An Organotypic Microcosm for the Pancreatic Tumor Microenvironment




Abstract
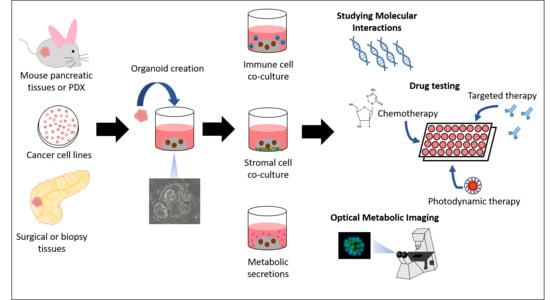
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Elucidating the Problem of Stromal Infiltration in Organoids
2.1. Stromal Interactions Lead to Increased Tumor Proliferation
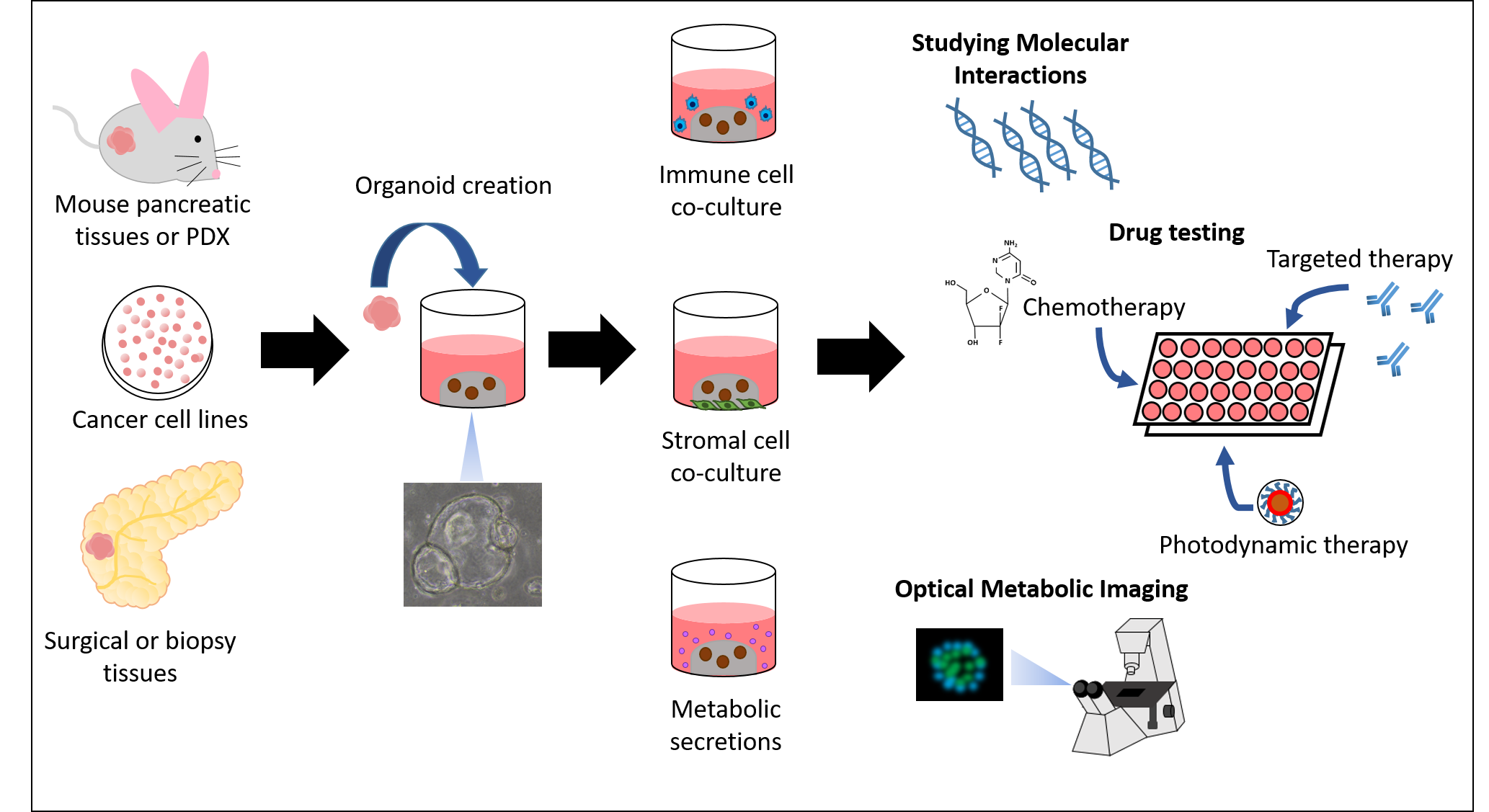
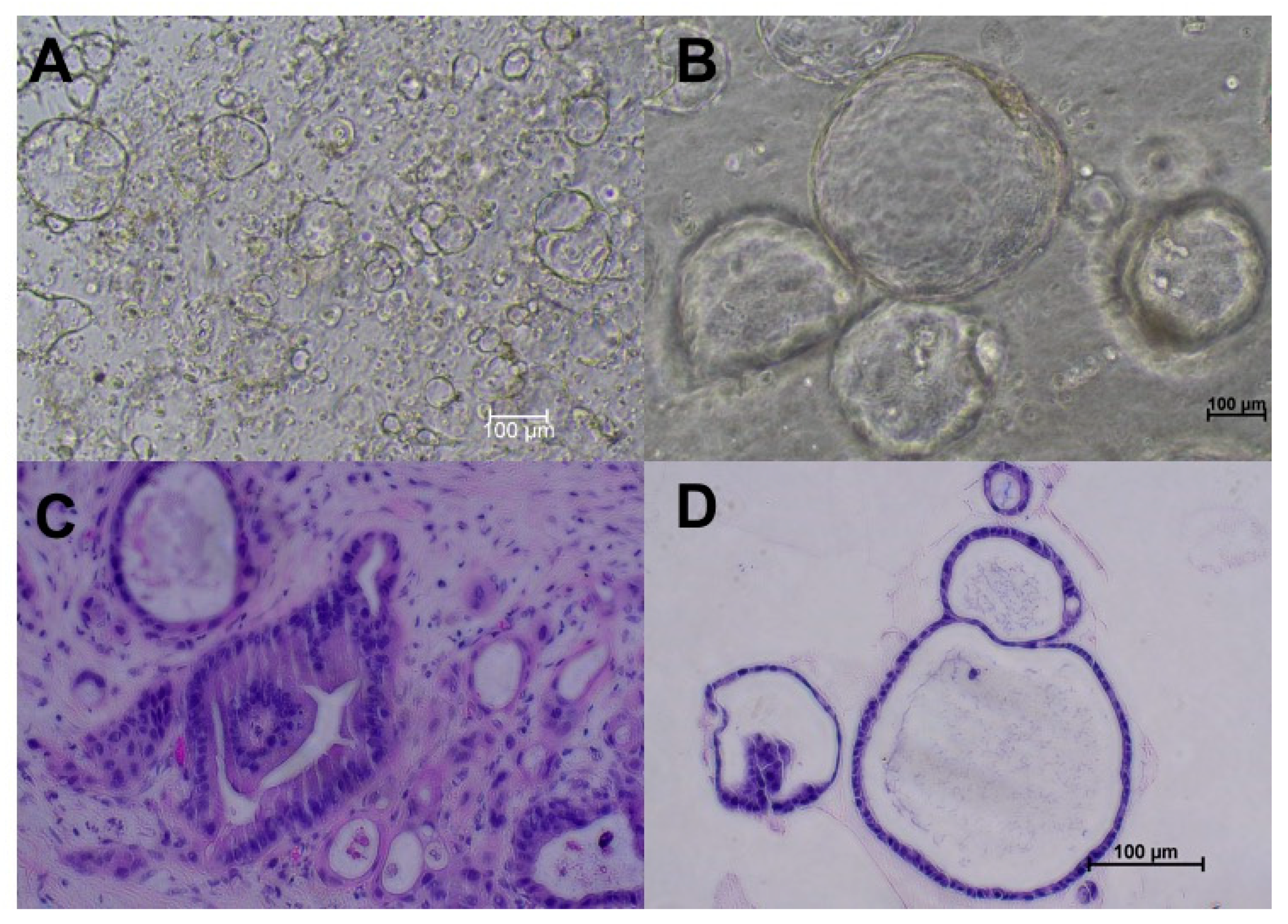
2.2. Culture of Mouse PDAC Organoids
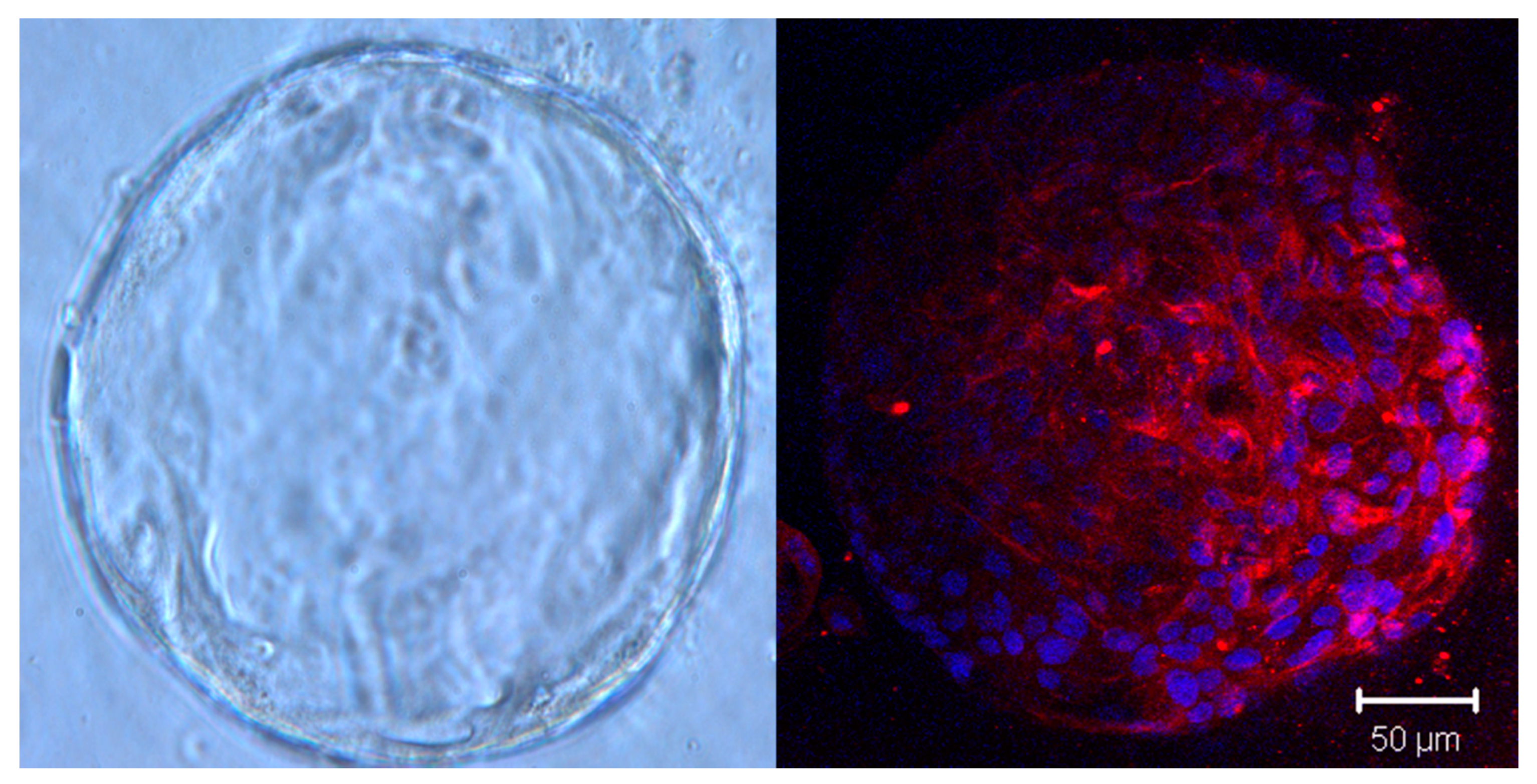
2.3. The Role of Organoid Fibroblasts in Modeling Tumor–Stroma Interactions
2.4. Efficient Drug Delivery to Overcome High Fibrosis in the Pancreas
3. Tracking Metabolic Transformations in PDAC Organoids
4. Immune Modeling in PDAC Organoids
4.1. Targeting an Immunosuppressive Tumor Microenvironment
4.2. Coculture of Immune Populations with PDAC Organoids
5. Biomimetic Organoid Culture
6. Limitations
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Howlander, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. SEER Cancer Statistics Review, 1975–2016; National Cancer Institute: Bethesda, MD, USA, 2019. Available online: https://seer.cancer.gov/csr/1975_2016/ (accessed on 18 February 2020).
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sleightholm, R.L.; Neilsen, B.K.; Li, J.; Steele, M.M.; Singh, R.K.; Hollingsworth, M.A.; Oupicky, D. Emerging roles of the CXCL12/CXCR4 axis in pancreatic cancer progression and therapy. Pharmacol. Ther. 2017, 179, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Broekgaarden, M.; Anbil, S.; Bulin, A.L.; Obaid, G.; Mai, Z.; Baglo, Y.; Rizvi, I.; Hasan, T. Modulation of redox metabolism negates cancer-associated fibroblasts-induced treatment resistance in a heterotypic 3D culture platform of pancreatic cancer. Biomaterials 2019, 222, e119421. [Google Scholar] [CrossRef]
- Upadhrasta, S.; Zheng, L. Strategies in developing immunotherapy for pancreatic cancer: Recognizing and correcting multiple immune “defects” in the tumor microenvironment. J. Clin. Med. 2019, 8, 1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, M.; Lin, M.; Moffitt, R.A.; Salazar, M.A.; Park, J.; Vacirca, J.; Huang, C.; Shroyer, K.R.; Choi, M.; Georgakis, G.V.; et al. Direct therapeutic targeting of immune checkpoint PD-1 in pancreatic cancer. Br. J. Cancer 2019, 120, 88–96. [Google Scholar] [CrossRef] [Green Version]
- Boj, S.F.; Hwang, C.I.; Baker, L.A.; Chio, I.I.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef] [Green Version]
- Tiriac, H.; Bucobo, J.C.; Tzimas, D.; Grewel, S.; Lacomb, J.F.; Rowehl, L.M.; Nagula, S.; Wu, M.; Kim, J.; Sasson, A.; et al. Successful creation of pancreatic cancer organoids by means of EUS-guided fine-needle biopsy sampling for personalized cancer treatment. Gastrointest. Endosc. 2018, 87, 1474–1480. [Google Scholar] [CrossRef]
- Tuveson, D.; Clevers, H. Cancer modeling meets human organoid technology. Science 2019, 364, 952–955. [Google Scholar] [CrossRef]
- Hohwieler, M.; Muller, M.; Frappart, P.O.; Heller, S. Pancreatic progenitors and organoids as a prerequisite to model pancreatic diseases and cancer. Stem Cells Int. 2019, 2019, e9301382. [Google Scholar] [CrossRef] [Green Version]
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinska, E.; Byrne, A.T.; Caldas, C.; Clarke, R.B.; de Jong, S.; Jonkers, J.; Maelandsmo, G.M.; et al. Patient-derived xenograft models: An emerging platform for translational cancer research. Cancer Discov. 2014, 4, 998–1013. [Google Scholar] [CrossRef] [Green Version]
- Tsai, S.; McOlash, L.; Palen, K.; Johnson, B.; Duris, C.; Yang, Q.; Dwinell, M.B.; Hunt, B.; Evans, D.B.; Gershan, J.; et al. Development of primary human pancreatic cancer organoids, matched stromal and immune cells and 3D tumor microenvironment models. BMC Cancer 2018, 18, e335. [Google Scholar] [CrossRef] [PubMed]
- Romero-Calvo, I.; Weber, C.R.; Ray, M.; Brown, M.; Kirby, K.; Nandi, R.K.; Long, T.M.; Sparrow, S.M.; Ugolkov, A.; Qiang, W.; et al. Human organoids share structural and genetic features with primary pancreatic adenocarcinoma tumors. Mol. Cancer Res. 2019, 17, 70–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bishehsari, F.; Zhang, L.; Barlass, U.; Preite, N.Z.; Turturro, S.; Najor, M.S.; Shetuni, B.B.; Zayas, J.P.; Mahdavinia, M.; Abukhdeir, A.M.; et al. KRAS mutation and epithelial-macrophage interplay in pancreatic neoplastic transformation. Int. J. Cancer 2018, 143, 1994–2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waters, A.M.; Der, C.J. KRAS: The critical driver and therapeutic target for pancreatic cancer. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.C.; Mansour, J.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, e6744. [Google Scholar] [CrossRef]
- Gendoo, D.M.A.; Denroche, R.E.; Zhang, A.; Radulovich, N.; Jang, G.H.; Lemire, M.; Fischer, S.; Chadwick, D.; Lungu, I.M.; Ibrahimov, E.; et al. Whole genomes define concordance of matched primary, xenograft, and organoid models of pancreas cancer. PLoS Comput. Biol. 2019, 15, e1006596. [Google Scholar] [CrossRef] [Green Version]
- Tiriac, H.; Belleau, P.; Engle, D.D.; Plenker, D.; Deschenes, A.; Somerville, T.D.D.; Froeling, F.E.M.; Burkhart, R.A.; Denroche, R.E.; Jang, G.H.; et al. Organoid profiling identifies common responders to chemotherapy in pancreatic cancer. Cancer Discov. 2018, 8, 1112–1129. [Google Scholar] [CrossRef] [Green Version]
- Coetzee, A.; Grose, R.; Kocher, H. Pancreatic cancer organotypic models. Curr. Top. Microbiol. Immunol. 2019. [Google Scholar] [CrossRef]
- Engle, D.D.; Tiriac, H.; Rivera, K.D.; Pommier, A.; Whalen, S.; Oni, T.E.; Alagesan, B.; Lee, E.J.; Yao, M.A.; Lucito, M.S.; et al. The glycan CA19-9 promotes pancreatitis and pancreatic cancer in mice. Science 2019, 364, 1156–1162. [Google Scholar] [CrossRef]
- Ardito, C.M.; Gruner, B.M.; Takeuchi, K.K.; Lubeseder-Martellato, C.; Teichmann, N.; Mazur, P.K.; Delgiorno, K.E.; Carpenter, E.S.; Halbrook, C.J.; Hall, J.C.; et al. EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell 2012, 22, 304–317. [Google Scholar] [CrossRef] [Green Version]
- Kaneta, Y.; Sato, T.; Hikiba, Y.; Sugimori, M.; Sue, S.; Kaneko, H.; Irie, K.; Sasaki, T.; Kondo, M.; Chuma, M.; et al. Loss of pancreatic E-cadherin causes pancreatitis-like changes and contributes to carcinogenesis. Cell. Mol. Gastroenterol. Hepatol. 2020, 9, 105–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obaid, G.; Bano, S.; Mallidi, S.; Broekgaarden, M.; Kuriakose, J.; Silber, Z.; Bulin, A.L.; Wang, Y.; Mai, Z.; Jin, W.; et al. Impacting pancreatic cancer therapy in heterotypic in vitro organoids and in vivo tumors with specificity-tuned, NIR-activable photoimmunonanoconjugates: Towards conquering desmoplasia? Nano Lett. 2019, 19, 7573–7587. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.R.; Cannon, A.; Thompson, C.; Santhamma, B.; Chavez-Riveros, A.; Bhatia, R.; Nair, H.B.; Nickisch, K.; Batra, S.K.; Kumar, S. Utilizing cell line-derived organoids to evaluate the efficacy of a novel LIFR-inhibitor, EC359 in targeting pancreatic tumor stroma. Genes Cancer 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, S.; Tiriac, H.; Sridharan, B.P.; Scampavia, L.; Madoux, F.; Seldin, J.; Souza, G.R.; Watson, D.; Tuveson, D.; Spicer, T.P. Advanced development of primary pancreatic organoid tumor models for high-throughput phenotypic drug screening. SLAS Discov. 2018, 23, 574–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koikawa, K.; Ohuchida, K.; Ando, Y.; Kibe, S.; Nakayama, H.; Takesue, S.; Endo, S.; Abe, T.; Okumura, T.; Iwamoto, C.; et al. Basement membrane destruction by pancreatic stellate cells leads to local invasion in pancreatic ductal adenocarcinoma. Cancer Lett. 2018, 425, 65–77. [Google Scholar] [CrossRef]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [Green Version]
- Fumagalli, A.; Drost, J.; Suijkerbuijk, S.J.; van Boxtel, R.; de Ligt, J.; Offerhaus, G.J.; Begthel, H.; Beerling, E.; Tan, E.H.; Sansom, O.J.; et al. Genetic dissection of colorectal cancer progression by orthotopic transplantation of engineered cancer organoids. Proc. Natl. Acad. Sci. USA 2017, 114, E2357–E2364. [Google Scholar] [CrossRef] [Green Version]
- Nanki, K.; Toshimitsu, K.; Takano, A.; Fujii, M.; Shimokawa, M.; Ohta, Y.; Matano, M.; Seino, T.; Nishikori, S.; Ishikawa, K.; et al. Divergent routes toward Wnt and R-spondin niche independency during human gastric carcinogenesis. Cell 2018, 174, 856–869. [Google Scholar] [CrossRef] [Green Version]
- Seino, T.; Kawasaki, S.; Shimokawa, M.; Tamagawa, H.; Toshimitsu, K.; Fujii, M.; Ohta, Y.; Matano, M.; Nanki, K.; Kawasaki, K.; et al. Human pancreatic tumor organoids reveal loss of stem cell niche factor dependence during disease progression. Cell Stem Cell 2018, 22, 454–467. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Srivastava, S.K.; Bhardwaj, A.; Owen, L.B.; Singh, A.P. CXCL12-CXCR4 signalling axis confers gemcitabine resistance to pancreatic cancer cells: A novel target for therapy. Br. J. Cancer 2010, 103, 1671–1679. [Google Scholar] [CrossRef] [Green Version]
- Walsh, A.J.; Castellanos, J.A.; Nagathihalli, N.S.; Merchant, N.B.; Skala, M.C. Optical imaging of drug-induced metabolism changes in murine and human pancreatic cancer organoids reveals heterogeneous drug response. Pancreas 2016, 45, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Cramer, G.M.; Jones, D.P.; El-Hamidi, H.; Celli, J.P. ECM composition and rheology regulate growth, motility, and response to photodynamic therapy in 3D models of pancreatic ductal adenocarcinoma. Mol. Cancer Res. 2017, 15, 15–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamanaka, Y.; Friess, H.; Kobrin, M.S.; Buchler, M.; Beger, H.G.; Korc, M. Coexpression of epidermal growth factor receptor and ligands in human pancreatic cancer is associated with enhanced tumor aggressiveness. Anticancer Res. 1993, 13, 565–569. [Google Scholar] [PubMed]
- Epstein Shochet, G.; Brook, E.; Eyal, O.; Edelstein, E.; Shitrit, D. Epidermal growth factor receptor paracrine upregulation in idiopathic pulmonary fibrosis fibroblasts is blocked by nintedanib. Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 316, L1025–L1034. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; Kalluri, R. A peek into cancer-associated fibroblasts: Origins, functions and translational impact. Dis. Models Mech. 2018, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chio, I.I.C.; Jafarnejad, S.M.; Ponz-Sarvise, M.; Park, Y.; Rivera, K.; Palm, W.; Wilson, J.; Sangar, V.; Hao, Y.; Ohlund, D.; et al. NRF2 promotes tumor maintenance by modulating mRNA translation in pancreatic cancer. Cell 2016, 166, 963–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative phosphorylation as an emerging target in cancer therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, E.; Kim, H.K.; Jee, J.; Hahn, S.; Jeong, S.; Yoo, J. Development of organoid-based drug metabolism model. Toxicol. Appl. Pharmacol. 2019, 385, e114790. [Google Scholar] [CrossRef]
- Walsh, A.J.; Cook, R.S.; Manning, H.C.; Hicks, D.J.; Lafontant, A.; Arteaga, C.L.; Skala, M.C. Optical metabolic imaging identifies glycolytic levels, subtypes, and early-treatment response in breast cancer. Cancer Res. 2013, 73, 6164–6174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [Green Version]
- Royal, R.E.; Levy, C.; Turner, K.; Mathur, A.; Hughes, M.; Kammula, U.S.; Sherry, R.M.; Topalian, S.L.; Yang, J.C.; Lowy, I.; et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J. Immunother. 2010, 33, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Lutz, E.R.; Wu, A.A.; Bigelow, E.; Sharma, R.; Mo, G.; Soares, K.; Solt, S.; Dorman, A.; Wamwea, A.; Yager, A.; et al. Immunotherapy converts nonimmunogenic pancreatic tumors into immunogenic foci of immune regulation. Cancer Immunol. Res. 2014, 2, 616–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goedegebuure, P.; Mitchem, J.B.; Porembka, M.R.; Tan, M.C.; Belt, B.A.; Wang-Gillam, A.; Gillanders, W.E.; Hawkins, W.G.; Linehan, D.C. Myeloid-derived suppressor cells: General characteristics and relevance to clinical management of pancreatic cancer. Curr. Cancer Drug Targets 2011, 11, 734–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Baas, P.; Kim, D.W.; Felip, E.; Perez-Gracia, J.L.; Han, J.Y.; Molina, J.; Kim, J.H.; Arvis, C.D.; Ahn, M.J.; et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): A randomised controlled trial. Lancet 2016, 387, 1540–1550. [Google Scholar] [CrossRef]
- Zhang, Y.S.; Duchamp, M.; Oklu, R.; Ellisen, L.W.; Langer, R.; Khademhosseini, A. Bioprinting the Cancer Microenvironment. ACS Biomater. Sci. Eng. 2016, 2, 1710–1721. [Google Scholar] [CrossRef] [Green Version]
- Boussommier-Calleja, A.; Li, R.; Chen, M.B.; Wong, S.C.; Kamm, R.D. Microfluidics: A new tool for modeling cancer-immune interactions. Trends Cancer 2016, 2, 6–19. [Google Scholar] [CrossRef] [Green Version]
- Peela, N.; Truong, D.; Saini, H.; Chu, H.; Mashaghi, S.; Ham, S.L.; Singh, S.; Tavana, H.; Mosadegh, B.; Nikkhah, M. Advanced biomaterials and bioengineering technologies to recapitulate the stepwise process of cancer metastasis. Biomaterials 2017, 133, 176–207. [Google Scholar] [CrossRef] [Green Version]
- Hakobyan, D.; Medina, C.; Dusserre, N.; Stachowicz, M.L.; Handschin, C.; Fricain, J.C.; Guillermet-Guibert, J.; Oliveira, H. Laser-assisted 3D bioprinting of exocrine pancreas spheroid models for cancer initiation study. Biofabrication 2020. [Google Scholar] [CrossRef]
- Murphy, S.V.; Atala, A. 3D bioprinting of tissues and organs. Nat. BIotechnol. 2014, 32, 773–785. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, S.K.; Khawar, I.A.; Jeong, S.Y.; Chung, S.; Kuh, H.J. Microfluidic co-culture of pancreatic tumor spheroids with stellate cells as a novel 3D model for investigation of stroma-mediated cell motility and drug resistance. J. Exp. Clin. Cancer Res. 2018, 37, e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Su, J.; Fu, X.; Zheng, L.; Yin, Z. Microfluidic device for primary tumor spheroid isolation. Exp. Hematol. Oncol. 2017, 6, e22. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, M.; Gao, M.; Pandalai, P.K.; Cavnar, M.J.; Kim, J. An Organotypic Microcosm for the Pancreatic Tumor Microenvironment. Cancers 2020, 12, 811. https://doi.org/10.3390/cancers12040811
Lin M, Gao M, Pandalai PK, Cavnar MJ, Kim J. An Organotypic Microcosm for the Pancreatic Tumor Microenvironment. Cancers. 2020; 12(4):811. https://doi.org/10.3390/cancers12040811
Chicago/Turabian StyleLin, Miranda, Mei Gao, Prakash K. Pandalai, Michael J. Cavnar, and Joseph Kim. 2020. "An Organotypic Microcosm for the Pancreatic Tumor Microenvironment" Cancers 12, no. 4: 811. https://doi.org/10.3390/cancers12040811
APA StyleLin, M., Gao, M., Pandalai, P. K., Cavnar, M. J., & Kim, J. (2020). An Organotypic Microcosm for the Pancreatic Tumor Microenvironment. Cancers, 12(4), 811. https://doi.org/10.3390/cancers12040811