Targeted Therapies: Friends or Foes for Patient’s NK Cell-Mediated Tumor Immune-Surveillance?

 , , and
, , and {kind=link}
{kind=link}
Abstract
:1. Introduction
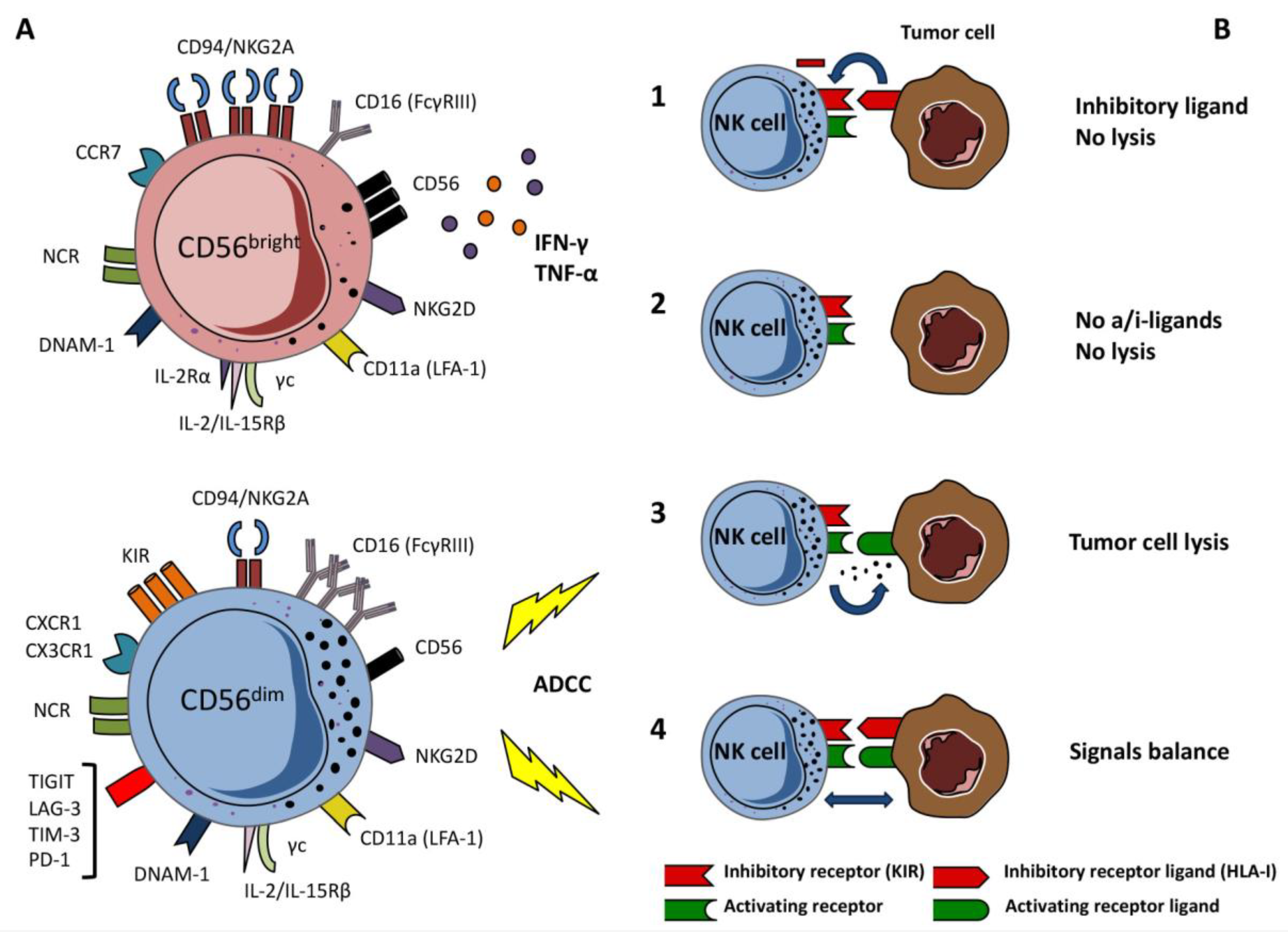
2. Natural Killer Cells
3. NK Cells and Hematological Malignancies
4. NK Cells and Solid Tumors
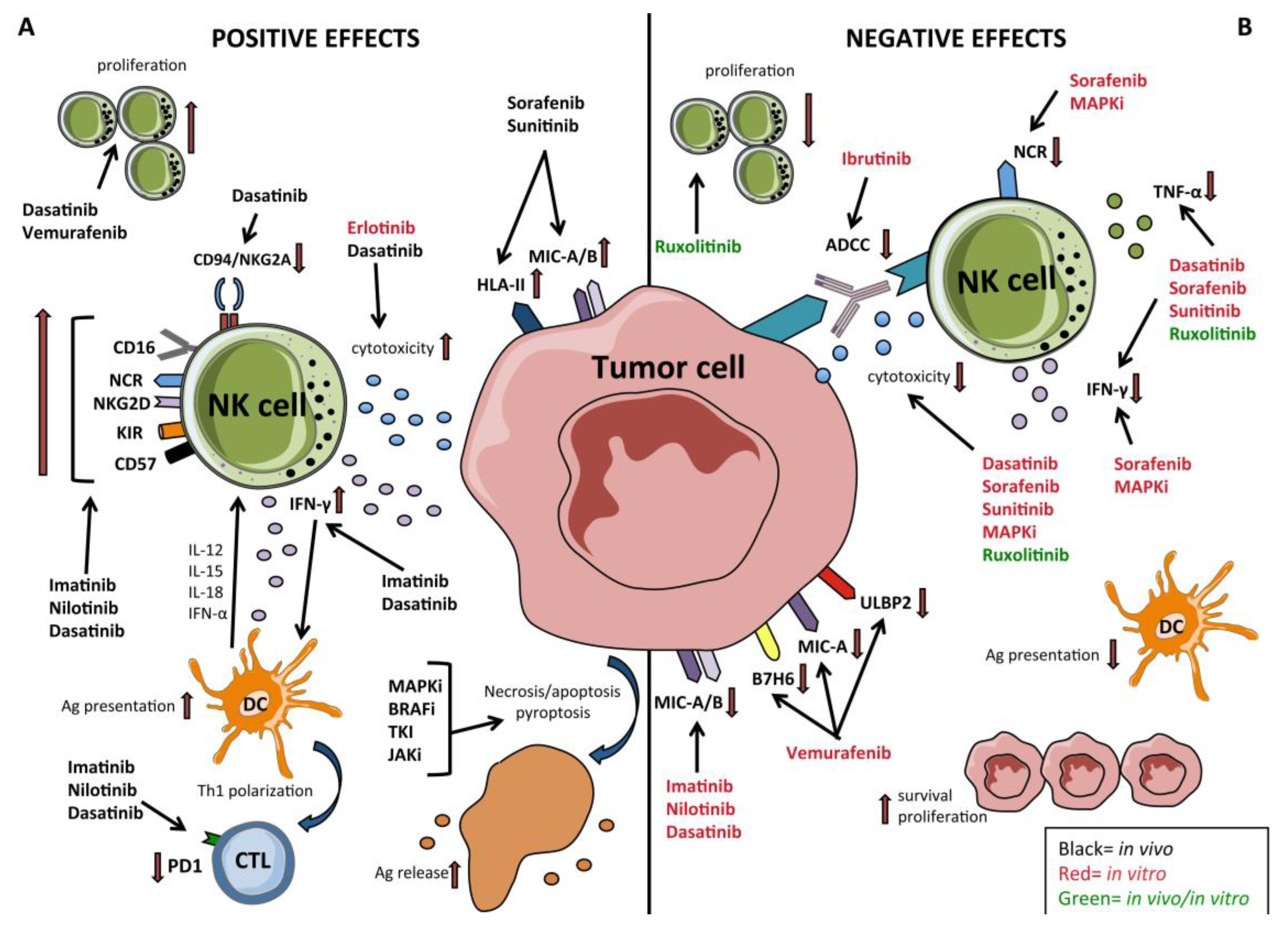
5. TKI—Targeted Therapy Effects on NK Cells in Hematological Malignancies and GIST
6. Targeted Therapies and Their Immunomodulatory Effects on NK Cells in Solid Tumors
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Haber, D.A.; Gray, N.S.; Baselga, J. The evolving war on cancer. Cell 2011, 145, 19–24. [Google Scholar] [CrossRef] [Green Version]
- Jabbour, E.; Cortes, J.; Ravandi, F.; O’Brien, S.; Kantarjian, H. Targeted therapies in hematology and their impact on patient care: Chronic and acute myeloid leukemia. Semin. Hematol. 2013, 50, 271–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.T.; Tan, Y.J.; Oon, C.E. Molecular targeted therapy: Treating cancer with specificity. Eur. J. Pharmacol. 2018, 834, 188–196. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, S.G.; Guilhot, F.; Larson, R.A.; Gathmann, I.; Baccarani, M.; Cervantes, F.; Cornelissen, J.J.; Fischer, T.; Hochhaus, A.; Hughes, T.; et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N. Engl. J. Med. 2003, 348, 994–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsimberidou, A.M. Targeted therapy in cancer. Cancer Chemother. Pharmacol. 2015, 76, 1113–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hehlmann, R. Innovation in hematology. Perspectives: CML 2016. Haematologica 2016, 101, 657–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Q.; Wang, H.; Yu, L.; Gale, R.P. Higher out-of-pocket expenses for tyrosine kinase-inhibitor therapy is associated with worse health-related quality-of-life in persons with chronic myeloid leukemia. J. Cancer Res. Clin. Oncol. 2017, 143, 2619–2630. [Google Scholar] [CrossRef]
- Vanneman, M.; Dranoff, G. Combining immunotherapy and targeted therapies in cancer treatment. Nat. Rev. Cancer 2012, 12, 237–251. [Google Scholar] [CrossRef] [Green Version]
- Kreutzman, A.; Porkka, K.; Mustjoki, S. Immunodulatory effects of Tyrosine Kinase Inhibitors. In International Trends in Immunity; Research Publisher Inc.: Santa Clara, CA, USA, 2013; Volume 1, pp. 22–32. [Google Scholar]
- Moretta, A.; Bottino, C.; Mingari, M.C.; Biassoni, R.; Moretta, L. What is a natural killer cell? Nat. Immunol. 2002, 3, 6–8. [Google Scholar] [CrossRef]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef]
- Pietra, G.; Vitale, C.; Pende, D.; Bertaina, A.; Moretta, F.; Falco, M.; Vacca, P.; Montaldo, E.; Cantoni, C.; Mingari, M.C.; et al. Human natural killer cells: News in the therapy of solid tumors and high-risk leukemias. Cancer Immunol. Immunother. 2016, 65, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Cantoni, C.; Huergo-Zapico, L.; Parodi, M.; Pedrazzi, M.; Mingari, M.C.; Moretta, A.; Sparatore, B.; Gonzalez, S.; Olive, D.; Bottino, C.; et al. NK Cells, Tumor Cell Transition, and Tumor Progression in Solid Malignancies: New Hints for NK-Based Immunotherapy? J. Immunol. Res. 2016, 2016, 4684268. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Yang, Y.; Liu, L.L.; Lundqvist, A. Strategies to Augment Natural Killer (NK) Cell Activity against Solid Tumors. Cancers 2019, 11, 1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vivier, E.; Raulet, D.H.; Moretta, A.; Caligiuri, M.A.; Zitvogel, L.; Lanier, L.L.; Yokoyama, W.M.; Ugolini, S. Innate or adaptive immunity? The example of natural killer cells. Science 2011, 331, 44–49. [Google Scholar] [CrossRef] [Green Version]
- Chiossone, L.; Dumas, P.Y.; Vienne, M.; Vivier, E. Natural killer cells and other innate lymphoid cells in cancer. Nat. Rev. Immunol. 2018, 18, 671–688. [Google Scholar] [CrossRef]
- Voskoboinik, I.; Whisstock, J.C.; Trapani, J.A. Perforin and granzymes: Function, dysfunction and human pathology. Nat. Rev. Immunol. 2015, 15, 388–400. [Google Scholar] [CrossRef]
- Fauriat, C.; Long, E.O.; Ljunggren, H.G.; Bryceson, Y.T. Regulation of human NK-cell cytokine and chemokine production by target cell recognition. Blood 2010, 115, 2167–2176. [Google Scholar] [CrossRef] [Green Version]
- Caligiuri, M.A. Human natural killer cells. Blood 2008, 112, 461–469. [Google Scholar] [CrossRef]
- Carrega, P.; Ferlazzo, G. Natural killer cell distribution and trafficking in human tissues. Front. Immunol. 2012, 3, 347. [Google Scholar] [CrossRef] [Green Version]
- Moretta, A. Natural killer cells and dendritic cells: Rendezvous in abused tissues. Nat. Rev. Immunol. 2002, 2, 957–964. [Google Scholar] [CrossRef]
- Riise, R.E.; Bernson, E.; Aurelius, J.; Martner, A.; Pesce, S.; Della Chiesa, M.; Marcenaro, E.; Bylund, J.; Hellstrand, K.; Moretta, L.; et al. TLR-Stimulated Neutrophils Instruct NK Cells To Trigger Dendritic Cell Maturation and Promote Adaptive T Cell Responses. J. Immunol. 2015, 195, 1121–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; Sun, C.; Tian, Z.; Xiao, W. NK cells in immunotolerant organs. Cell Mol. Immunol. 2013, 10, 202–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moretta, L.; Bottino, C.; Pende, D.; Vitale, M.; Mingari, M.C.; Moretta, A. Different checkpoints in human NK-cell activation. Trends Immunol. 2004, 25, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Botet, M.; Perez-Villar, J.J.; Carretero, M.; Rodriguez, A.; Melero, I.; Bellon, T.; Llano, M.; Navarro, F. Structure and function of the CD94 C-type lectin receptor complex involved in recognition of HLA class I molecules. Immunol. Rev. 1997, 155, 165–174. [Google Scholar] [CrossRef]
- Braud, V.M.; Allan, D.S.; O’Callaghan, C.A.; Soderstrom, K.; D’Andrea, A.; Ogg, G.S.; Lazetic, S.; Young, N.T.; Bell, J.I.; Phillips, J.H.; et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature 1998, 391, 795–799. [Google Scholar] [CrossRef]
- Long, E.O. Regulation of immune responses through inhibitory receptors. Annu. Rev. Immunol. 1999, 17, 875–904. [Google Scholar] [CrossRef]
- Anfossi, N.; Andre, P.; Guia, S.; Falk, C.S.; Roetynck, S.; Stewart, C.A.; Breso, V.; Frassati, C.; Reviron, D.; Middleton, D.; et al. Human NK cell education by inhibitory receptors for MHC class I. Immunity 2006, 25, 331–342. [Google Scholar] [CrossRef]
- Moretta, A.; Bottino, C.; Vitale, M.; Pende, D.; Cantoni, C.; Mingari, M.C.; Biassoni, R.; Moretta, L. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu. Rev. Immunol. 2001, 19, 197–223. [Google Scholar] [CrossRef]
- Bottino, C.; Castriconi, R.; Moretta, L.; Moretta, A. Cellular ligands of activating NK receptors. Trends Immunol. 2005, 26, 221–226. [Google Scholar] [CrossRef]
- Kruse, P.H.; Matta, J.; Ugolini, S.; Vivier, E. Natural cytotoxicity receptors and their ligands. Immunol. Cell Biol. 2014, 92, 221–229. [Google Scholar] [CrossRef]
- Parodi, M.; Favoreel, H.; Candiano, G.; Gaggero, S.; Sivori, S.; Mingari, M.C.; Moretta, L.; Vitale, M.; Cantoni, C. NKp44-NKp44 Ligand Interactions in the Regulation of Natural Killer Cells and Other Innate Lymphoid Cells in Humans. Front. Immunol. 2019, 10, 719. [Google Scholar] [CrossRef]
- Raulet, D.H. Roles of the NKG2D immunoreceptor and its ligands. Nat. Rev. Immunol. 2003, 3, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Meinke, S.; Watzl, C. NK cell cytotoxicity mediated by 2B4 and NTB-A is dependent on SAP acting downstream of receptor phosphorylation. Front. Immunol. 2013, 4, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welte, S.; Kuttruff, S.; Waldhauer, I.; Steinle, A. Mutual activation of natural killer cells and monocytes mediated by NKp80-AICL interaction. Nat. Immunol. 2006, 7, 1334–1342. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Kim, H.S. Targeting Checkpoint Receptors and Molecules for Therapeutic Modulation of Natural Killer Cells. Front. Immunol. 2018, 9, 2041. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Correa, B.; Valhondo, I.; Hassouneh, F.; Lopez-Sejas, N.; Pera, A.; Bergua, J.M.; Arcos, M.J.; Banas, H.; Casas-Aviles, I.; Duran, E.; et al. DNAM-1 and the TIGIT/PVRIG/TACTILE Axis: Novel Immune Checkpoints for Natural Killer Cell-Based Cancer Immunotherapy. Cancers 2019, 11, 877. [Google Scholar] [CrossRef] [Green Version]
- Ndhlovu, L.C.; Lopez-Verges, S.; Barbour, J.D.; Jones, R.B.; Jha, A.R.; Long, B.R.; Schoeffler, E.C.; Fujita, T.; Nixon, D.F.; Lanier, L.L. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood 2012, 119, 3734–3743. [Google Scholar] [CrossRef] [Green Version]
- Bryceson, Y.T.; Ljunggren, H.G.; Long, E.O. Minimal requirement for induction of natural cytotoxicity and intersection of activation signals by inhibitory receptors. Blood 2009, 114, 2657–2666. [Google Scholar] [CrossRef] [Green Version]
- Bryceson, Y.T.; March, M.E.; Barber, D.F.; Ljunggren, H.G.; Long, E.O. Cytolytic granule polarization and degranulation controlled by different receptors in resting NK cells. J. Exp. Med. 2005, 202, 1001–1012. [Google Scholar] [CrossRef]
- Moretta, A.; Sivori, S.; Vitale, M.; Pende, D.; Morelli, L.; Augugliaro, R.; Bottino, C.; Moretta, L. Existence of both inhibitory (p58) and activatory (p50) receptors for HLA-C molecules in human natural killer cells. J. Exp. Med. 1995, 182, 875–884. [Google Scholar] [CrossRef]
- Chewning, J.H.; Gudme, C.N.; Hsu, K.C.; Selvakumar, A.; Dupont, B. KIR2DS1-positive NK cells mediate alloresponse against the C2 HLA-KIR ligand group in vitro. J. Immunol. 2007, 179, 854–868. [Google Scholar] [CrossRef] [PubMed]
- Beziat, V.; Liu, L.L.; Malmberg, J.A.; Ivarsson, M.A.; Sohlberg, E.; Bjorklund, A.T.; Retiere, C.; Sverremark-Ekstrom, E.; Traherne, J.; Ljungman, P.; et al. NK cell responses to cytomegalovirus infection lead to stable imprints in the human KIR repertoire and involve activating KIRs. Blood 2013, 121, 2678–2688. [Google Scholar] [CrossRef] [PubMed]
- Guma, M.; Budt, M.; Saez, A.; Brckalo, T.; Hengel, H.; Angulo, A.; Lopez-Botet, M. Expansion of CD94/NKG2C+ NK cells in response to human cytomegalovirus-infected fibroblasts. Blood 2006, 107, 3624–3631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foley, B.; Cooley, S.; Verneris, M.R.; Pitt, M.; Curtsinger, J.; Luo, X.; Lopez-Verges, S.; Lanier, L.L.; Weisdorf, D.; Miller, J.S. Cytomegalovirus reactivation after allogeneic transplantation promotes a lasting increase in educated NKG2C+ natural killer cells with potent function. Blood 2012, 119, 2665–2674. [Google Scholar] [CrossRef]
- Graef, T.; Moesta, A.K.; Norman, P.J.; Abi-Rached, L.; Vago, L.; Older Aguilar, A.M.; Gleimer, M.; Hammond, J.A.; Guethlein, L.A.; Bushnell, D.A.; et al. KIR2DS4 is a product of gene conversion with KIR3DL2 that introduced specificity for HLA-A*11 while diminishing avidity for HLA-C. J. Exp. Med. 2009, 206, 2557–2572. [Google Scholar] [CrossRef] [Green Version]
- Carrega, P.; Bonaccorsi, I.; Di Carlo, E.; Morandi, B.; Paul, P.; Rizzello, V.; Cipollone, G.; Navarra, G.; Mingari, M.C.; Moretta, L.; et al. CD56(bright)perforin(low) noncytotoxic human NK cells are abundant in both healthy and neoplastic solid tissues and recirculate to secondary lymphoid organs via afferent lymph. J. Immunol. 2014, 192, 3805–3815. [Google Scholar] [CrossRef] [Green Version]
- Bjorkstrom, N.K.; Riese, P.; Heuts, F.; Andersson, S.; Fauriat, C.; Ivarsson, M.A.; Bjorklund, A.T.; Flodstrom-Tullberg, M.; Michaelsson, J.; Rottenberg, M.E.; et al. Expression patterns of NKG2A, KIR, and CD57 define a process of CD56dim NK-cell differentiation uncoupled from NK-cell education. Blood 2010, 116, 3853–3864. [Google Scholar] [CrossRef] [Green Version]
- De Maria, A.; Bozzano, F.; Cantoni, C.; Moretta, L. Revisiting human natural killer cell subset function revealed cytolytic CD56(dim)CD16+ NK cells as rapid producers of abundant IFN-gamma on activation. Proc. Natl. Acad. Sci. USA 2011, 108, 728–732. [Google Scholar] [CrossRef] [Green Version]
- Trinchieri, G. Biology of natural killer cells. Adv. Immunol. 1989, 47, 187–376. [Google Scholar] [CrossRef]
- Pende, D.; Rivera, P.; Marcenaro, S.; Chang, C.C.; Biassoni, R.; Conte, R.; Kubin, M.; Cosman, D.; Ferrone, S.; Moretta, L.; et al. Major histocompatibility complex class I-related chain A and UL16-binding protein expression on tumor cell lines of different histotypes: Analysis of tumor susceptibility to NKG2D-dependent natural killer cell cytotoxicity. Cancer Res. 2002, 62, 6178–6186. [Google Scholar]
- Pende, D.; Spaggiari, G.M.; Marcenaro, S.; Martini, S.; Rivera, P.; Capobianco, A.; Falco, M.; Lanino, E.; Pierri, I.; Zambello, R.; et al. Analysis of the receptor-ligand interactions in the natural killer-mediated lysis of freshly isolated myeloid or lymphoblastic leukemias: Evidence for the involvement of the Poliovirus receptor (CD155) and Nectin-2 (CD112). Blood 2005, 105, 2066–2073. [Google Scholar] [CrossRef]
- Carbone, E.; Neri, P.; Mesuraca, M.; Fulciniti, M.T.; Otsuki, T.; Pende, D.; Groh, V.; Spies, T.; Pollio, G.; Cosman, D.; et al. HLA class I, NKG2D, and natural cytotoxicity receptors regulate multiple myeloma cell recognition by natural killer cells. Blood 2005, 105, 251–258. [Google Scholar] [CrossRef] [Green Version]
- Cebo, C.; Da Rocha, S.; Wittnebel, S.; Turhan, A.G.; Abdelali, J.; Caillat-Zucman, S.; Bourhis, J.H.; Chouaib, S.; Caignard, A. The decreased susceptibility of Bcr/Abl targets to NK cell-mediated lysis in response to imatinib mesylate involves modulation of NKG2D ligands, GM1 expression, and synapse formation. J. Immunol. 2006, 176, 864–872. [Google Scholar] [CrossRef] [Green Version]
- Schlegel, P.; Ditthard, K.; Lang, P.; Mezger, M.; Michaelis, S.; Handgretinger, R.; Pfeiffer, M. NKG2D Signaling Leads to NK Cell Mediated Lysis of Childhood AML. J. Immunol. Res. 2015, 2015, 473175. [Google Scholar] [CrossRef] [Green Version]
- Carlsten, M.; Baumann, B.C.; Simonsson, M.; Jadersten, M.; Forsblom, A.M.; Hammarstedt, C.; Bryceson, Y.T.; Ljunggren, H.G.; Hellstrom-Lindberg, E.; Malmberg, K.J. Reduced DNAM-1 expression on bone marrow NK cells associated with impaired killing of CD34+ blasts in myelodysplastic syndrome. Leukemia 2010, 24, 1607–1616. [Google Scholar] [CrossRef]
- Godal, R.; Bachanova, V.; Gleason, M.; McCullar, V.; Yun, G.H.; Cooley, S.; Verneris, M.R.; McGlave, P.B.; Miller, J.S. Natural killer cell killing of acute myelogenous leukemia and acute lymphoblastic leukemia blasts by killer cell immunoglobulin-like receptor-negative natural killer cells after NKG2A and LIR-1 blockade. Biol. Blood Marrow Transplant. J. Am. Soc. Blood Marrow Transplant. 2010, 16, 612–621. [Google Scholar] [CrossRef] [Green Version]
- Marcenaro, E.; Carlomagno, S.; Pesce, S.; Della Chiesa, M.; Moretta, A.; Sivori, S. Role of alloreactive KIR2DS1(+) NK cells in haploidentical hematopoietic stem cell transplantation. J. Leukoc. Biol. 2011, 90, 661–667. [Google Scholar] [CrossRef]
- Venstrom, J.M.; Dupont, B.; Hsu, K.C.; Pittari, G.; Gooley, T.A.; Chewning, J.H.; Spellman, S.; Haagenson, M.; Gallagher, M.M.; Malkki, M.; et al. Donor activating KIR2DS1 in leukemia. N. Engl. J. Med. 2014, 371, 2042. [Google Scholar] [CrossRef]
- Verfaillie, C.; Kay, N.; Miller, W.; McGlave, P. Diminished A-LAK cytotoxicity and proliferation accompany disease progression in chronic myelogenous leukemia. Blood 1990, 76, 401–408. [Google Scholar] [CrossRef] [Green Version]
- Pierson, B.A.; Miller, J.S. CD56+bright and CD56+dim natural killer cells in patients with chronic myelogenous leukemia progressively decrease in number, respond less to stimuli that recruit clonogenic natural killer cells, and exhibit decreased proliferation on a per cell basis. Blood 1996, 88, 2279–2287. [Google Scholar] [CrossRef] [Green Version]
- Kiladjian, J.J.; Bourgeois, E.; Lobe, I.; Braun, T.; Visentin, G.; Bourhis, J.H.; Fenaux, P.; Chouaib, S.; Caignard, A. Cytolytic function and survival of natural killer cells are severely altered in myelodysplastic syndromes. Leukemia 2006, 20, 463–470. [Google Scholar] [CrossRef] [Green Version]
- Costello, R.T.; Sivori, S.; Marcenaro, E.; Lafage-Pochitaloff, M.; Mozziconacci, M.J.; Reviron, D.; Gastaut, J.A.; Pende, D.; Olive, D.; Moretta, A. Defective expression and function of natural killer cell-triggering receptors in patients with acute myeloid leukemia. Blood 2002, 99, 3661–3667. [Google Scholar] [CrossRef]
- Fauriat, C.; Moretta, A.; Olive, D.; Costello, R.T. Defective killing of dendritic cells by autologous natural killer cells from acute myeloid leukemia patients. Blood 2005, 106, 2186–2188. [Google Scholar] [CrossRef]
- Khaznadar, Z.; Boissel, N.; Agaugue, S.; Henry, G.; Cheok, M.; Vignon, M.; Geromin, D.; Cayuela, J.M.; Castaigne, S.; Pautas, C.; et al. Defective NK Cells in Acute Myeloid Leukemia Patients at Diagnosis Are Associated with Blast Transcriptional Signatures of Immune Evasion. J. Immunol. 2015, 195, 2580–2590. [Google Scholar] [CrossRef] [Green Version]
- Stringaris, K.; Sekine, T.; Khoder, A.; Alsuliman, A.; Razzaghi, B.; Sargeant, R.; Pavlu, J.; Brisley, G.; de Lavallade, H.; Sarvaria, A.; et al. Leukemia-induced phenotypic and functional defects in natural killer cells predict failure to achieve remission in acute myeloid leukemia. Haematologica 2014, 99, 836–847. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Correa, B.; Gayoso, I.; Bergua, J.M.; Casado, J.G.; Morgado, S.; Solana, R.; Tarazona, R. Decreased expression of DNAM-1 on NK cells from acute myeloid leukemia patients. Immunol. Cell Biol. 2012, 90, 109–115. [Google Scholar] [CrossRef]
- Khaznadar, Z.; Henry, G.; Setterblad, N.; Agaugue, S.; Raffoux, E.; Boissel, N.; Dombret, H.; Toubert, A.; Dulphy, N. Acute myeloid leukemia impairs natural killer cells through the formation of a deficient cytotoxic immunological synapse. Eur. J. Immunol. 2014, 44, 3068–3080. [Google Scholar] [CrossRef]
- Chan, C.J.; Martinet, L.; Gilfillan, S.; Souza-Fonseca-Guimaraes, F.; Chow, M.T.; Town, L.; Ritchie, D.S.; Colonna, M.; Andrews, D.M.; Smyth, M.J. The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat. Immunol. 2014, 15, 431–438. [Google Scholar] [CrossRef]
- Coles, S.J.; Wang, E.C.; Man, S.; Hills, R.K.; Burnett, A.K.; Tonks, A.; Darley, R.L. CD200 expression suppresses natural killer cell function and directly inhibits patient anti-tumor response in acute myeloid leukemia. Leukemia 2011, 25, 792–799. [Google Scholar] [CrossRef] [Green Version]
- Hadadi, L.; Hafezi, M.; Amirzargar, A.A.; Sharifian, R.A.; Abediankenari, S.; Asgarian-Omran, H. Dysregulated Expression of Tim-3 and NKp30 Receptors on NK Cells of Patients with Chronic Lymphocytic Leukemia. Oncol. Res. Treat. 2019, 42, 202–208. [Google Scholar] [CrossRef]
- Rabinovich, G.A.; Gabrilovich, D.; Sotomayor, E.M. Immunosuppressive strategies that are mediated by tumor cells. Annu. Rev. Immunol. 2007, 25, 267–296. [Google Scholar] [CrossRef] [Green Version]
- Romero, A.I.; Thoren, F.B.; Brune, M.; Hellstrand, K. NKp46 and NKG2D receptor expression in NK cells with CD56dim and CD56bright phenotype: Regulation by histamine and reactive oxygen species. Br. J. Haematol. 2006, 132, 91–98. [Google Scholar] [CrossRef]
- Szczepanski, M.J.; Szajnik, M.; Welsh, A.; Foon, K.A.; Whiteside, T.L.; Boyiadzis, M. Interleukin-15 enhances natural killer cell cytotoxicity in patients with acute myeloid leukemia by upregulating the activating NK cell receptors. Cancer Immunol. Immunother. 2010, 59, 73–79. [Google Scholar] [CrossRef]
- Curti, A.; Trabanelli, S.; Salvestrini, V.; Baccarani, M.; Lemoli, R.M. The role of indoleamine 2,3-dioxygenase in the induction of immune tolerance: Focus on hematology. Blood 2009, 113, 2394–2401. [Google Scholar] [CrossRef]
- Della Chiesa, M.; Carlomagno, S.; Frumento, G.; Balsamo, M.; Cantoni, C.; Conte, R.; Moretta, L.; Moretta, A.; Vitale, M. The tryptophan catabolite L-kynurenine inhibits the surface expression of NKp46- and NKG2D-activating receptors and regulates NK-cell function. Blood 2006, 108, 4118–4125. [Google Scholar] [CrossRef]
- Hilpert, J.; Grosse-Hovest, L.; Grunebach, F.; Buechele, C.; Nuebling, T.; Raum, T.; Steinle, A.; Salih, H.R. Comprehensive analysis of NKG2D ligand expression and release in leukemia: Implications for NKG2D-mediated NK cell responses. J. Immunol. 2012, 189, 1360–1371. [Google Scholar] [CrossRef] [Green Version]
- Clayton, A.; Mitchell, J.P.; Court, J.; Linnane, S.; Mason, M.D.; Tabi, Z. Human tumor-derived exosomes down-modulate NKG2D expression. J. Immunol. 2008, 180, 7249–7258. [Google Scholar] [CrossRef] [Green Version]
- Reiners, K.S.; Topolar, D.; Henke, A.; Simhadri, V.R.; Kessler, J.; Sauer, M.; Bessler, M.; Hansen, H.P.; Tawadros, S.; Herling, M.; et al. Soluble ligands for NK cell receptors promote evasion of chronic lymphocytic leukemia cells from NK cell anti-tumor activity. Blood 2013, 121, 3658–3665. [Google Scholar] [CrossRef]
- Chretien, A.S.; Fauriat, C.; Orlanducci, F.; Galseran, C.; Rey, J.; Bouvier Borg, G.; Gautherot, E.; Granjeaud, S.; Hamel-Broza, J.F.; Demerle, C.; et al. Natural Killer Defective Maturation Is Associated with Adverse Clinical Outcome in Patients with Acute Myeloid Leukemia. Front. Immunol. 2017, 8, 573. [Google Scholar] [CrossRef]
- Schepers, K.; Campbell, T.B.; Passegue, E. Normal and leukemic stem cell niches: Insights and therapeutic opportunities. Cell Stem Cell 2015, 16, 254–267. [Google Scholar] [CrossRef] [Green Version]
- Duan, C.W.; Shi, J.; Chen, J.; Wang, B.; Yu, Y.H.; Qin, X.; Zhou, X.C.; Cai, Y.J.; Li, Z.Q.; Zhang, F.; et al. Leukemia propagating cells rebuild an evolving niche in response to therapy. Cancer Cell 2014, 25, 778–793. [Google Scholar] [CrossRef] [Green Version]
- Vasold, J.; Wagner, M.; Drolle, H.; Deniffel, C.; Kutt, A.; Oostendorp, R.; Sironi, S.; Rieger, C.; Fiegl, M. The bone marrow microenvironment is a critical player in the NK cell response against acute myeloid leukaemia in vitro. Leuk. Res. 2015, 39, 257–262. [Google Scholar] [CrossRef]
- Vitale, C.; Ambrosini, P.; Montaldo, E.; Ballerini, F.; Moretta, L.; Mingari, M.C. IL-1beta-releasing human acute myeloid leukemia blasts modulate natural killer cell differentiation from CD34+ precursors. Haematologica 2015, 100, e42–e45. [Google Scholar] [CrossRef] [Green Version]
- Ambrosini, P.; Loiacono, F.; Conte, R.; Moretta, L.; Vitale, C.; Mingari, M.C. IL-1beta inhibits ILC3 while favoring NK-cell maturation of umbilical cord blood CD34(+) precursors. Eur. J. Immunol. 2015, 45, 2061–2071. [Google Scholar] [CrossRef]
- Scoville, S.D.; Nalin, A.P.; Chen, L.; Chen, L.; Zhang, M.H.; McConnell, K.; Beceiro Casas, S.; Ernst, G.; Traboulsi, A.A.; Hashi, N.; et al. Human AML activates the aryl hydrocarbon receptor pathway to impair NK cell development and function. Blood 2018, 132, 1792–1804. [Google Scholar] [CrossRef]
- Bottcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Reis e Sousa, C. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 2018, 172, 1022–1037.e14. [Google Scholar] [CrossRef] [Green Version]
- Barry, K.C.; Hsu, J.; Broz, M.L.; Cueto, F.J.; Binnewies, M.; Combes, A.J.; Nelson, A.E.; Loo, K.; Kumar, R.; Rosenblum, M.D.; et al. A natural killer-dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments. Nat. Med. 2018, 24, 1178–1191. [Google Scholar] [CrossRef]
- Coca, S.; Perez-Piqueras, J.; Martinez, D.; Colmenarejo, A.; Saez, M.A.; Vallejo, C.; Martos, J.A.; Moreno, M. The prognostic significance of intratumoral natural killer cells in patients with colorectal carcinoma. Cancer 1997, 79, 2320–2328. [Google Scholar] [CrossRef]
- Ishigami, S.; Natsugoe, S.; Tokuda, K.; Nakajo, A.; Che, X.; Iwashige, H.; Aridome, K.; Hokita, S.; Aikou, T. Prognostic value of intratumoral natural killer cells in gastric carcinoma. Cancer 2000, 88, 577–583. [Google Scholar] [CrossRef]
- Villegas, F.R.; Coca, S.; Villarrubia, V.G.; Jimenez, R.; Chillon, M.J.; Jareno, J.; Zuil, M.; Callol, L. Prognostic significance of tumor infiltrating natural killer cells subset CD57 in patients with squamous cell lung cancer. Lung Cancer 2002, 35, 23–28. [Google Scholar] [CrossRef]
- Schleypen, J.S.; Von Geldern, M.; Weiss, E.H.; Kotzias, N.; Rohrmann, K.; Schendel, D.J.; Falk, C.S.; Pohla, H. Renal cell carcinoma-infiltrating natural killer cells express differential repertoires of activating and inhibitory receptors and are inhibited by specific HLA class I allotypes. Int. J. Cancer 2003, 106, 905–912. [Google Scholar] [CrossRef]
- Rusakiewicz, S.; Semeraro, M.; Sarabi, M.; Desbois, M.; Locher, C.; Mendez, R.; Vimond, N.; Concha, A.; Garrido, F.; Isambert, N.; et al. Immune infiltrates are prognostic factors in localized gastrointestinal stromal tumors. Cancer Res. 2013, 73, 3499–3510. [Google Scholar] [CrossRef] [Green Version]
- Mamessier, E.; Sylvain, A.; Bertucci, F.; Castellano, R.; Finetti, P.; Houvenaeghel, G.; Charaffe-Jaufret, E.; Birnbaum, D.; Moretta, A.; Olive, D. Human breast tumor cells induce self-tolerance mechanisms to avoid NKG2D-mediated and DNAM-mediated NK cell recognition. Cancer Res. 2011, 71, 6621–6632. [Google Scholar] [CrossRef] [Green Version]
- Platonova, S.; Cherfils-Vicini, J.; Damotte, D.; Crozet, L.; Vieillard, V.; Validire, P.; Andre, P.; Dieu-Nosjean, M.C.; Alifano, M.; Regnard, J.F.; et al. Profound coordinated alterations of intratumoral NK cell phenotype and function in lung carcinoma. Cancer Res. 2011, 71, 5412–5422. [Google Scholar] [CrossRef] [Green Version]
- Delahaye, N.F.; Rusakiewicz, S.; Martins, I.; Menard, C.; Roux, S.; Lyonnet, L.; Paul, P.; Sarabi, M.; Chaput, N.; Semeraro, M.; et al. Alternatively spliced NKp30 isoforms affect the prognosis of gastrointestinal stromal tumors. Nat. Med. 2011, 17, 700–707. [Google Scholar] [CrossRef]
- Zhang, Q.; Bi, J.; Zheng, X.; Chen, Y.; Wang, H.; Wu, W.; Wang, Z.; Wu, Q.; Peng, H.; Wei, H.; et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 2018, 19, 723–732. [Google Scholar] [CrossRef]
- Zheng, Y.; Li, Y.; Lian, J.; Yang, H.; Li, F.; Zhao, S.; Qi, Y.; Zhang, Y.; Huang, L. TNF-alpha-induced Tim-3 expression marks the dysfunction of infiltrating natural killer cells in human esophageal cancer. J. Transl. Med. 2019, 17, 165. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zhu, J.; Gu, H.; Yuan, Y.; Zhang, B.; Zhu, D.; Zhou, J.; Zhu, Y.; Chen, W. The Clinical Significance of Abnormal Tim-3 Expression on NK Cells from Patients with Gastric Cancer. Immunol. Investig. 2015, 44, 578–589. [Google Scholar] [CrossRef]
- Xu, L.; Huang, Y.; Tan, L.; Yu, W.; Chen, D.; Lu, C.; He, J.; Wu, G.; Liu, X.; Zhang, Y. Increased Tim-3 expression in peripheral NK cells predicts a poorer prognosis and Tim-3 blockade improves NK cell-mediated cytotoxicity in human lung adenocarcinoma. Int. Immunopharmacol. 2015, 29, 635–641. [Google Scholar] [CrossRef]
- Da Silva, I.P.; Gallois, A.; Jimenez-Baranda, S.; Khan, S.; Anderson, A.C.; Kuchroo, V.K.; Osman, I.; Bhardwaj, N. Reversal of NK-cell exhaustion in advanced melanoma by Tim-3 blockade. Cancer Immunol. Res. 2014, 2, 410–422. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Liu, L.; Huang, Q.; Liu, H.; Huang, M.; Wang, J.; Wen, H.; Lin, R.; Qu, K.; Li, K.; et al. Accumulation of Tumor-Infiltrating CD49a(+) NK Cells Correlates with Poor Prognosis for Human Hepatocellular Carcinoma. Cancer Immunol. Res. 2019, 7, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Beldi-Ferchiou, A.; Lambert, M.; Dogniaux, S.; Vely, F.; Vivier, E.; Olive, D.; Dupuy, S.; Levasseur, F.; Zucman, D.; Lebbe, C.; et al. PD-1 mediates functional exhaustion of activated NK cells in patients with Kaposi sarcoma. Oncotarget 2016, 7, 72961–72977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabellini, G.; Benassi, M.; Marcenaro, E.; Coltrini, D.; Patrizi, O.; Ricotta, D.; Rampinelli, F.; Moretta, A.; Parolini, S. Primitive neuroectodermal tumor in an ovarian cystic teratoma: Natural killer and neuroblastoma cell analysis. Case Rep. Oncol. 2014, 7, 70–78. [Google Scholar] [CrossRef]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef] [Green Version]
- De Andrade, L.F.; Lu, Y.; Luoma, A.; Ito, Y.; Pan, D.; Pyrdol, J.W.; Yoon, C.H.; Yuan, G.C.; Wucherpfennig, K.W. Discovery of specialized NK cell populations infiltrating human melanoma metastases. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Ghiringhelli, F.; Menard, C.; Terme, M.; Flament, C.; Taieb, J.; Chaput, N.; Puig, P.E.; Novault, S.; Escudier, B.; Vivier, E.; et al. CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor-beta-dependent manner. J. Exp. Med. 2005, 202, 1075–1085. [Google Scholar] [CrossRef]
- Li, T.; Yang, Y.; Hua, X.; Wang, G.; Liu, W.; Jia, C.; Tai, Y.; Zhang, Q.; Chen, G. Hepatocellular carcinoma-associated fibroblasts trigger NK cell dysfunction via PGE2 and IDO. Cancer Lett. 2012, 318, 154–161. [Google Scholar] [CrossRef]
- Li, T.; Yi, S.; Liu, W.; Jia, C.; Wang, G.; Hua, X.; Tai, Y.; Zhang, Q.; Chen, G. Colorectal carcinoma-derived fibroblasts modulate natural killer cell phenotype and antitumor cytotoxicity. Med. Oncol. 2013, 30, 663. [Google Scholar] [CrossRef]
- Balsamo, M.; Scordamaglia, F.; Pietra, G.; Manzini, C.; Cantoni, C.; Boitano, M.; Queirolo, P.; Vermi, W.; Facchetti, F.; Moretta, A.; et al. Melanoma-associated fibroblasts modulate NK cell phenotype and antitumor cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 20847–20852. [Google Scholar] [CrossRef] [Green Version]
- Pietra, G.; Manzini, C.; Rivara, S.; Vitale, M.; Cantoni, C.; Petretto, A.; Balsamo, M.; Conte, R.; Benelli, R.; Minghelli, S.; et al. Melanoma cells inhibit natural killer cell function by modulating the expression of activating receptors and cytolytic activity. Cancer Res. 2012, 72, 1407–1415. [Google Scholar] [CrossRef] [Green Version]
- Hoskin, D.W.; Mader, J.S.; Furlong, S.J.; Conrad, D.M.; Blay, J. Inhibition of T cell and natural killer cell function by adenosine and its contribution to immune evasion by tumor cells (Review). Int. J. Oncol. 2008, 32, 527–535. [Google Scholar] [CrossRef] [Green Version]
- Bottino, C.; Dondero, A.; Bellora, F.; Moretta, L.; Locatelli, F.; Pistoia, V.; Moretta, A.; Castriconi, R. Natural killer cells and neuroblastoma: Tumor recognition, escape mechanisms, and possible novel immunotherapeutic approaches. Front. Immunol. 2014, 5, 56. [Google Scholar] [CrossRef] [Green Version]
- Krockenberger, M.; Dombrowski, Y.; Weidler, C.; Ossadnik, M.; Honig, A.; Hausler, S.; Voigt, H.; Becker, J.C.; Leng, L.; Steinle, A.; et al. Macrophage migration inhibitory factor contributes to the immune escape of ovarian cancer by down-regulating NKG2D. J. Immunol. 2008, 180, 7338–7348. [Google Scholar] [CrossRef]
- Gubbels, J.A.; Felder, M.; Horibata, S.; Belisle, J.A.; Kapur, A.; Holden, H.; Petrie, S.; Migneault, M.; Rancourt, C.; Connor, J.P.; et al. MUC16 provides immune protection by inhibiting synapse formation between NK and ovarian tumor cells. Mol. Cancer 2010, 9, 11. [Google Scholar] [CrossRef] [Green Version]
- Groh, V.; Wu, J.; Yee, C.; Spies, T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature 2002, 419, 734–738. [Google Scholar] [CrossRef]
- Kaiser, B.K.; Yim, D.; Chow, I.T.; Gonzalez, S.; Dai, Z.; Mann, H.H.; Strong, R.K.; Groh, V.; Spies, T. Disulphide-isomerase-enabled shedding of tumour-associated NKG2D ligands. Nature 2007, 447, 482–486. [Google Scholar] [CrossRef]
- Rosental, B.; Brusilovsky, M.; Hadad, U.; Oz, D.; Appel, M.Y.; Afergan, F.; Yossef, R.; Rosenberg, L.A.; Aharoni, A.; Cerwenka, A.; et al. Proliferating cell nuclear antigen is a novel inhibitory ligand for the natural cytotoxicity receptor NKp44. J. Immunol. 2011, 187, 5693–5702. [Google Scholar] [CrossRef]
- Balsamo, M.; Manzini, C.; Pietra, G.; Raggi, F.; Blengio, F.; Mingari, M.C.; Varesio, L.; Moretta, L.; Bosco, M.C.; Vitale, M. Hypoxia downregulates the expression of activating receptors involved in NK-cell-mediated target cell killing without affecting ADCC. Eur. J. Immunol. 2013, 43, 2756–2764. [Google Scholar] [CrossRef]
- Saglio, G.; Jabbour, E. First-line therapy for chronic phase CML: Selecting the optimal BCR-ABL1-targeted TKI. Leuk. Lymphoma 2018, 59, 1523–1538. [Google Scholar] [CrossRef]
- El Fakih, R.; Jabbour, E.; Ravandi, F.; Hassanein, M.; Anjum, F.; Ahmed, S.; Kantarjian, H. Current paradigms in the management of Philadelphia chromosome positive acute lymphoblastic leukemia in adults. Am. J. Hematol. 2018, 93, 286–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiyoi, H. Flt3 Inhibitors: Recent Advances and Problems for Clinical Application. Nagoya J. Med. Sci. 2015, 77, 7–17. [Google Scholar] [PubMed]
- Fernandez, S.; Desplat, V.; Villacreces, A.; Guitart, A.V.; Milpied, N.; Pigneux, A.; Vigon, I.; Pasquet, J.M.; Dumas, P.Y. Targeting Tyrosine Kinases in Acute Myeloid Leukemia: Why, Who and How? Int. J. Mol. Sci. 2019, 20, 3429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A. Myelofibrosis with myeloid metaplasia. N. Engl. J. Med. 2000, 342, 1255–1265. [Google Scholar] [CrossRef] [Green Version]
- Percy, M.J.; McMullin, M.F. The V617F JAK2 mutation and the myeloproliferative disorders. Hematol. Oncol. 2005, 23, 91–93. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Corless, C.L.; Demetri, G.D.; Blanke, C.D.; von Mehren, M.; Joensuu, H.; McGreevey, L.S.; Chen, C.J.; Van den Abbeele, A.D.; Druker, B.J.; et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2003, 21, 4342–4349. [Google Scholar] [CrossRef]
- Hemming, M.L.; Heinrich, M.C.; Bauer, S.; George, S. Translational insights into gastrointestinal stromal tumor and current clinical advances. Ann. Oncol. 2018, 29, 2037–2045. [Google Scholar] [CrossRef]
- Vuky, J.; Isacson, C.; Fotoohi, M.; dela Cruz, J.; Otero, H.; Picozzi, V.; Malpass, T.; Aboulafia, D.; Jacobs, A. Phase II trial of imatinib (Gleevec) in patients with metastatic renal cell carcinoma. Investig. New Drugs 2006, 24, 85–88. [Google Scholar] [CrossRef]
- Bauman, J.E.; Eaton, K.D.; Martins, R.G. Antagonism of platelet-derived growth factor receptor in non small cell lung cancer: Rationale and investigations. Clin. Cancer Res. An Off. J. Am. Assoc. Cancer Res. 2007, 13, s4632–s4636. [Google Scholar] [CrossRef] [Green Version]
- Pane, F.; Intrieri, M.; Quintarelli, C.; Izzo, B.; Muccioli, G.C.; Salvatore, F. BCR/ABL genes and leukemic phenotype: From molecular mechanisms to clinical correlations. Oncogene 2002, 21, 8652–8667. [Google Scholar] [CrossRef]
- Eide, C.A.; O’Hare, T. Chronic myeloid leukemia: Advances in understanding disease biology and mechanisms of resistance to tyrosine kinase inhibitors. Curr. Hematol. Malig. Rep. 2015, 10, 158–166. [Google Scholar] [CrossRef] [Green Version]
- Byrd, J.C.; Brown, J.R.; O’Brien, S.; Barrientos, J.C.; Kay, N.E.; Reddy, N.M.; Coutre, S.; Tam, C.S.; Mulligan, S.P.; Jaeger, U.; et al. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N. Engl. J. Med. 2014, 371, 213–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, A.W.; Davids, M.S.; Pagel, J.M.; Kahl, B.S.; Puvvada, S.D.; Gerecitano, J.F.; Kipps, T.J.; Anderson, M.A.; Brown, J.R.; Gressick, L.; et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2016, 374, 311–322. [Google Scholar] [CrossRef]
- Woyach, J.A.; Furman, R.R.; Liu, T.M.; Ozer, H.G.; Zapatka, M.; Ruppert, A.S.; Xue, L.; Li, D.H.; Steggerda, S.M.; Versele, M.; et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N. Engl. J. Med. 2014, 370, 2286–2294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herling, C.D.; Abedpour, N.; Weiss, J.; Schmitt, A.; Jachimowicz, R.D.; Merkel, O.; Cartolano, M.; Oberbeck, S.; Mayer, P.; Berg, V.; et al. Clonal dynamics towards the development of venetoclax resistance in chronic lymphocytic leukemia. Nat. Commun. 2018, 9, 727. [Google Scholar] [CrossRef] [PubMed]
- Krieg, S.; Ullrich, E. Novel immune modulators used in hematology: Impact on NK cells. Front. Immunol. 2012, 3, 388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlsten, M.; Jaras, M. Natural Killer Cells in Myeloid Malignancies: Immune Surveillance, NK Cell Dysfunction, and Pharmacological Opportunities to Bolster the Endogenous NK Cells. Front. Immunol. 2019, 10, 2357. [Google Scholar] [CrossRef] [Green Version]
- Zitvogel, L.; Rusakiewicz, S.; Routy, B.; Ayyoub, M.; Kroemer, G. Immunological off-target effects of imatinib. Nat. Rev. Clin. Oncol. 2016, 13, 431–446. [Google Scholar] [CrossRef]
- McLornan, D.P.; Khan, A.A.; Harrison, C.N. Immunological Consequences of JAK Inhibition: Friend or Foe? Curr. Hematol. Malig. Rep. 2015, 10, 370–379. [Google Scholar] [CrossRef]
- Schonberg, K.; Rudolph, J.; Vonnahme, M.; Parampalli Yajnanarayana, S.; Cornez, I.; Hejazi, M.; Manser, A.R.; Uhrberg, M.; Verbeek, W.; Koschmieder, S.; et al. JAK Inhibition Impairs NK Cell Function in Myeloproliferative Neoplasms. Cancer Res. 2015, 75, 2187–2199. [Google Scholar] [CrossRef] [Green Version]
- Kohrt, H.E.; Sagiv-Barfi, I.; Rafiq, S.; Herman, S.E.; Butchar, J.P.; Cheney, C.; Zhang, X.; Buggy, J.J.; Muthusamy, N.; Levy, R.; et al. Ibrutinib antagonizes rituximab-dependent NK cell-mediated cytotoxicity. Blood 2014, 123, 1957–1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salih, J.; Hilpert, J.; Placke, T.; Grunebach, F.; Steinle, A.; Salih, H.R.; Krusch, M. The BCR/ABL-inhibitors imatinib, nilotinib and dasatinib differentially affect NK cell reactivity. Int. J. Cancer 2010, 127, 2119–2128. [Google Scholar] [CrossRef] [PubMed]
- Houshmand, M.; Blanco, T.M.; Circosta, P.; Yazdi, N.; Kazemi, A.; Saglio, G.; Zarif, M.N. Bone marrow microenvironment: The guardian of leukemia stem cells. World J. Stem Cells 2019, 11, 476–490. [Google Scholar] [CrossRef] [PubMed]
- Appel, S.; Balabanov, S.; Brummendorf, T.H.; Brossart, P. Effects of imatinib on normal hematopoiesis and immune activation. Stem Cells 2005, 23, 1082–1088. [Google Scholar] [CrossRef] [PubMed]
- Damele, L.; Montaldo, E.; Moretta, L.; Vitale, C.; Mingari, M.C. Effect of Tyrosin Kinase Inhibitors on NK Cell and ILC3 Development and Function. Front. Immunol. 2018, 9, 2433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christiansson, L.; Soderlund, S.; Mangsbo, S.; Hjorth-Hansen, H.; Hoglund, M.; Markevarn, B.; Richter, J.; Stenke, L.; Mustjoki, S.; Loskog, A.; et al. The tyrosine kinase inhibitors imatinib and dasatinib reduce myeloid suppressor cells and release effector lymphocyte responses. Mol. Cancer Ther. 2015, 14, 1181–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, A.; Nishikawa, H.; Noguchi, S.; Sugiyama, D.; Morikawa, H.; Takeuchi, Y.; Ha, D.; Shigeta, N.; Kitawaki, T.; Maeda, Y.; et al. Tyrosine kinase inhibitor imatinib augments tumor immunity by depleting effector regulatory T cells. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Hayashi, Y.; Nakamae, H.; Katayama, T.; Nakane, T.; Koh, H.; Nakamae, M.; Hirose, A.; Hagihara, K.; Terada, Y.; Nakao, Y.; et al. Different immunoprofiles in patients with chronic myeloid leukemia treated with imatinib, nilotinib or dasatinib. Leuk. Lymphoma 2012, 53, 1084–1089. [Google Scholar] [CrossRef]
- Mustjoki, S.; Auvinen, K.; Kreutzman, A.; Rousselot, P.; Hernesniemi, S.; Melo, T.; Lahesmaa-Korpinen, A.M.; Hautaniemi, S.; Bouchet, S.; Molimard, M.; et al. Rapid mobilization of cytotoxic lymphocytes induced by dasatinib therapy. Leukemia 2013, 27, 914–924. [Google Scholar] [CrossRef] [Green Version]
- El Missiry, M.; Adnan Awad, S.; Rajala, H.L.; Al-Samadi, A.; Ekblom, M.; Markevan, B.; Astrand-Grundstrom, I.; Wold, M.; Svedahl, E.R.; Juhl, B.R.; et al. Assessment of bone marrow lymphocytic status during tyrosine kinase inhibitor therapy and its relation to therapy response in chronic myeloid leukaemia. J. Cancer Res. Clin. Oncol. 2016, 142, 1041–1050. [Google Scholar] [CrossRef] [Green Version]
- Binotto, G.; Frison, L.; Boscaro, E.; Zambello, R.; Lessi, F.; Parolo, A.; Piazza, F.; Bertorelle, R.; Bonaldi, L.; Semenzato, G. Comparative Analysis of NK Receptor and T-Cell Receptor Repertoires in Patients with Chronic Myeloid Leukemia Treated with Different Tyrosine Kinase Inhibitors. Blood 2014, 124, 5508. [Google Scholar] [CrossRef]
- Chang, M.C.; Cheng, H.I.; Hsu, K.; Hsu, Y.N.; Kao, C.W.; Chang, Y.F.; Lim, K.H.; Chen, C.G. NKG2A Down-Regulation by Dasatinib Enhances Natural Killer Cytotoxicity and Accelerates Effective Treatment Responses in Patients With Chronic Myeloid Leukemia. Front. Immunol. 2018, 9, 3152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, A.; Clarson, J.; Tang, C.; Vidovic, L.; White, D.L.; Hughes, T.P.; Yong, A.S. CML patients with deep molecular responses to TKI have restored immune effectors and decreased PD-1 and immune suppressors. Blood 2017, 129, 1166–1176. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.; Rea, D.; Lipton, J.H. Treatment-free remission with first- and second-generation tyrosine kinase inhibitors. Am. J. Hematol. 2019, 94, 346–357. [Google Scholar] [CrossRef]
- Shanmuganathan, N.; Hughes, T.P. Molecular monitoring in CML: How deep? How often? How should it influence therapy? Blood 2018, 132, 2125–2133. [Google Scholar] [CrossRef] [Green Version]
- Hughes, A.; Yong, A.S.M. Immune Effector Recovery in Chronic Myeloid Leukemia and Treatment-Free Remission. Front. Immunol. 2017, 8, 469. [Google Scholar] [CrossRef] [Green Version]
- Clapp, G.D.; Lepoutre, T.; Nicolini, F.E.; Levy, D. BCR-ABL transcript variations in chronic phase chronic myelogenous leukemia patients on imatinib first-line: Possible role of the autologous immune system. Oncoimmunology 2016, 5, e1122159. [Google Scholar] [CrossRef] [Green Version]
- Ilander, M.; Olsson-Stromberg, U.; Schlums, H.; Guilhot, J.; Bruck, O.; Lahteenmaki, H.; Kasanen, T.; Koskenvesa, P.; Soderlund, S.; Hoglund, M.; et al. Increased proportion of mature NK cells is associated with successful imatinib discontinuation in chronic myeloid leukemia. Leukemia 2017, 31, 1108–1116. [Google Scholar] [CrossRef]
- Imagawa, J.; Tanaka, H.; Okada, M.; Nakamae, H.; Hino, M.; Murai, K.; Ishida, Y.; Kumagai, T.; Sato, S.; Ohashi, K.; et al. Discontinuation of dasatinib in patients with chronic myeloid leukaemia who have maintained deep molecular response for longer than 1 year (DADI trial): A multicentre phase 2 trial. Lancet Haematol. 2015, 2, e528–e535. [Google Scholar] [CrossRef]
- Rea, D.; Henry, G.; Khaznadar, Z.; Etienne, G.; Guilhot, F.; Nicolini, F.; Guilhot, J.; Rousselot, P.; Huguet, F.; Legros, L.; et al. Natural killer-cell counts are associated with molecular relapse-free survival after imatinib discontinuation in chronic myeloid leukemia: The IMMUNOSTIM study. Haematologica 2017, 102, 1368–1377. [Google Scholar] [CrossRef]
- Kumagai, T.; Nakaseko, C.; Nishiwaki, K.; Yoshida, C.; Ohashi, K.; Takezako, N.; Takano, H.; Kouzai, Y.; Murase, T.; Matsue, K.; et al. Dasatinib cessation after deep molecular response exceeding 2 years and natural killer cell transition during dasatinib consolidation. Cancer Sci. 2018, 109, 182–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ureshino, H.; Shindo, T.; Kojima, H.; Kusunoki, Y.; Miyazaki, Y.; Tanaka, H.; Saji, H.; Kawaguchi, A.; Kimura, S. Allelic Polymorphisms of KIRs and HLAs Predict Favorable Responses to Tyrosine Kinase Inhibitors in CML. Cancer Immunol. Res. 2018, 6, 745–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marin, D.; Gabriel, I.H.; Ahmad, S.; Foroni, L.; de Lavallade, H.; Clark, R.; O’Brien, S.; Sergeant, R.; Hedgley, C.; Milojkovic, D.; et al. KIR2DS1 genotype predicts for complete cytogenetic response and survival in newly diagnosed chronic myeloid leukemia patients treated with imatinib. Leukemia 2012, 26, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Yeung, D.T.; Tang, C.; Vidovic, L.; White, D.L.; Branford, S.; Hughes, T.P.; Yong, A.S. KIR2DL5B genotype predicts outcomes in CML patients treated with response-directed sequential imatinib/nilotinib strategy. Blood 2015, 126, 2720–2723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menard, C.; Blay, J.Y.; Borg, C.; Michiels, S.; Ghiringhelli, F.; Robert, C.; Nonn, C.; Chaput, N.; Taieb, J.; Delahaye, N.F.; et al. Natural killer cell IFN-gamma levels predict long-term survival with imatinib mesylate therapy in gastrointestinal stromal tumor-bearing patients. Cancer Res. 2009, 69, 3563–3569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rusakiewicz, S.; Perier, A.; Semeraro, M.; Pitt, J.M.; Pogge von Strandmann, E.; Reiners, K.S.; Aspeslagh, S.; Piperoglou, C.; Vely, F.; Ivagnes, A.; et al. NKp30 isoforms and NKp30 ligands are predictive biomarkers of response to imatinib mesylate in metastatic GIST patients. Oncoimmunology 2017, 6, e1137418. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Tiacci, E.; Trifonov, V.; Schiavoni, G.; Holmes, A.; Kern, W.; Martelli, M.P.; Pucciarini, A.; Bigerna, B.; Pacini, R.; Wells, V.A.; et al. BRAF mutations in hairy-cell leukemia. N. Engl. J. Med. 2011, 364, 2305–2315. [Google Scholar] [CrossRef] [Green Version]
- Xing, M. BRAF mutation in thyroid cancer. Endocr. Relat. Cancer 2005, 12, 245–262. [Google Scholar] [CrossRef] [Green Version]
- Maurer, G.; Tarkowski, B.; Baccarini, M. Raf kinases in cancer-roles and therapeutic opportunities. Oncogene 2011, 30, 3477–3488. [Google Scholar] [CrossRef] [Green Version]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaherty, K.T.; Smalley, K.S. Preclinical and clinical development of targeted therapy in melanoma: Attention to schedule. Pigment. Cell Melanoma Res. 2009, 22, 529–531. [Google Scholar] [CrossRef] [PubMed]
- Spagnolo, F.; Ghiorzo, P.; Queirolo, P. Overcoming resistance to BRAF inhibition in BRAF-mutated metastatic melanoma. Oncotarget 2014, 5, 10206–10221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med. 2012, 367, 1694–1703. [Google Scholar] [CrossRef] [Green Version]
- Larkin, J.; Ascierto, P.A.; Dreno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandala, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef] [Green Version]
- Sumimoto, H.; Imabayashi, F.; Iwata, T.; Kawakami, Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J. Exp. Med. 2006, 203, 1651–1656. [Google Scholar] [CrossRef] [Green Version]
- Knight, D.A.; Ngiow, S.F.; Li, M.; Parmenter, T.; Mok, S.; Cass, A.; Haynes, N.M.; Kinross, K.; Yagita, H.; Koya, R.C.; et al. Host immunity contributes to the anti-melanoma activity of BRAF inhibitors. J. Clin. Investig. 2013, 123, 1371–1381. [Google Scholar] [CrossRef]
- Ferrari de Andrade, L.; Ngiow, S.F.; Stannard, K.; Rusakiewicz, S.; Kalimutho, M.; Khanna, K.K.; Tey, S.K.; Takeda, K.; Zitvogel, L.; Martinet, L.; et al. Natural killer cells are essential for the ability of BRAF inhibitors to control BRAFV600E-mutant metastatic melanoma. Cancer Res. 2014, 74, 7298–7308. [Google Scholar] [CrossRef] [Green Version]
- Manzini, C.; Vene, R.; Cossu, I.; Gualco, M.; Zupo, S.; Dono, M.; Spagnolo, F.; Queirolo, P.; Moretta, L.; Mingari, M.C.; et al. Cytokines can counteract the inhibitory effect of MEK-i on NK-cell function. Oncotarget 2016, 7, 60858–60871. [Google Scholar] [CrossRef] [Green Version]
- Schilling, B.; Sondermann, W.; Zhao, F.; Griewank, K.G.; Livingstone, E.; Sucker, A.; Zelba, H.; Weide, B.; Trefzer, U.; Wilhelm, T.; et al. Differential influence of vemurafenib and dabrafenib on patients’ lymphocytes despite similar clinical efficacy in melanoma. Ann. Oncol. 2014, 25, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wei, S.; Xu, X.; Jiang, Y.; Xue, L.; Jiang, P.; Wang, J. Sorafenib attenuated the function of natural killer cells infiltrated in HCC through inhibiting ERK1/2. Int. Immunopharmacol. 2019, 76, 105855. [Google Scholar] [CrossRef] [PubMed]
- Bottos, A.; Gotthardt, D.; Gill, J.W.; Gattelli, A.; Frei, A.; Tzankov, A.; Sexl, V.; Wodnar-Filipowicz, A.; Hynes, N.E. Decreased NK-cell tumour immunosurveillance consequent to JAK inhibition enhances metastasis in breast cancer models. Nat. Commun. 2016, 7, 12258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Cobo, S.; Pieper, N.; Campos-Silva, C.; Garcia-Cuesta, E.M.; Reyburn, H.T.; Paschen, A.; Vales-Gomez, M. Impaired NK cell recognition of vemurafenib-treated melanoma cells is overcome by simultaneous application of histone deacetylase inhibitors. Oncoimmunology 2018, 7, e1392426. [Google Scholar] [CrossRef]
- Furfaro, A.L.; Ottonello, S.; Loi, G.; Cossu, I.; Piras, S.; Spagnolo, F.; Queirolo, P.; Marinari, U.M.; Moretta, L.; Pronzato, M.A.; et al. HO-1 downregulation favors BRAF(V600) melanoma cell death induced by Vemurafenib/PLX4032 and increases NK recognition. Int. J. Cancer 2020, 146, 1950–1962. [Google Scholar] [CrossRef]
- Dominguez, C.; Tsang, K.Y.; Palena, C. Short-term EGFR blockade enhances immune-mediated cytotoxicity of EGFR mutant lung cancer cells: Rationale for combination therapies. Cell Death Dis. 2016, 7, e2380. [Google Scholar] [CrossRef]
- Robert, L.; Ribas, A.; Hu-Lieskovan, S. Combining targeted therapy with immunotherapy. Can 1+1 equal more than 2? Semin. Immunol. 2016, 28, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Hughes, P.E.; Caenepeel, S.; Wu, L.C. Targeted Therapy and Checkpoint Immunotherapy Combinations for the Treatment of Cancer. Trends Immunol. 2016, 37, 462–476. [Google Scholar] [CrossRef]
- Vitale, M.; Della Chiesa, M.; Carlomagno, S.; Pende, D.; Arico, M.; Moretta, L.; Moretta, A. NK-dependent DC maturation is mediated by TNFalpha and IFNgamma released upon engagement of the NKp30 triggering receptor. Blood 2005, 106, 566–571. [Google Scholar] [CrossRef]
- Marcenaro, E.; Carlomagno, S.; Pesce, S.; Moretta, A.; Sivori, S. Bridging innate NK cell functions with adaptive immunity. Adv. Exp. Med. Biol. 2011, 780, 45–55. [Google Scholar] [CrossRef]
- Karachaliou, N.; Gonzalez-Cao, M.; Sosa, A.; Berenguer, J.; Bracht, J.W.P.; Ito, M.; Rosell, R. The combination of checkpoint immunotherapy and targeted therapy in cancer. Ann. Transl. Med. 2017, 5, 388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Correa, B.; Lopez-Sejas, N.; Duran, E.; Labella, F.; Alonso, C.; Solana, R.; Tarazona, R. Modulation of NK cells with checkpoint inhibitors in the context of cancer immunotherapy. Cancer Immunol. Immunother. 2019, 68, 861–870. [Google Scholar] [CrossRef] [PubMed]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Damele, L.; Ottonello, S.; Mingari, M.C.; Pietra, G.; Vitale, C. Targeted Therapies: Friends or Foes for Patient’s NK Cell-Mediated Tumor Immune-Surveillance? Cancers 2020, 12, 774. https://doi.org/10.3390/cancers12040774
Damele L, Ottonello S, Mingari MC, Pietra G, Vitale C. Targeted Therapies: Friends or Foes for Patient’s NK Cell-Mediated Tumor Immune-Surveillance? Cancers. 2020; 12(4):774. https://doi.org/10.3390/cancers12040774
Chicago/Turabian StyleDamele, Laura, Selene Ottonello, Maria Cristina Mingari, Gabriella Pietra, and Chiara Vitale. 2020. "Targeted Therapies: Friends or Foes for Patient’s NK Cell-Mediated Tumor Immune-Surveillance?" Cancers 12, no. 4: 774. https://doi.org/10.3390/cancers12040774
APA StyleDamele, L., Ottonello, S., Mingari, M. C., Pietra, G., & Vitale, C. (2020). Targeted Therapies: Friends or Foes for Patient’s NK Cell-Mediated Tumor Immune-Surveillance? Cancers, 12(4), 774. https://doi.org/10.3390/cancers12040774