Ensartinib (X-396) Effectively Modulates Pharmacokinetic Resistance Mediated by ABCB1 and ABCG2 Drug Efflux Transporters and CYP3A4 Biotransformation Enzyme

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
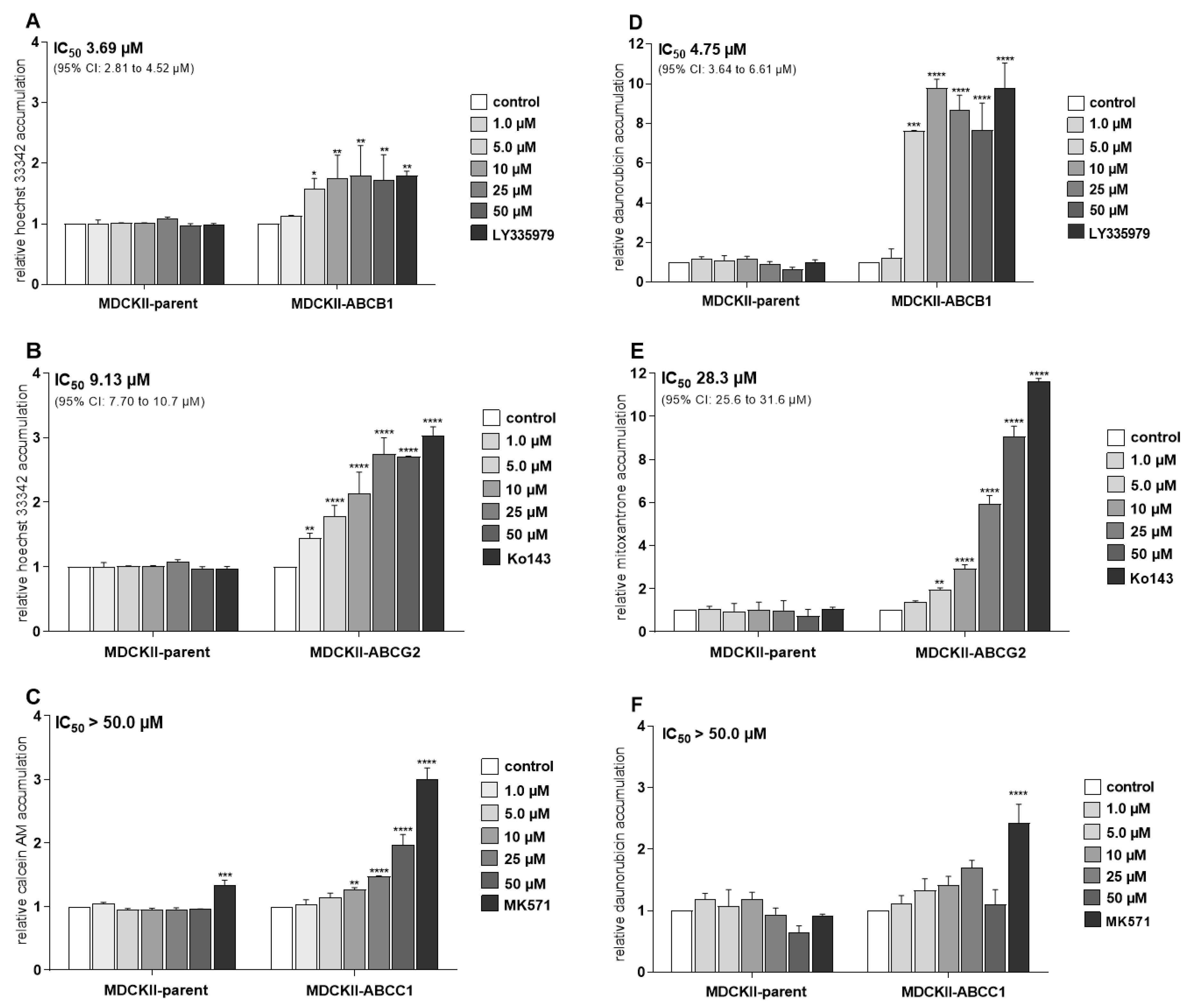
2.1. Ensartinib Potently Inhibits ABCB1 and ABCG2 Transporters in MDCKII Sublines
2.2. Ensartinib Blocks the Activity of Several Clinically Important CYP Isoforms
2.3. Inhibition of CYP3A4 in Intact HepG2-CYP3A4 Cells by Ensartinib
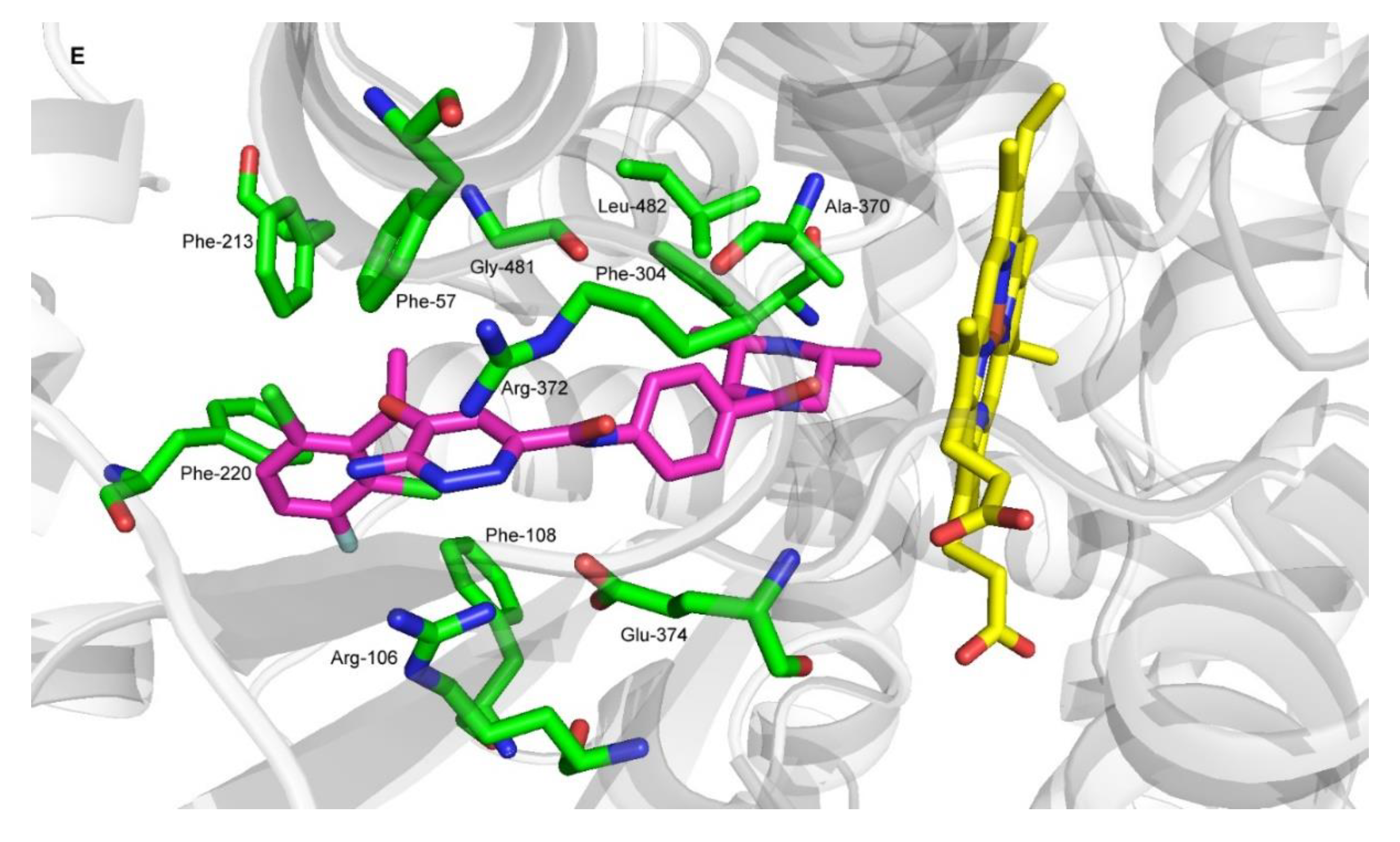
2.4. Molecular Docking of Ensartinib into ABCB1, ABCG2 and CYP3A4
2.5. Modulatory Effects of Ensartinib on ABC Transporter-Mediated Cytostatic MDR
2.6. Ensartinib Attenuates CYP3A4-Mediated Docetaxel Resistance
2.7. Ensartinib Is a Substrate of ABCB1, But Not of the ABCG2 and ABCC1 Transporters
2.8. ABCB1, ABCG2, and ABCC1 Transporters Do Not Confer Resistance to Ensartinib
2.9. Changes in ABCB1, ABCG2, ABCC1, and CYP1A2, CYP3A4, CYP2B6 Expression Following Exposure to Ensartinib
3. Discussion
4. Materials and Methods
4.1. Reagents and Chemicals
4.2. Cell Culture
4.3. Cellular Accumulation Assay with Hoechst 33342 and Calcein AM
4.4. Cellular Accumulation Assay with Daunorubicin and Mitoxantrone
4.5. Inhibitory Assay for Human Recombinant CYP Isoforms
4.6. Inhibition of CYP3A4 in Intact HepG2-CYP3A4 Cells
4.7. Molecular Docking Simulations
4.8. ABCB1 ATPase Assay
4.9. MTT Proliferation Assay
4.10. XTT Proliferation Assay
4.11. Drug Combination Assays
4.12. MDCKII Monolayer Transport Assay
4.13. UHPLC-MS/MS Analysis
4.14. Gene Induction Studies
4.15. Statistical and Data Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosland, G.V.; Engelsen, A.S. Novel points of attack for targeted cancer therapy. Basic Clin. Pharmacol. Toxicol. 2015, 116, 9–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, A.; Gadgeel, S. The Current Landscape of Anaplastic Lymphoma Kinase (ALK) in Non-Small Cell Lung Cancer: Emerging Treatment Paradigms and Future Directions. Target. Oncol. 2017, 12, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Spitaleri, G.; Passaro, A.; de Marinis, F. Ensartinib (X-396) a novel drug for anaplastic lymphoma kinasepositive non-small cell lung cancer patients: We need smart trials to avoid wasting good bullets. Chin. Clin. Oncol. 2018. [Google Scholar] [CrossRef]
- Fan, J.; Fong, T.; Xia, Z.; Zhang, J.; Luo, P. The efficacy and safety of ALK inhibitors in the treatment of ALK-positive non-small cell lung cancer: A network meta-analysis. Cancer Med. 2018, 7, 4993–5005. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Ludwig, J.; Xia, D.; Szakacs, G. Defeating drug resistance in cancer. Discov. Med. 2006, 6, 18–23. [Google Scholar]
- Staud, F.; Cerveny, L.; Ceckova, M. Pharmacotherapy in pregnancy; effect of ABC and SLC transporters on drug transport across the placenta and fetal drug exposure. J. Drug Target. 2012, 20, 736–763. [Google Scholar] [CrossRef]
- Szakacs, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef]
- Müller, F.; Fromm, M.F. Transporter-mediated drug–drug interactions. Pharmacogenomics 2011, 12, 1017–1037. [Google Scholar] [CrossRef]
- European Medicines Agency. Guideline on the Investigation of Drug Interactions. CPMP/EWP/560/95/Rev. 1 Corr. 2**; 2012. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-drug-interactions_en.pdf (accessed on 22 September 2019).
- Food and Drug Administration. In Vitro Metabolism- and Transporter-Mediated Drug—Drug Interaction Studies Guidance for Industry; Clinical Pharmacology: Silver Spring, MD, USA, 2017.
- Benedetti, M.S.; Whomsley, R.; Poggesi, I.; Cawello, W.; Mathy, F.X.; Delporte, M.L.; Papeleu, P.; Watelet, J.B. Drug metabolism and pharmacokinetics. Drug Metab. Rev. 2009, 41, 344–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michael, M.; Doherty, M.M. Tumoral drug metabolism: Overview and its implications for cancer therapy. J. Clin. Oncol. 2005, 23, 205–229. [Google Scholar] [CrossRef] [PubMed]
- Rochat, B. Importance of influx and efflux systems and xenobiotic metabolizing enzymes in intratumoral disposition of anticancer agents. Curr. Cancer Drug Targets 2009, 9, 652–674. [Google Scholar] [CrossRef] [PubMed]
- Vadlapatla, R.K.; Vadlapudi, A.D.; Pal, D.; Mitra, A.K. Mechanisms of drug resistance in cancer chemotherapy: Coordinated role and regulation of efflux transporters and metabolizing enzymes. Curr. Pharm. Des. 2013, 19, 7126–7140. [Google Scholar] [CrossRef] [PubMed]
- Oyama, T.; Kagawa, N.; Kunugita, N.; Kitagawa, K.; Ogawa, M.; Yamaguchi, T.; Suzuki, R.; Kinaga, T.; Yashima, Y.; Ozaki, S.; et al. Expression of cytochrome P450 in tumor tissues and its association with cancer development. Front. Biosci. 2004, 9, 1967–1976. [Google Scholar] [CrossRef] [Green Version]
- Bugde, P.; Biswas, R.; Merien, F.; Lu, J.; Liu, D.X.; Chen, M.; Zhou, S.; Li, Y. The therapeutic potential of targeting ABC transporters to combat multi-drug resistance. Expert Opin. Ther. Targets 2017, 21, 511–530. [Google Scholar] [CrossRef]
- Kathawala, R.J.; Gupta, P.; Ashby, C.R., Jr.; Chen, Z.S. The modulation of ABC transporter-mediated multidrug resistance in cancer: A review of the past decade. Drug Resist. Updates 2015, 18, 1–17. [Google Scholar] [CrossRef]
- Beretta, G.L.; Cassinelli, G.; Pennati, M.; Zuco, V.; Gatti, L. Overcoming ABC transporter-mediated multidrug resistance: The dual role of tyrosine kinase inhibitors as multitargeting agents. Eur. J. Med. Chem. 2017, 142, 271–289. [Google Scholar] [CrossRef]
- Hofman, J.; Ahmadimoghaddam, D.; Hahnova, L.; Pavek, P.; Ceckova, M.; Staud, F. Olomoucine II and purvalanol A inhibit ABCG2 transporter in vitro and in situ and synergistically potentiate cytostatic effect of mitoxantrone. Pharmacol. Res. 2012, 65, 312–319. [Google Scholar] [CrossRef]
- Sorf, A.; Hofman, J.; Kucera, R.; Staud, F.; Ceckova, M. Ribociclib shows potential for pharmacokinetic drug-drug interactions being a substrate of ABCB1 and potent inhibitor of ABCB1, ABCG2 and CYP450 isoforms in vitro. Biochem. Pharmacol. 2018, 154, 10–17. [Google Scholar] [CrossRef]
- Cihalova, D.; Hofman, J.; Ceckova, M.; Staud, F. Purvalanol A, olomoucine II and roscovitine inhibit ABCB1 transporter and synergistically potentiate cytotoxic effects of daunorubicin in vitro. PLoS ONE 2013, 8, e83467. [Google Scholar] [CrossRef] [PubMed]
- Cihalova, D.; Ceckova, M.; Kucera, R.; Klimes, J.; Staud, F. Dinaciclib, a cyclin-dependent kinase inhibitor, is a substrate of human ABCB1 and ABCG2 and an inhibitor of human ABCC1 in vitro. Biochem. Pharmacol. 2015, 98, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Cihalova, D.; Staud, F.; Ceckova, M. Interactions of cyclin-dependent kinase inhibitors AT-7519, flavopiridol and SNS-032 with ABCB1, ABCG2 and ABCC1 transporters and their potential to overcome multidrug resistance in vitro. Cancer Chemother. Pharmacol. 2015, 76, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Hofman, J.; Sorf, A.; Vagiannis, D.; Sucha, S.; Novotna, E.; Kammerer, S.; Kupper, J.H.; Ceckova, M.; Staud, F. Interactions of Alectinib with Human ATP-Binding Cassette Drug Efflux Transporters and Cytochrome P450 Biotransformation Enzymes: Effect on Pharmacokinetic Multidrug Resistance. Drug Metab. Dispos. 2019, 47, 699–709. [Google Scholar] [CrossRef]
- Hofman, J.; Sorf, A.; Vagiannis, D.; Sucha, S.; Kammerer, S.; Kupper, J.H.; Chen, S.; Guo, L.; Ceckova, M.; Staud, F. Brivanib Exhibits Potential for Pharmacokinetic Drug-Drug Interactions and the Modulation of Multidrug Resistance through the Inhibition of Human ABCG2 Drug Efflux Transporter and CYP450 Biotransformation Enzymes. Mol. Pharm. 2019, 16, 4436–4450. [Google Scholar] [CrossRef]
- Ferreira, R.J.; Ferreira, M.J.; dos Santos, D.J. Molecular docking characterizes substrate-binding sites and efflux modulation mechanisms within P-glycoprotein. J. Chem. Inf. Model. 2013, 53, 1747–1760. [Google Scholar] [CrossRef]
- Horn, L.; Infante, J.R.; Reckamp, K.L.; Blumenschein, G.R.; Leal, T.A.; Waqar, S.N.; Gitlitz, B.J.; Sanborn, R.E.; Whisenant, J.G.; Du, L.; et al. Ensartinib (X-396) in ALK-Positive Non-Small Cell Lung Cancer: Results from a First-in-Human Phase I/II, Multicenter Study. Clin. Cancer Res. 2018, 24, 2771–2779. [Google Scholar] [CrossRef] [Green Version]
- Fang, W.F.; Ma, Y.X.; Huang, J.J.; Zhang, Y.; Zou, B.Y.; Zhao, H.Y. Ensartinib (X-396), a second-generation ALK TKI, in Chinese ALK-positive non-small cell lung cancer: A phase I, dose-escalation study. J. Clin. Oncol. 2018, 36, e21122. [Google Scholar] [CrossRef]
- Bruno, R.; Hille, D.; Riva, A.; Vivier, N.; ten Bokkel Huinnink, W.W.; van Oosterom, A.T.; Kaye, S.B.; Verweij, J.; Fossella, F.V.; Valero, V.; et al. Population pharmacokinetics/pharmacodynamics of docetaxel in phase II studies in patients with cancer. J. Clin. Oncol. 1998, 16, 187–196. [Google Scholar] [CrossRef]
- Lovly, C.M.; Heuckmann, J.M.; de Stanchina, E.; Chen, H.; Thomas, R.K.; Liang, C.; Pao, W. Insights into ALK-driven cancers revealed through development of novel ALK tyrosine kinase inhibitors. Cancer Res. 2011, 71, 4920–4931. [Google Scholar] [CrossRef] [Green Version]
- Mayne, C.G.; Arcario, M.J.; Mahinthichaichan, P.; Baylon, J.L.; Vermaas, J.V.; Navidpour, L.; Wen, P.C.; Thangapandian, S.; Tajkhorshid, E. The cellular membrane as a mediator for small molecule interaction with membrane proteins. Biochim. Biophys. Acta 2016, 1858, 2290–2304. [Google Scholar] [CrossRef] [PubMed]
- Bayat Mokhtari, R.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.J.; Zhang, X.; Cheng, C.; Wang, F.; Wang, X.K.; Liang, Y.J.; To, K.K.; Zhou, W.; Huang, H.B.; Fu, L.W. Crizotinib (PF-02341066) reverses multidrug resistance in cancer cells by inhibiting the function of P-glycoprotein. Br. J. Pharmacol. 2012, 166, 1669–1683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Zhang, X.; Wang, F.; Wang, X.; Yang, K.; Xu, M.; To, K.K.; Li, Q.; Fu, L. Effect of ceritinib (LDK378) on enhancement of chemotherapeutic agents in ABCB1 and ABCG2 overexpressing cells in vitro and in vivo. Oncotarget 2015, 6, 44643–44659. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Chen, Y.; To, K.K.; Wang, F.; Li, D.; Chen, L.; Fu, L. Alectinib (CH5424802) antagonizes ABCB1- and ABCG2-mediated multidrug resistance in vitro, in vivo and ex vivo. Exp. Mol. Med. 2017, 49, e303. [Google Scholar] [CrossRef]
- Wu, S.; Fu, L. Tyrosine kinase inhibitors enhanced the efficacy of conventional chemotherapeutic agent in multidrug resistant cancer cells. Mol. Cancer 2018, 17, 25. [Google Scholar] [CrossRef]
- Cygalova, L.H.; Hofman, J.; Ceckova, M.; Staud, F. Transplacental pharmacokinetics of glyburide, rhodamine 123, and BODIPY FL prazosin: Effect of drug efflux transporters and lipid solubility. J. Pharmacol. Exp. Ther. 2009, 331, 1118–1125. [Google Scholar] [CrossRef] [Green Version]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef]
- Elkind, N.B.; Szentpetery, Z.; Apati, A.; Ozvegy-Laczka, C.; Varady, G.; Ujhelly, O.; Szabo, K.; Homolya, L.; Varadi, A.; Buday, L.; et al. Multidrug transporter ABCG2 prevents tumor cell death induced by the epidermal growth factor receptor inhibitor Iressa (ZD1839, Gefitinib). Cancer Res. 2005, 65, 1770–1777. [Google Scholar] [CrossRef] [Green Version]
- Ujhelly, O.; Ozvegy, C.; Varady, G.; Cervenak, J.; Homolya, L.; Grez, M.; Scheffer, G.; Roos, D.; Bates, S.E.; Varadi, A.; et al. Application of a human multidrug transporter (ABCG2) variant as selectable marker in gene transfer to progenitor cells. Hum. Gene Ther. 2003, 14, 403–412. [Google Scholar] [CrossRef]
- Hollo, Z.; Homolya, L.; Hegedus, T.; Sarkadi, B. Transport properties of the multidrug resistance-associated protein (MRP) in human tumour cells. FEBS Lett. 1996, 383, 99–104. [Google Scholar] [CrossRef] [Green Version]
- Xuan, J.; Chen, S.; Ning, B.; Tolleson, W.H.; Guo, L. Development of HepG2-derived cells expressing cytochrome P450s for assessing metabolism-associated drug-induced liver toxicity. Chem. Biol. Interact. 2016, 255, 63–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herzog, N.; Hansen, M.; Miethbauer, S.; Schmidtke, K.U.; Anderer, U.; Lupp, A.; Sperling, S.; Seehofer, D.; Damm, G.; Scheibner, K.; et al. Primary-like human hepatocytes genetically engineered to obtain proliferation competence display hepatic differentiation characteristics in monolayer and organotypical spheroid cultures. Cell Biol. Int. 2016, 40, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, S.D.; Vivares, A.; Klieber, S.; Hewitt, N.J.; Muenst, B.; Heinz, S.; Walles, H.; Braspenning, J. Applicability of second-generation upcyte(R) human hepatocytes for use in CYP inhibition and induction studies. Pharmacol. Res. Perspect. 2015, 3, e00161. [Google Scholar] [CrossRef] [PubMed]
- Zinc Database. Available online: http://zinc.docking.org (accessed on 15 October 2019).
- Protein Data Bank. Available online: http://www.rcsb.org (accessed on 2 November 2019).
- Kim, Y.; Chen, J. Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation. Science 2018, 359, 915–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Jaimes, K.F.; Aller, S.G. Refined structures of mouse P-glycoprotein. Protein Sci. 2014, 23, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Manolaridis, I.; Jackson, S.M.; Taylor, N.M.I.; Kowal, J.; Stahlberg, H.; Locher, K.P. Cryo-EM structures of a human ABCG2 mutant trapped in ATP-bound and substrate-bound states. Nature 2018, 563, 426–430. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.M.; Manolaridis, I.; Kowal, J.; Zechner, M.; Taylor, N.M.I.; Bause, M.; Bauer, S.; Bartholomaeus, R.; Bernhardt, G.; Koenig, B.; et al. Structural basis of small-molecule inhibition of human multidrug transporter ABCG2. Nat. Struct. Mol. Biol. 2018, 25, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Ekroos, M.; Sjogren, T. Structural basis for ligand promiscuity in cytochrome P450 3A4. Proc. Natl. Acad. Sci. USA 2006, 103, 13682–13687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Drug(s) | IC50 1 (µM) | 95% CI (µM) | RR2 |
|---|---|---|---|---|
| MDCKII-parent | ||||
| daunorubicin | 0.947 | (0.814–1.07) | ||
| mitoxantrone | 1.59 | (1.39–1.64) | ||
| daunorubicin + ensartinib | 0.673 ns | (0.551–0.743) | 1.41 | |
| mitoxantrone + ensartinib | 0.955 ns | (0.746–1.13) | 1.66 | |
| MDCKII-ABCB1 | ||||
| daunorubicin | 11.9 | (10.7–12.9) | ||
| daunorubicin + ensartinib | 1.06 **** | (0.743–1.22) | 11.2 | |
| MDCKII-ABCG2 | ||||
| mitoxantrone | 9.98 | (8.91–11.5) | ||
| mitoxantrone + ensartinib | 1.62 *** | (1.22–1.98) | 6.16 | |
| HL60-parent | ||||
| daunorubicin | 0.0531 | (0.0473–0.0589) | ||
| mitoxantrone | 0.0305 | (0.230–0.0370) | ||
| daunorubicin + ensartinib | 0.0221 ns | (0.0169–0.0340) | 2.40 | |
| mitoxantrone + ensartinib | 0.0089 ns | (0.0059–0.0119) | 3.43 | |
| HL60-ABCB1 | ||||
| daunorubicin | 2.39 | (1.61–2.53) | ||
| daunorubicin + ensartinib | 0.108 **** | (0.0664–0.2248) | 22.1 | |
| HL60-ABCG2 | ||||
| mitoxantrone | 0.666 | (0.520–0.823) | ||
| mitoxantrone + ensartinib | 0.0359 **** | (0.0197–0.0603) | 18.6 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vagiannis, D.; Novotna, E.; Skarka, A.; Kammerer, S.; Küpper, J.-H.; Chen, S.; Guo, L.; Staud, F.; Hofman, J. Ensartinib (X-396) Effectively Modulates Pharmacokinetic Resistance Mediated by ABCB1 and ABCG2 Drug Efflux Transporters and CYP3A4 Biotransformation Enzyme. Cancers 2020, 12, 813. https://doi.org/10.3390/cancers12040813
Vagiannis D, Novotna E, Skarka A, Kammerer S, Küpper J-H, Chen S, Guo L, Staud F, Hofman J. Ensartinib (X-396) Effectively Modulates Pharmacokinetic Resistance Mediated by ABCB1 and ABCG2 Drug Efflux Transporters and CYP3A4 Biotransformation Enzyme. Cancers. 2020; 12(4):813. https://doi.org/10.3390/cancers12040813
Chicago/Turabian StyleVagiannis, Dimitrios, Eva Novotna, Adam Skarka, Sarah Kammerer, Jan-Heiner Küpper, Si Chen, Lei Guo, Frantisek Staud, and Jakub Hofman. 2020. "Ensartinib (X-396) Effectively Modulates Pharmacokinetic Resistance Mediated by ABCB1 and ABCG2 Drug Efflux Transporters and CYP3A4 Biotransformation Enzyme" Cancers 12, no. 4: 813. https://doi.org/10.3390/cancers12040813