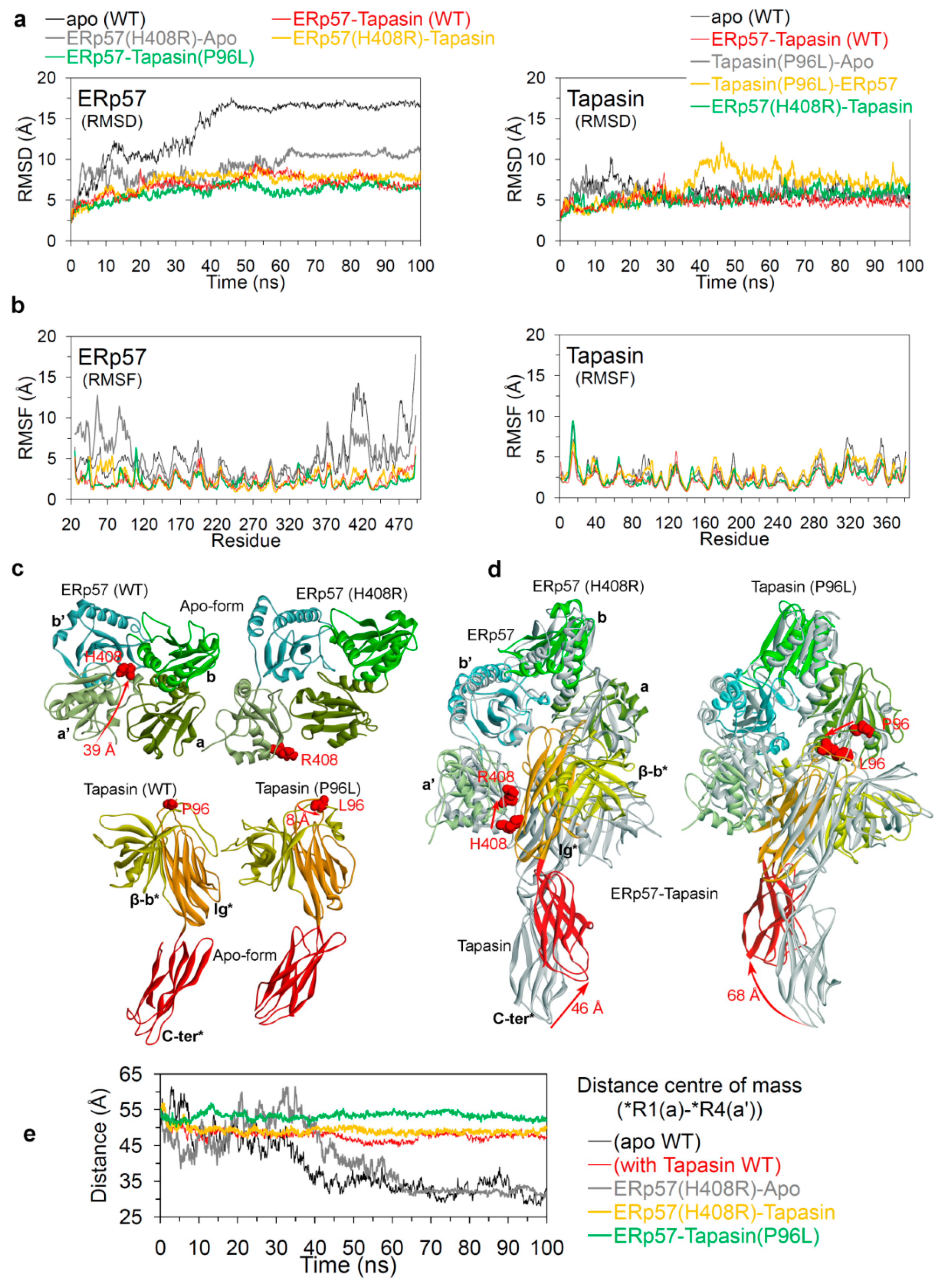
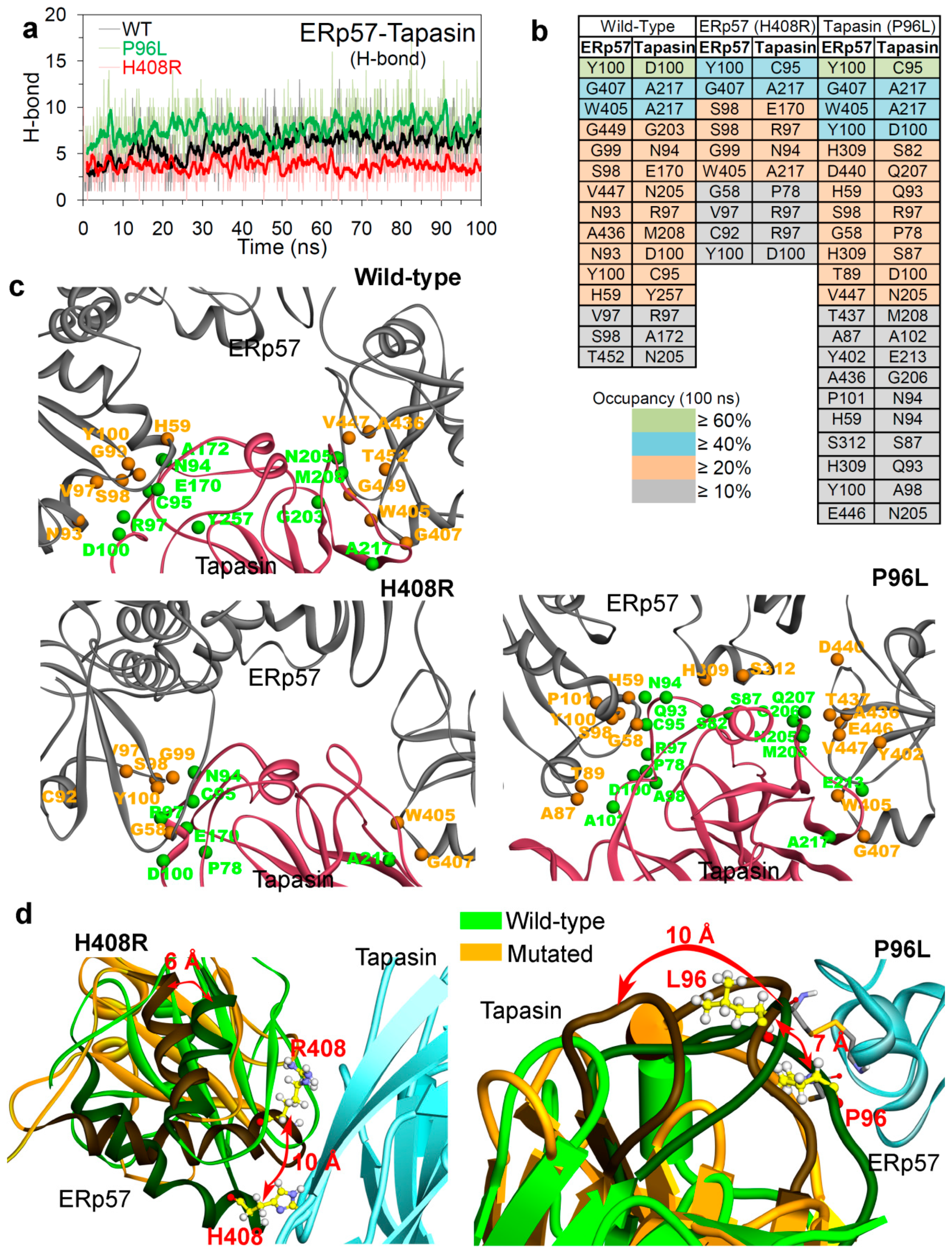
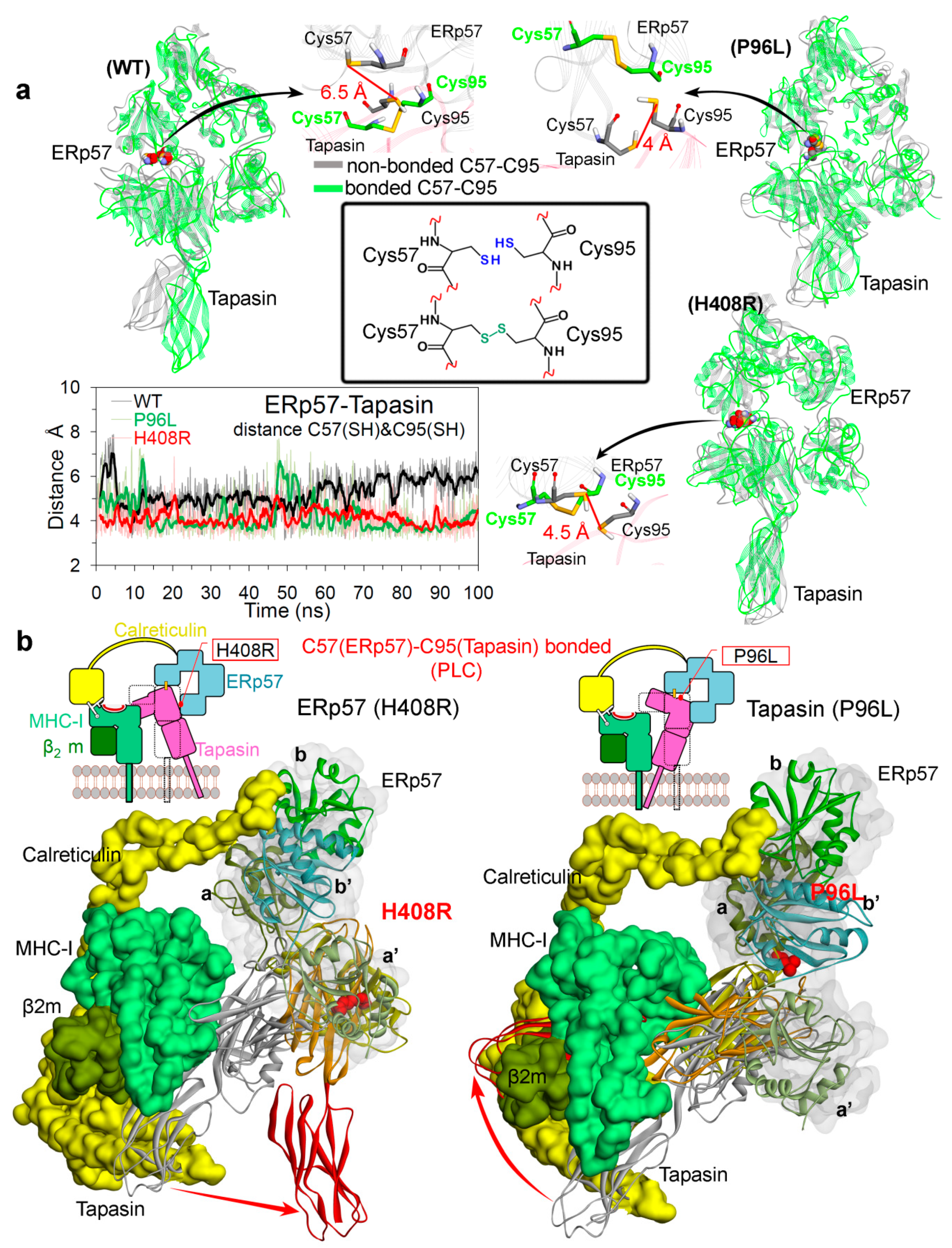
2.2. Insights into the Effects of Cancer Variants at the ERp57-Tapasin Interface
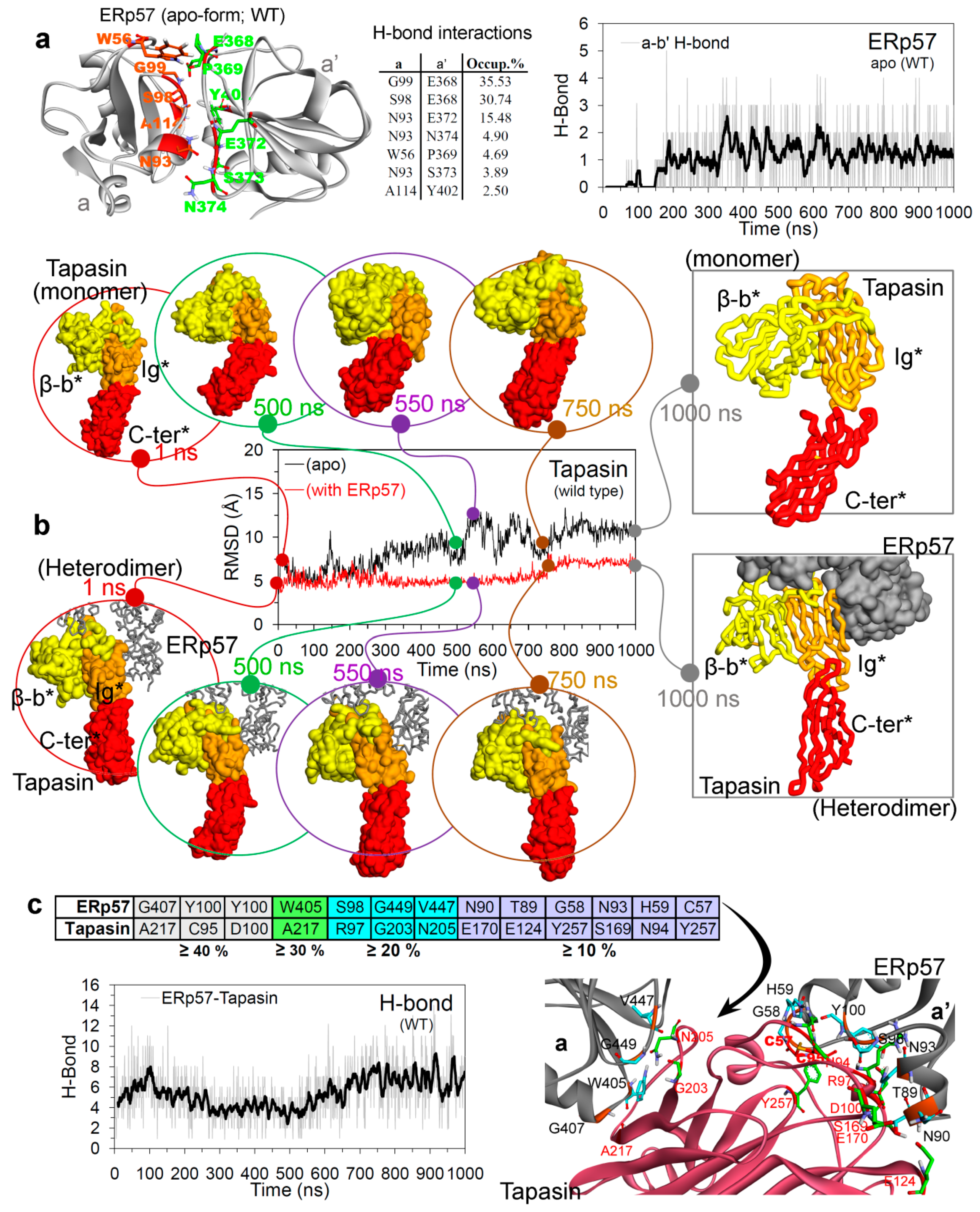
Hydrogen Bond Interactions and the Conformation Dynamics
3. Material and Methods
3.1. Protein Preparation
3.2. In Silico Mutation and Molecular Dynamics Simulation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Reeves, E.; James, E. Antigen processing and immune regulation in the response to tumours. Immunology 2017, 150, 16–24. [Google Scholar] [CrossRef] [Green Version]
- Vesely, M.D.; Schreiber, R.D. Cancer immunoediting: Antigens, mechanisms, and implications to cancer immunotherapy. Ann. N. Y. Acad. Sci. 2013, 1284, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehnert, E.; Tampé, R. Structure and dynamics of antigenic peptides in complex with TAP. Front. Immunol. 2017, 8, e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blees, A.; Januliene, D.; Hofmann, T.; Koller, N.; Schmidt, C.; Trowitzsch, S.; Moeller, A.; Tampé, R. Structure of the human MHC-I peptide-loading complex. Nature 2017, 551, 525–528. [Google Scholar] [CrossRef] [PubMed]
- Purcell, A.W.; Elliott, T. Molecular machinations of the MHC-I peptide loading complex. Curr. Opin. Immunol. 2008, 20, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, M.; Del Cid, N.; Rizvi, S.M.; Peters, L.R. MHC class I assembly: Out and about. Trends Immunol. 2008, 29, 436–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulpke, S.; Tampé, R. The MHC I loading complex: A multitasking machinery in adaptive immunity. Trends Biochem. Sci. 2013, 38, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Ortmann, B. A Critical role for Tapasin in the assembly and function of multimeric MHC Class I-TAP complexes. Science 1997, 277, 1306–1309. [Google Scholar] [CrossRef] [Green Version]
- Hearn, A.; York, I.A.; Rock, K.L. The specificity of trimming of MHC class I-presented peptides in the endoplasmic reticulum. J. Immunol. 2009, 183, 5526–5536. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, A.L.; Cascio, P.; Saric, T.; Rock, K.L. The importance of the proteasome and subsequent proteolytic steps in the generation of antigenic peptides. Mol. Immunol. 2002, 39, 147–164. [Google Scholar] [CrossRef]
- Rock, K.L.; York, I.A.; Saric, T.; Goldberg, A.L. Protein degradation and the generation of MHC class I-presented peptides. Adv. Immunol. 2002, 80, 1–70. [Google Scholar] [PubMed]
- Momburg, F.; Roelse, J.; Hammerling, G.J.; Neefjes, J.J. Peptide size selection by the major histocompatibility complex-encoded peptide transporter. J. Exp. Med. 1994, 179, 1613–1623. [Google Scholar] [CrossRef] [PubMed]
- Lauvau, G.; Kakimi, K.; Niedermann, G.; Ostankovitch, M.; Yotnda, P.; Firat, H.; Chisari, F.V.; van Endert, P.M. Human transporters associated with antigen processing (TAPs) select epitope precursor peptides for processing in the endoplasmic reticulum and presentation to T cells. J. Exp. Med. 1999, 190, 1227–1240. [Google Scholar] [CrossRef] [Green Version]
- Knuehl, C.; Spee, P.; Ruppert, T.; Kuckelkorn, U.; Henklein, P.; Neefjes, J.; Kloetzel, P.M. The murine cytomegalovirus pp89 immunodominant H-2Ld epitope is generated and translocated into the endoplasmic reticulum as an 11-mer precursor peptide. J. Immunol. 2001, 167, 1515–1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neisig, A.; Roelse, J.; Sijts, A.J.; Ossendorp, F.; Feltkamp, M.C.; Kast, W.M.; Melief, C.J.; Neefjes, J.J. Major differences in transporter associated with antigen presentation (TAP)-dependent translocation of MHC class I-presentable peptides and the effect of flanking sequences. J. Immunol. 1995, 154, 1273–1279. [Google Scholar] [PubMed]
- Shastri, N.; Cardinaud, S.; Schwab, S.R.; Serwold, T.; Kunisawa, J. All the peptides that fit: The beginning, the middle, and the end of the MHC class I antigen-processing pathway. Immunol. Rev. 2005, 207, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Rock, K.L.; York, I.A.; Goldberg, A.L. Post-proteasomal antigen processing for major histocompatibility complex class I presentation. Nat. Immunol. 2004, 5, 670–677. [Google Scholar] [CrossRef] [PubMed]
- Apcher, S.; Prado, M.R.; Fahraeus, R. The source of MHC class I presented peptides and its implications. Curr. Opin. Immunol. 2016, 40, 117–122. [Google Scholar] [CrossRef]
- Blum, J.S.; Wearsch, P.A. Cresswell, P. Pathways of antigen processing. Annu. Rev. Immunol. 2013, 31, 443–473. [Google Scholar] [CrossRef] [Green Version]
- Ortmann, B.; Androlewicz, M.J.; Cresswell, P. MHC class l/β2-microglobulin complexes associate with TAP transporters before peptide binding. Nature 1994, 368, 864–867. [Google Scholar] [CrossRef]
- Sadasivan, B.; Lehner, P.J.; Ortmann, B.; Spies, T.; Cresswell, P. Roles for calreticulin and a novel glycoprotein, Tapasin, in the interaction of MHC Class I molecules with TAP. Immunity 1996, 5, 103–114. [Google Scholar] [CrossRef] [Green Version]
- Dong, G.; Wearsch, P.A.; Peaper, D.R.; Cresswell, P.; Reinisch, K.M. Insights into MHC class I peptide loading from the structure of the tapasin-ERp57 thiol oxidoreductase heterodimer. Immunity 2009, 30, 21–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garbi, N.; Hämmerling, G.; Tanaka, S. Interaction of ERp57 and tapasin in the generation of MHC class I–peptide complexes. Curr. Opin. Immunol. 2007, 19, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Peaper, D.R.; Wearsch, P.A.; Cresswell, P. Tapasin and ERp57 form a stable disulfide-linked dimer within the MHC class I peptide-loading complex. EMBO J. 2005, 24, 3613–3623. [Google Scholar] [CrossRef] [Green Version]
- Howarth, M.; Williams, A.; Tolstrup, A.B.; Elliott, T. Tapasin enhances MHC class I peptide presentation according to peptide half-life. Proc. Natl. Acad. Sci. USA 2004, 101, 11737–11742. [Google Scholar] [CrossRef] [Green Version]
- Garbi, N.; Tanaka, S.; Momburg, F.; Hämmerling, G.J. Impaired assembly of the major histocompatibility complex class I peptide-loading complex in mice deficient in the oxidoreductase ERp57. Nat. Immunol. 2006, 7, 93–102. [Google Scholar] [CrossRef]
- Kienast, A.; Preuss, M.; Winkler, M.; Dick, T.P. Redox regulation of peptide receptivity of major histocompatibility complex class I molecules by ERp57 and tapasin. Nat. Immunol. 2007, 8, 864–872. [Google Scholar] [CrossRef]
- Wearsch, P.A.; Cresswell, P. Selective loading of high-affinity peptides onto major histocompatibility complex class I molecules by the tapasin-ERp57 heterodimer. Nat. Immunol. 2007, 8, 873–881. [Google Scholar] [CrossRef]
- Dick, T.P.; Bangia, N.; Peaper, D.R.; Cresswell, P. Disulfide Bond Isomerization and the Assembly of MHC Class I-Peptide Complexes. Immunity 2002, 16, 87–98. [Google Scholar] [CrossRef] [Green Version]
- Turnquist, H.R.; Petersen, J.L.; Vargas, S.E.; Mcilhaney, M.M.; Bedows, E.; Mayer, W.E.; Grandea, A.G.; Van Kaer, L.; Solheim, J.C. The Ig-Like domain of Tapasin influences intermolecular interactions. J. Immunol. 2004, 172, 2976–2984. [Google Scholar] [CrossRef] [Green Version]
- Peaper, D.R.; Cresswell, P. The redox activity of ERp57 is not essential for its functions in MHC class I peptide loading. Proc. Natl. Acad. Sci. USA 2008, 105, 10477–10482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helenius, A.; Aebi, M. Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 2004, 73, 1019–1049. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Zheng, L.; Goncearenco, A.; Panchenko, A.R.; Li, M. Computational approaches to prioritize cancer driver missense mutations. Int. J. Mol. Sci. 2018, 19, 2113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Magliery, T.J. Protein stability: Computation, sequence statistics, and new experimental methods. Curr. Opin. Struct. Biol. 2015, 33, 161–168. [Google Scholar] [CrossRef] [Green Version]
- Ellgaard, L.; Ruddock, L.W. The human protein disulphide isomerase family: Substrate interactions and functional properties. EMBO Rep. 2005, 6, 28–32. [Google Scholar] [CrossRef]
- Tian, G.; Xiang, S.; Noiva, R.; Lennarz, W.J.; Schindelin, H. The crystal structure of yeast protein disulfide isomerase suggests cooperativity between its active sites. Cell 2006, 124, 61–73. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Li, W.; Ren, J.; Fang, J.; Ke, H.; Gong, W.; Feng, W.; Wang, C.C. Structural insights into the redox-regulated dynamic conformations of human protein disulfide isomerase. Antioxid. Redox Signal. 2013, 19, 36–45. [Google Scholar] [CrossRef] [Green Version]
- Chiu, J.; Hogg, P.J. Allosteric disulfides: Sophisticated molecular structures enabling flexible protein regulation. J. Biol. Chem. 2019, 294, 2949–2960. [Google Scholar] [CrossRef] [Green Version]
- Maattanen, P.; Kozlov, G.; Gehring, K.; Thomas, D.Y. ERp57 and PDI: Multifunctional protein disulfide isomerases with similar domain architectures but differing substrate-partner associations. Biochem. Cell Biol. 2006, 84, 881–889. [Google Scholar] [CrossRef]
- Karamzadeh, R.; Karimi-Jafari, M.H.; Sharifi-Zarchi, A.; Chitsaz, H.; Salekdeh, G.H.; Moosavi-Movahedi, A.A. Machine learning and network analysis of molecular dynamics trajectories reveal two chains of Red/Ox-specific residue interactions in human protein disulfide isomerase. Sci. Rep. 2017, 7, e3666. [Google Scholar] [CrossRef] [Green Version]
- Serve, O.; Kamiya, Y.; Maeno, A.; Nakano, M.; Murakami, C.; Sasakawa, H.; Yamaguchi, Y.; Harada, T.; Kurimoto, E.; Yagi-Utsumi, M.; et al. Redox-dependent domain rearrangement of protein disulfide isomerase coupled with exposure of its substrate-binding hydrophobic surface. J. Mol. Biol. 2010, 396, 361–374. [Google Scholar] [CrossRef]
- Yang, S.; Wang, X.; Cui, L.; Ding, X.; Niu, L.; Yang, F.; Wang, C.; Wang, C.C.; Lou, J. Compact conformations of human protein disulfide isomerase. PLoS ONE 2014, 9, e103472. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wang, X.; Wang, C.C. Protein disulfide-isomerase, a folding catalyst and a redox-regulated chaperone. Free Radic. Biol. Med. 2015, 83, 305–313. [Google Scholar] [CrossRef]
- Kozlov, G.; Maattanen, P.; Schrag, J.D.; Pollock, S.; Cygler, M.; Nagar, B.; Thomas, D.Y.; Gehring, K. Crystal structure of the bb’ domains of the protein disulfide isomerase ERp57. Structure 2006, 14, 1331–1339. [Google Scholar] [CrossRef]
- Dror, R.O.; Jensen, M.Ø.; Borhani, D.W.; Shaw, D.E. Exploring atomic resolution physiology on a femtosecond to millisecond timescale using molecular dynamics simulations. J. Gen. Physiol. 2010, 135, 555–562. [Google Scholar] [CrossRef]
- Fisette, O.; Wingbermühle, S.; Tampé, R.; Schäfer, L.V. Molecular mechanism of peptide editing in the tapasin-MHC I complex. Sci. Rep. 2016, 6, e19085. [Google Scholar] [CrossRef] [Green Version]
- Rose, P.W.; Beran, B.; Bi, C.; Bluhm, W.F.; Dimitropoulos, D.; Goodsell, D.S.; Prlić, A.; Quesada, M.; Quinn, G.B.; Westbrook, J.D. The RCSB Protein Data Bank: Redesigned website and web services. Nucleic Acids Res. 2010, 39, D392–D401. [Google Scholar] [CrossRef] [Green Version]
- Eriksen, D.T.; Lian, J.Z.; Zhao, H.M. Protein design for pathway engineering. J. Struct. Biol. 2014, 185, 234–242. [Google Scholar] [CrossRef] [Green Version]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; Van Der Spoel, D.; et al. GROMACS 4.5: A High-Throughput and Highly Parallel Open Source Molecular Simulation Toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A Message-Passing Parallel Molecular Dynamics Implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Schuler, L.D.; Daura, X.; Gunsteren, W.F. An improved GROMOS96 force field for aliphatic hydrocarbons in the condensed phase. J. Comput. Chem. 2001, 22, 1205–1218. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, e014101. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Gunsteren, W.F.; Berendsen, H.J. A Leap-frog algorithm for stochastic dynamics. Mol. Simul. 1988, 1, 173–185. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Padariya, M.; Kalathiya, U.; Houston, D.R.; Alfaro, J.A. Recognition Dynamics of Cancer Mutations on the ERp57-Tapasin Interface. Cancers 2020, 12, 737. https://doi.org/10.3390/cancers12030737
Padariya M, Kalathiya U, Houston DR, Alfaro JA. Recognition Dynamics of Cancer Mutations on the ERp57-Tapasin Interface. Cancers. 2020; 12(3):737. https://doi.org/10.3390/cancers12030737
Chicago/Turabian StylePadariya, Monikaben, Umesh Kalathiya, Douglas R. Houston, and Javier Antonio Alfaro. 2020. "Recognition Dynamics of Cancer Mutations on the ERp57-Tapasin Interface" Cancers 12, no. 3: 737. https://doi.org/10.3390/cancers12030737