Therapy-Induced Senescence: An “Old” Friend Becomes the Enemy

 ,
, 
Abstract
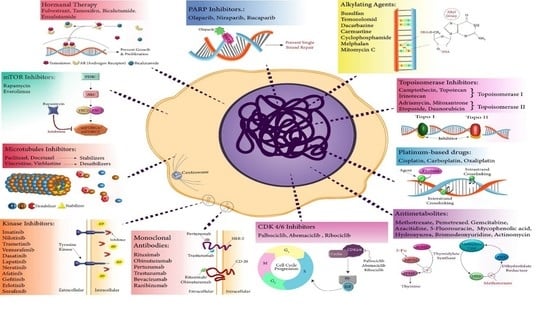
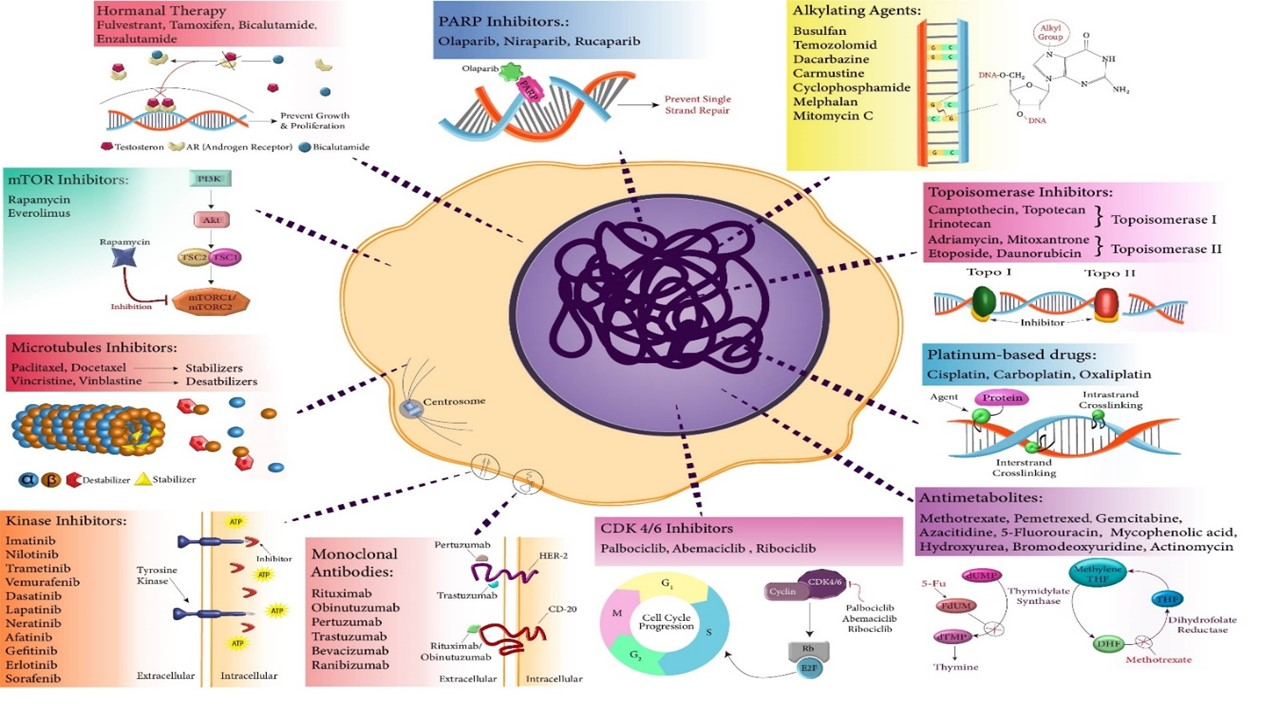
:
1. Introduction
2. Senescence as an Established Response to an Array of Anticancer Chemotherapeutics In Vitro and In Vivo
2.1. Topoisomerase Poisons/Inhibitors
2.2. Alkylating Agents
2.3. Platinum-Based Drugs
2.4. Antimetabolites
2.5. Microtubule Inhibitors
2.6. Hormonal Therapy
2.7. Kinase Inhibitors
2.8. mTOR Inhibitors
2.9. Monoclonal Antibodies
2.10. CDK 4/6 Inhibitors
2.11. Aurora Kinase Inhibitors
2.12. PARP Inhibitors
3. Unfavorable Outcomes of Therapy-Induced Senescence and Its Contribution to Cancer Recurrence
3.1. Evidence for the Reversibility of Therapy-Induced Senescence (TIS)
3.2. Deleterious Effects of the Accumulation of Senescent Cells on Outcomes of Cancer Therapy
4. Clearance of Senescent Tumor Cells as A New Approach to Prevent or Delay Cancer Relapse
4.1. Dasatinib + Quercetin
4.2. Navitoclax (ABT263)
4.3. Fisetin
4.4. Metformin
4.5. Panobinostat
4.6. Autophagy Modulators
4.7. Fibrates
4.8. Cardiac Glycosides
5. Conclusions
Funding
Conflicts of Interest
References
- Hayflick, L.; Moorhead, P.S. The Serial Cultivation of Human Diploid Cell Strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Hayflick, L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 1965, 636, 614–636. [Google Scholar] [CrossRef]
- Shay, J.W.; Woodring, E. Wright Hayflick, his limit, and cellular ageing. Nat. Rev. Mol. Cell Biol. 2000, 1, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. Geroconversion: Irreversible step to cellular senescence. Cell Cycle 2014, 13, 3628–3635. [Google Scholar] [CrossRef]
- Childs, B.G.; Durik, M.; Baker, D.J.; Deursen, J.M. Van Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2016, 21, 1424–1435. [Google Scholar] [CrossRef] [Green Version]
- Campisi, J. Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol. 2001, 11, 27–31. [Google Scholar] [CrossRef] [Green Version]
- Hinds, P.; Pietruska, J. Senescence and tumor suppression. F1000Research 2017, 6, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Collado, M.; Gil, J.; Efeyan, A.; Guerra, C.; Schuhmacher, A.J.; Barradas, M.; Benguría, A.; Zaballos, A.; Flores, J.M.; Barbacid, M.; et al. Tumour biology: Senescence in premalignant tumours. Nature 2005, 436, 642. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef] [Green Version]
- Michaloglou, C.; Vredeveld, L.C.W.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; Van Der Horst, C.M.A.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005, 436, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.W.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-Induced Senescence Relayed by an Interleukin-Dependent Inflammatory Network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotarelo, C.L.; Schad, A.; Kirkpatrick, C.J.; Sleeman, J.P.; Springer, E.; Schmidt, M.; Thaler, S. Detection of cellular senescence within human invasive breast carcinomas distinguishes different breast tumor subtypes. Oncotarget 2016, 7, 74846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garré, M.; Giovanni Nuciforo, P.; Bensimon, A.; et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006, 444, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Stroikin, Y.; Dalen, H.; Brunk, U.T.; Terman, A. Testing the “garbage” accumulation theory of ageing: Mitotic activity protects cells from death induced by inhibition of autophagy. Biogerontology 2005, 6, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Baker, D.J.; Wijshake, T.; Conover, C.A.; Campisi, J.; Van Deursen, J.M. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 2016, 354, 472–477. [Google Scholar] [CrossRef]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef]
- Martin, J.A.; Buckwalter, J.A. Roles of articular cartilage aging and chondrocyte senescence in the pathogenesis of osteoarthritis. Iowa Orthop. J. 2001, 21, 1–7. [Google Scholar]
- Kritsilis, M.; Rizou, S.V.; Koutsoudaki, P.N.; Evangelou, K.; Gorgoulis, V.G.; Papadopoulos, D. Ageing, cellular senescence and neurodegenerative disease. Int. J. Mol. Sci. 2018, 19, 2937. [Google Scholar] [CrossRef] [Green Version]
- Ogrodnik, M.; Zhu, Y.; Langhi, L.G.P.; Tchkonia, T.; Krüger, P.; Fielder, E.; Victorelli, S.; Ruswhandi, R.A.; Giorgadze, N.; Pirtskhalava, T.; et al. Obesity-Induced Cellular Senescence Drives Anxiety and Impairs Neurogenesis. Cell Metab. 2019, 29, 1061–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valentijn, F.A.; Falke, L.L.; Nguyen, T.Q.; Goldschmeding, R. Cellular senescence in the aging and diseased kidney. J. Cell Commun. Signal. 2018, 12, 69–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campisi, J. Aging, Cellular Senescence, and Cancer. Annu. Rev. Physiol. 2014, 75, 685–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roninson, I.B. Tumor cell senescence in cancer treatment. Cancer Res. 2003, 63, 2705–2715. [Google Scholar]
- Lee, S.; Lee, J.S. Cellular senescence: A promising strategy for cancer therapy. BMB Rep. 2019, 52, 35–41. [Google Scholar] [CrossRef] [Green Version]
- Nardella, C.; Clohessy, J.G.; Alimonti, A.; Pandolfi, P.P. Pro-senescence therapy for cancer treatment. Nat. Rev. Cancer 2011, 11, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Saleh, T.; Tyutyunyk-Massey, L.; Murray, G.F.; Alotaibi, M.R.; Kawale, A.S.; Elsayed, Z.; Henderson, S.C.; Yakovlev, V.; Elmore, L.W.; Toor, A.; et al. Tumor cell escape from therapy-induced senescence. Biochem. Pharmacol. 2019, 162, 202–212. [Google Scholar] [CrossRef]
- Demaria, M.; Leary, M.N.O.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017, 7, 165–177. [Google Scholar] [CrossRef] [Green Version]
- Shay, J.W.; Bacchetti, S. A survey of telomerase activity in human cancer. Eur. J. Cancer Part A 1997, 33, 787–791. [Google Scholar] [CrossRef]
- Wang, X.; Wong, S.C.H.; Pan, J.; Cells, C.; Tsao, S.W.; Fung, K.H.Y.; Kwong, D.L.W.; Sham, J.S.T.; Nicholls, J.M. Evidence of Cisplatin-induced Senescent-like Growth Arrest in Nasopharyngeal Carcinoma Cells Advances in Brief Growth Arrest in Nasopharyngeal. Cancer Res. 1998, 58, 5019–5022. [Google Scholar]
- Chang, B.D.; Broude, E.V.; Dokmanovic, M.; Zhu, H.; Ruth, A.; Xuan, Y.; Kandel, E.S.; Lausch, E.; Christov, K.; Roninson, I.B. A senescence-like phenotype distinguishes tumor cells that undergo terminal proliferation arrest after exposure to anticancer agents. Cancer Res. 1999, 59, 3761–3767. [Google Scholar] [PubMed]
- Gewirtz, D.A.; Holt, S.E.; Elmore, L.W. Accelerated senescence: An emerging role in tumor cell response to chemotherapy and radiation. Biochem. Pharmacol. 2008, 76, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Poele, R.H.; Okorokov, A.L.; Jardine, L.; Cummings, J.; Joel, S.P. DNA Damage Is Able to Induce Senescence in Tumor Cells in Vitro and in Vivo. Cancer Res. 2002, 62, 1876–1883. [Google Scholar]
- Ewald, J.A.; Desotelle, J.A.; Wilding, G.; Jarrard, D.F. Therapy-Induced Senescence in Cancer. J. Natl. Cancer Inst. 2010, 102, 1536–1546. [Google Scholar] [CrossRef] [Green Version]
- Elmore, L.W.; Rehder, C.W.; Di, X.; McChesney, P.A.; Jackson-cook, C.K.; Gewirtz, D.A.; Holt, S.E. Adriamycin-induced senescence in breast tumor cells involves functional p53 and telomere dysfunction. J. Biol. Chem. 2002, 277, 35509–35515. [Google Scholar] [CrossRef] [Green Version]
- Su, D.; Zhu, S.; Han, X.; Feng, Y.; Huang, H.; Ren, G.; Pan, L.; Zhang, Y.; Lu, J.; Huang, B. BMP4-Smad signaling pathway mediates adriamycin-induced premature senescence in lung cancer cells. J. Biol. Chem. 2009, 284, 12153–12164. [Google Scholar] [CrossRef] [Green Version]
- Chang, B.D.; Xuan, Y.; Broude, E.V.; Zhu, H.; Schott, B.; Fang, J.; Roninson, I.B. Role of p53 and p21(waf1/cip1) in senescence-like terminal proliferation arrest induced in human tumor cells by chemotherapeutic drugs. Oncogene 1999, 18, 4808–4818. [Google Scholar] [CrossRef] [Green Version]
- You, R.; Dai, J.; Zhang, P.; Barding, G.A.; Raftery, D. Dynamic metabolic response to adriamycin-induced senescence in breast cancer cells. Metabolites 2018, 8, 95. [Google Scholar] [CrossRef] [Green Version]
- Mosieniak, G.; Sliwinska, M.A.; Alster, O.; Strzeszewska, A.; Sunderland, P.; Piechota, M.; Was, H.; Sikora, E. Polyploidy Formation in Doxorubicin-Treated Cancer Cells Can Favor Escape from Senescence. Neoplasia 2015, 17, 882–893. [Google Scholar] [CrossRef] [Green Version]
- Schwarze, S.R.; Fu, V.X.; Desotelle, J.A.; Kenowski, M.L.; Jarrard, D.F. The identification of senescence-specific genes during the induction of senescence in prostate cancer cells. Neoplasia 2005, 7, 816–823. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.Y.; Lin, P.M.; Liu, Y.C.; Hsiao, H.H.; Yang, W.C.; Hsu, J.F.; Hsu, C.M.; Lin, S.F. Induction of cellular senescence by doxorubicin is associated with upregulated miR-375 and induction of autophagy in K562 cells. PLoS ONE 2012, 7, e37205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, Z.; Zhang, Y.; Liu, L.; Guo, X.; Qin, J.; Cui, G. Mesenchymal stem cells derived from different origins have unique sensitivities to different chemotherapeutic agents. Cell Biol. Int. 2012, 36, 857–862. [Google Scholar] [CrossRef] [PubMed]
- Bojko, A.; Czarnecka-Herok, J.; Charzynska, A.; Dabrowski, M.; Sikora, E. Diversity of the Senescence Phenotype of Cancer Cells Treated with Chemotherapeutic Agents. Cells 2019, 8, 1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansilla, S.; Piña, B.; Portugal, J. Daunorubicin-induced variations in gene transcription: Commitment to proliferation arrest, senescence and apoptosis. Biochem. J. 2003, 372, 703–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagano, T.; Nakano, M.; Nakashima, A.; Onishi, K.; Yamao, S.; Enari, M.; Kikkawa, U.; Kamada, S. Identification of cellular senescence-specific genes by comparative transcriptomics. Sci. Rep. 2016, 22, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yosef, R.; Pilpel, N.; Papismadov, N.; Gal, H.; Ovadya, Y.; Vadai, E.; Miller, S.; Porat, Z.; Ben-Dor, S.; Krizhanovsky, V. p21 maintains senescent cell viability under persistent DNA damage response by restraining JNK and caspase signaling. EMBO J. 2017, 36, 2280–2295. [Google Scholar] [CrossRef]
- Yang, H.; Wang, H.; Ren, U.; Chen, Q.; Chena, Z.J. CGAS is essential for cellular senescence. Proc. Natl. Acad. Sci. USA 2017, 114, E4612–E4620. [Google Scholar] [CrossRef] [Green Version]
- Gu, L.; Kitamura, M. Sensitive Detection and Monitoring of Senescence-Associated Secretory Phenotype by SASP-RAP Assay. PLoS ONE 2012, 7, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Decaup, E.; Jean, C.; Laurent, C.; Gravelle, P.; Fruchon, S.; Capilla, F.; Marrot, A.; Al Saati, T.; Frenois, F.X.; Laurent, G.; et al. Anti-tumor activity of obinutuzumab and rituximab in a follicular lymphoma 3D model. Blood Cancer J. 2013, 3, e131. [Google Scholar] [CrossRef] [Green Version]
- Coppe, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Munoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the p53 Tumor Suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef]
- Zhao, H.; Halicka, H.D.; Traganos, F.; Jorgensen, E.; Darzynkiewicz, Z. New biomarkers probing depth of cell senescence assessed by laser scanning cytometry. Cytom. Part A 2010, 77, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Wei, W.; Dunaway, S.; Darnowski, J.W.; Calabresi, P.; Sedivy, J.; Hendrickson, E.A.; Balan, K.V.; Pantazis, P.; Wyche, J.H. Role of p21 in apoptosis and senescence of human colon cancer cells treated with camptothecin. J. Biol. Chem. 2002, 277, 17154–17160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Zhang, S.; Song, J.; Sun, K.; Zong, C.; Zhao, Q.; Liu, W.; Li, R.; Wu, M.; Wei, L. Autophagy inhibition switches low-dose camptothecin-induced premature senescence to apoptosis in human colorectal cancer cells. Biochem. Pharmacol. 2014, 90, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Velichko, A.K.; Petrova, N.V.; Razin, S.V.; Kantidze, O.L. Mechanism of heat stress-induced cellular senescence elucidates the exclusive vulnerability of early S-phase cells to mild genotoxic stress. Nucleic Acids Res. 2015, 43, 6309–6320. [Google Scholar] [CrossRef] [Green Version]
- Taschner-mandl, S.; Schwarz, M.; Blaha, J.; Kauer, M.; Kromp, F.; Frank, N.; Rifatbegovic, F.; Weiss, T.; Ladenstein, R.; Hohenegger, M.; et al. Metronomic topotecan impedes tumor growth of MYCN-amplified neuroblastoma cells in vitro and in vivo by therapy induced senescence. Oncotarget 2015, 7, 3571–3586. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Guo, W.J.; Zhang, X.W.; Cai, X.; Tian, S.; Li, J. Cetuximab enhances the activities of irinotecan on gastric cancer cell lines through downregulating the EGFR pathway upregulated by irinotecan. Cancer Chemother. Pharmacol. 2011, 68, 871–878. [Google Scholar] [CrossRef]
- Probin, V.; Wang, Y.; Zhou, D. Busulfan-induced senescence is dependent on ROS production upstream of the MAPK pathway. Free Radic. Biol. Med. 2007, 42, 1858–1865. [Google Scholar] [CrossRef] [Green Version]
- Mei, Q.; Li, F.; Quan, H.; Liu, Y.; Xu, H. Busulfan inhibits growth of human osteosarcoma through miR-200 family microRNAs in vitro and in vivo. Cancer Sci. 2014, 105, 755–762. [Google Scholar] [CrossRef] [Green Version]
- Probin, V.; Wang, Y.; Bai, A.; Zhou, D. Busulfan selectively induces cellular senescence but not apoptosis in WI38 fibroblasts via a p53-independent but extracellular signal-regulated kinase-p38 mitogen-activated protein kinase-dependent mechanism. J. Pharmacol. Exp. Ther. 2006, 319, 551–560. [Google Scholar] [CrossRef]
- Cells, M.H.; Meng, A.; Wang, Y.; Zant, G.V.; Zhou, D. Ionizing Radiation and Busulfan Induce Premature Senescence in Murine Bone Marrow Hematopoietic Cells. Cancer Res. 2003, 63, 5414–5419. [Google Scholar]
- Kipper, F.C.; Silva, A.O.; Marc, A.L.; Confortin, G.; Junqueira, A.V.; Neto, E.P.; Lenz, G. Vinblastine and antihelmintic mebendazole potentiate temozolomide in resistant gliomas. Investig. New Drugs 2018, 36, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Papait, R.; Magrassi, L.; Rigamonti, D.; Cattaneo, E. Temozolomide and carmustine cause large-scale heterochromatin reorganization in glioma cells. Biochem. Biophys. Res. Commun. 2009, 379, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Aasland, D.; Gotzinger, L.; Hauck, L.; Berte, N.; Meyer, J.; Effenberger, M.; Schneider, S.; Reuber, E.E.; Roos, W.P.; Tomicic, M.T.; et al. Temozolomide induces senescence and repression of DNA repair pathways in glioblastoma cells via activation of ATR–Chk1, p21, and NF-kB. Cancer Res. 2019, 79, 99–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Shi, B.; Zheng, H.; Min, L.; Yang, J.; Li, X.; Liao, X.; Huang, W.; Zhang, M.; Xu, S.; et al. Senescence-associated secretory factors induced by cisplatin in melanoma cells promote non-senescent melanoma cell growth through activation of the ERK1/2-RSK1 pathway. Cell Death Dis. 2018, 9, 260. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, C.A.; Fridman, J.S.; Yang, M.; Lee, S.; Baranov, E.; Hoffman, R.M.; Lowe, S.W. A senescence program controlled by p53 and p16INK4acontributes to the outcome of cancer therapy. Cell 2002, 109, 335–346. [Google Scholar] [CrossRef] [Green Version]
- Antonangeli, F.; Soriani, A.; Ricci, B.; Ponzetta, A.; Benigni, G.; Morrone, S.; Bernardini, G.; Santoni, A. Natural killer cell recognition of in vivo drug-induced senescent multiple myeloma cells. Oncoimmunology 2016, 5, 1–11. [Google Scholar] [CrossRef] [Green Version]
- McKenna, E.; Traganos, F.; Zhao, H.; Darzynkiewicz, Z. Persistent DNA damage caused by low levels of mitomycin C induces irreversible cell senescence. Cell Cycle 2012, 11, 3132–3140. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Wang, W.; Dong, H.; Li, Y.; Li, L.I.; Han, L.; Han, Z.; Wang, S.; Ma, D.; Wang, H. Cisplatin-induced senescence in ovarian cancer cells is mediated by GRP78. Oncol. Rep. 2014, 31, 2525–2534. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, K.; Rahman, M.; Rahman, M.T.; Nakamura, K.; Sato, E.; Katagiri, H.; Ishibashi, T.; Ishikawa, M.; Iida, K.; Razia, S.; et al. Nucleus accumbens-1/GADD45GIP1 axis mediates cisplatin resistance through cellular senescence in ovarian cancer. Oncol. Lett. 2017, 13, 4713–4719. [Google Scholar] [CrossRef] [Green Version]
- Qu, K.; Lin, T.; Wang, Z.; Liu, S.; Chang, H.; Xu, X.; Meng, F.; Zhou, L.; Wei, J.; Tai, M.; et al. Reactive oxygen species generation is essential for cisplatin induced accelerated senescence in hepatocellular carcinoma. Front. Med. China 2014, 8, 227–235. [Google Scholar] [CrossRef]
- Roberson, R.S.; Kussick, S.J.; Vallieres, E.; Chen, S.J.; Wu, D.Y. Escape from therapy-induced accelerated cellular senescence in p53-null lung cancer cells and in human lung cancers. Cancer Res. 2005, 65, 2795–2803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seignez, C.; Martin, A.; Rollet, C.E.; Racoeur, C.; Scagliarini, A.; Jeannin, J.F.; Bettaieb, A.; Paul, C. Senescence of tumor cells induced by oxaliplatin increases the efficiency of a lipid A immunotherapy via the recruitment of neutrophils. Oncotarget 2014, 5, 11442–11451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, K.; Xu, X.; Liu, C.; Wu, Q.; Wei, J.; Meng, F.; Zhou, L.; Wang, Z.; Lei, L.; Liu, P. Negative regulation of transcription factor FoxM1 by p53 enhances oxaliplatin-induced senescence in hepatocellular carcinoma. Cancer Lett. 2013, 331, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Dabrowska, M.; Skoneczny, M.; Uram, L.; Rode, W. Methotrexate-induced senescence of human colon cancer cells depends on p53 acetylation, but not genomic aberrations. Anticancer Drugs 2019, 30, 374–382. [Google Scholar] [CrossRef]
- Dabrowska, M.; Skoneczny, M.; Rode, W. Functional gene expression profile underlying methotrexate-induced senescence in human colon cancer cells. Tumor Biol. 2011, 32, 965–976. [Google Scholar] [CrossRef] [Green Version]
- Hattangadi, D.K.; DeMasters, G.A.; Walker, T.D.; Jones, K.R.; Di, X.; Newsham, I.F.; Gewirtz, D.A. Influence of p53 and caspase 3 activity on cell death and senescence in response to methotrexate in the breast tumor cell. Biochem. Pharmacol. 2004, 68, 1699–1708. [Google Scholar] [CrossRef]
- Ge, H.; Ke, J.; Xu, N.; Li, H.; Gong, J.; Li, X.; Song, Y.; Zhu, H.; Bai, C. Dexamethasone alleviates pemetrexed-induced senescence in Non-Small-Cell Lung Cancer. Food Chem. Toxicol. 2018, 119, 86–97. [Google Scholar] [CrossRef]
- Tanino, R.; Tsubata, Y.; Harashima, N.; Harada, M.; Isobe, T. Novel drug-resistance mechanisms of pemetrexed-treated nonsmall cell lung cancer. Oncotarget 2018, 9, 16807–16821. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Baba, T.; Mukaida, N. Gemcitabine induces cell senescence in human pancreatic cancer cell lines. Biochem. Biophys. Res. Commun. 2016, 477, 515–519. [Google Scholar] [CrossRef] [Green Version]
- Modrak, D.E.; Leon, E.; Goldenberg, D.M.; Gold, D.V. Ceramide regulates gemcitabine-induced senescence and apoptosis in human pancreatic cancer cell lines. Mol. Cancer Res. 2009, 7, 890–896. [Google Scholar] [CrossRef] [Green Version]
- Putri, J.F.; Widodo, N.; Sakamoto, K.; Kaul, S.C.; Wadhwa, R. Induction of senescence in cancer cells by 5′-Aza-2′-deoxycytidine: Bioinformatics and experimental insights to its targets. Comput. Biol. Chem. 2017, 70, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Zhang, L.; Lin, J.; Huang, H.; Shi, B.; Lin, X.; Huang, Z.; Wang, C.; Qiu, J.; Wei, X. Hypermethylation of the HIC1 promoter and aberrant expression of HIC1/SIRT1 contribute to the development of thyroid papillary carcinoma. Oncotarget 2016, 7, 84416–84427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moolmuang, B.; Singhirunnusorn, P.; Ruchirawat, M. Effects of 5-aza-2′-deoxycytidine, bromodeoxyuridine, interferons and hydrogen peroxide on cellular senescence in cholangiocarcinoma cells. Asian Pac. J. Cancer Prev. 2016, 17, 957–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bu, X.; Le, C.; Jia, F.; Guo, X.; Zhang, L.; Zhang, B.; Wu, M.; Wei, L. Synergistic effect of mTOR inhibitor rapamycin and fluorouracil in inducing apoptosis and cell senescence in hepatocarcinoma cells. Cancer Biol. Ther. 2008, 7, 392–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milczarek, M.; Wiktorska, K.; Mielczarek, L.; Koronkiewicz, M.; Dąbrowska, A.; Lubelska, K.; Matosiuk, D.; Chilmonczyk, Z. Autophagic cell death and premature senescence: New mechanism of 5-fluorouracil and sulforaphane synergistic anticancer effect in MDA-MB-231 triple negative breast cancer cell line. Food Chem. Toxicol. 2018, 111, 1–8. [Google Scholar] [CrossRef]
- Drullion, C.; Lagarde, V.; Gioia, R.; Legembre, P.; Priault, M.; Cardinaud, B.; Lippert, E.; Mahon, F.-X.; Pasquet, J.-M. Mycophenolic Acid Overcomes Imatinib and Nilotinib Resistance of Chronic Myeloid Leukemia Cells by Apoptosis or a Senescent-Like Cell Cycle Arrest. Leuk. Res. Treat. 2012, 2012, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Narath, R.; Ambros, I.M.; Kowalska, A.; Bozsaky, E.; Boukamp, P.; Ambros, P.F. Induction of senescence in MYCN amplified neuroblastoma cell lines by hydroxyurea. Genes Chromosomes Cancer 2007, 46, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Robles, S.J.; Adami, G.R. Agents that cause DNA double strand breaks lead to p16INK4a enrichment and the premature senescence of normal fibrolasts. Oncogene 1998, 16, 1113–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khongkow, P.; Gomes, A.R.; Gong, C.; Man, E.P.S.; Tsang, J.W.H.; Zhao, F.; Monteiro, L.J.; Coombes, R.C.; Medema, R.H.; Khoo, U.S.; et al. Paclitaxel targets FOXM1 to regulate KIF20A in mitotic catastrophe and breast cancer paclitaxel resistance. Oncogene 2016, 35, 990–1002. [Google Scholar] [CrossRef] [Green Version]
- Di Mitri, D.; Toso, A.; Chen, J.J.; Sarti, M.; Pinton, S.; Jost, T.R.; D’Antuono, R.; Montani, E.; Garcia-Escudero, R.; Guccini, I.; et al. Tumour-infiltrating Gr-1 + myeloid cells antagonize senescence in cancer. Nature 2014, 515, 134–137. [Google Scholar] [CrossRef]
- Groth-Pedersen, L.; Ostenfeld, M.S.; Høyer-Hansen, M.; Nylandsted, J.; Jäättelä, M. Vincristine induces dramatic lysosomal changes and sensitizes cancer cells to lysosome-destabilizing siramesine. Cancer Res. 2007, 67, 2217–2225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.H.; Kang, B.S.; Bae, Y.S. Premature senescence in human breast cancer and colon cancer cells by tamoxifen-mediated reactive oxygen species generation. Life Sci. 2014, 97, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, R.; Miller, K.E.; Maiorano, J.N.; Termuhlen, P.M.; Gao, Y.; Steven, J. Berberich Novel senescence associated gene, YPEL3, is repressed by estrogen in ER+ mammary tumor cells and required for tamoxifen-induced cellular senescence. Int. J. Cancer 2012, 130, 2291–2299. [Google Scholar] [CrossRef]
- Dolfi, S.C.; Jäger, A.V.; Medina, D.J.; Haffty, B.G.; Yang, J.M.; Hirshfield, K.M. Fulvestrant treatment alters MDM2 protein turnover and sensitivity of human breast carcinoma cells to chemotherapeutic drugs. Cancer Lett. 2014, 350, 52–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.; Pepowski, B.; Takahashi, S.; Kron, S.J. A cmap-enabled gene expression signature-matching approach identifies small-molecule inducers of accelerated cell senescence. BMC Genom. 2019, 20, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burton, D.G.A.; Giribaldi, M.G.; Munoz, A.; Halvorsen, K.; Patel, A.; Jorda, M.; Perez-Stable, C.; Rai, P. Androgen Deprivation-Induced Senescence Promotes Outgrowth of Androgen-Refractory Prostate Cancer Cells. PLoS ONE 2013, 8, e68003. [Google Scholar] [CrossRef]
- Kawata, H.; Kamiakito, T.; Nakaya, T.; Komatsubara, M. Stimulation of cellular senescent processes, including secretory phenotypes and anti-oxidant responses, after androgen deprivation therapy in human prostate cancer. J. Steroid Biochem. Mol. Biol. 2017, 165, 219–227. [Google Scholar] [CrossRef]
- Barakat, D.J.; Zhang, J.; Barberi, T.; Denmeade, S.R.; Friedman, A.D. CCAAT/Enhancer binding protein β controls androgen-deprivation-induced senescence in prostate cancer cells. Oncogene 2015, 34, 5912–5922. [Google Scholar] [CrossRef] [Green Version]
- Ewald, J.A.; Joshua, A.D.; Church, D.R.; Yang, B.; Hyang, W.; Laurila, T.A.; Jarrard, D.F. Androgen Deprivation Induces Senescence Characteristics in Prostate Cancer Cells In vitro and In vivo. Prostate 2016, 73, 337–345. [Google Scholar] [CrossRef] [Green Version]
- Drullion, C.; Trégoat, C.; Lagarde, V.; Tan, S.; Gioia, R.; Priault, M.; Djavaheri-Mergny, M.; Brisson, A.; Auberger, P.; Mahon, F.X.; et al. Apoptosis and autophagy have opposite roles on imatinib-induced K562 leukemia cell senescence. Cell Death Dis. 2012, 3, e373. [Google Scholar] [CrossRef]
- Sun, R.; Bao, M.Y.; Long, X.; Yuan, Y.; Wu, M.M.; Li, X.; Bao, J.K. Metabolic gene NR4A1 as a potential therapeutic target for non-smoking female non-small cell lung cancer patients. Thorac. Cancer 2019, 10, 715–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartman, M.L.; Sztiller-Sikorska, M.; Czyz, M. Whole-exome sequencing reveals novel genetic variants associated with diverse phenotypes of melanoma cells. Mol. Carcinog. 2019, 58, 588–602. [Google Scholar] [CrossRef] [PubMed]
- Ruscetti, M.; Leibold, J.; Bott, M.J.; Fennell, M.; Kulick, A.; Salgado, N.R.; Chen, C.C.; Ho, Y.; Sanchez-Rivera, F.J.; Feucht, J.; et al. NK cell–mediated cytotoxicity contributes to tumor control by a cytostatic drug combination. Science 2018, 362, 1416–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Li, N.; Jiang, W.; Deng, W.; Ye, R.; Xu, C.; Qiao, Y.; Sharma, A.; Zhang, M.; Hung, M.C.; et al. Mutant LKB1 confers enhanced radiosensitization in combination with trametinib in KRAS-mutant non–small cell lung cancer. Clin. Cancer Res. 2018, 24, 5744–5756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krayem, M.; Najem, A.; Journe, F.; Morandini, R.; Sales, F.; Awada, A.; Ghanem, G.E. Acquired resistance to BRAFi reverses senescence-like phenotype in mutant BRAF melanoma. Oncotarget 2018, 9, 31888–31903. [Google Scholar] [CrossRef]
- Haferkamp, S.; Borst, A.; Adam, C.; Becker, T.M.; Motschenbacher, S.; Windhövel, S.; Hufnagel, A.L.; Houben, R.; Meierjohann, S. Vemurafenib induces senescence features in melanoma cells. J. Investig. Dermatol. 2013, 133, 1601–1609. [Google Scholar] [CrossRef] [Green Version]
- Sen, B.; Peng, S.; Tang, X.; Erickson, H.S.; Galindo, H.; Mazumdar, T.; Stewart, D.J.; Wistuba, I.; Johnson, F.M. Kinase Impaired BRAF Mutations Confer Lung Cancer Sensitivity to Dasatinib. Sci. Transl. Med. 2013, 4, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Peng, S.; Sen, B.; Mazumdar, T.; Byers, L.A.; Diao, L.; Wang, J.; Tong, P.; Giri, U.; Heymach, J.V.; Kadara, H.N.; et al. Dasatinib induces DNA damage and activates DNA repair pathways leading to senescence in non-small cell lung cancer cell lines with kinase-inactivating BRAF mutations. Oncotarget 2016, 7, 565–579. [Google Scholar] [CrossRef] [Green Version]
- McDermott, M.S.J.; Conlon, N.; Browne, B.C.; Szabo, A.; Synnott, N.C.; O’brien, N.A.; Duffy, M.J.; Crown, J.; O’donovan, N. HER2-targeted tyrosine kinase inhibitors cause therapy-induced-senescence in breast cancer cells. Cancers 2019, 11, 197. [Google Scholar] [CrossRef] [Green Version]
- Sugita, S.; Ito, K.; Yamashiro, Y.; Moriya, S.; Che, X.-F.; Yokoyama, T.; Hiramoto, M.; Miyazawa, K. EGFR-independent autophagy induction with gefitinib and enhancement of its cytotoxic effect by targeting autophagy with clarithromycin in non-small cell lung cancer cells. Biochem. Biophys. Res. Commun. 2015, 461, 28–34. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Morsbach, F.; Sander, D.; Gheorghiu, L.; Nanda, A.; Benes, C.; Kriegs, M.; Krause, M.; Dikomey, E.; Baumann, M.; et al. EGF Receptor Inhibition Radiosensitizes NSCLC Cells By Inducing Senescence In Cells Sustaining DNA Double-Strand Breaks. Cancer Res. 2011, 71, 6261–6269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bollard, J.; Miguela, V.; Ruiz De Galarreta, M.; Venkatesh, A.; Bian, C.B.; Roberto, M.P.; Tovar, V.; Sia, D.; Molina-Sánchez, P.; Nguyen, C.B.; et al. Palbociclib (PD-0332991), a selective CDK4/6 inhibitor, restricts tumour growth in preclinical models of hepatocellular carcinoma. Gut 2017, 66, 1286–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ota, H.; Eto, M.; Ako, J.; Ogawa, S.; Iijima, K.; Akishita, M.; Ouchi, Y. Sirolimus and everolimus induce endothelial cellular senescence via sirtuin 1 down-regulation: Therapeutic implication of cilostazol after drug-eluting stent implantation. J. Am. Coll. Cardiol. 2009, 53, 2298–2305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Däbritz, J.H.M.; Yu, Y.; Milanovic, M.; Schönlein, M.; Rosenfeldt, M.T.; Dörr, J.R.; Kaufmann, A.M.; Dörken, B.; Schmitt, C.A. CD20-targeting immunotherapy promotes cellular senescence in B-cell lymphoma. Mol. Cancer Ther. 2016, 15, 1074–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosemblit, C.; Datta, J.; Lowenfeld, L.; Xu, S.; Basu, A.; Kodumudi, K.; Wiener, D.; Czerniecki, B.J. Oncodriver inhibition and CD4+ Th1 cytokines cooperate through Stat1 activation to induce tumor senescence and apoptosis in HER2+ and triple negative breast cancer: Implications for combining immune and targeted therapies. Oncotarget 2018, 9, 23058–23077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasan, M.R.; Ho, S.H.Y.; Owen, D.A.; Tai, I.T. Inhibition of VEGF induces cellular senescence in colorectal cancer cells. Int. J. Cancer 2011, 129, 2115–2123. [Google Scholar] [CrossRef] [PubMed]
- Schottler, J.; Randoll, N.; Lucius, R.; Caliebe, A.; Roider, J.; Klettner, A. Long-term treatment with anti-VEGF does not induce cell aging in primary retinal pigment epithelium. Exp. Eye Res. 2018, 171, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Michaud, K.; Solomon, D.A.; Oermann, E.; Kim, J.; Zhong, W.; Prados, M.D.; Ozawa, T.; James, C.D.; Waldman, T. Pharmacologic Inhibition of Cyclin-Dependent Kinases 4 and 6 Arrests the Growth of Glioblastoma Multiforme Intracranial Xenografts. Cancer Res. 2010, 70, 3228–3239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, L.; Ke, N.; Hydbring, P.; Choi, Y.J.; Widlund, H.R.; Chick, J.M.; Zhai, H.; Vidal, M.; Gygi, S.P.; Braun, P.; et al. A Systematic Screen for CDK4/6 Substrates Links FOXM1 Phosphorylation to Senescence Suppression in Cancer Cells. Cancer Cell 2011, 20, 620–634. [Google Scholar] [CrossRef] [Green Version]
- Kovatcheva, M.; Liu, D.D.; Dickson, M.A.; Klein, M.E.; Connor, O.; Wilder, F.O.; Socci, N.D.; Tap, W.D.; Gary, K.; Singer, S.; et al. MDM2 turnover and expression of ATRX determine the choice between quiescence and senescence in response to CDK4 inhibition. Oncotarget 2015, 6, 8226. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, A.; Lee, E.K.; Diehl, J.A. Induction of Therapeutic Senescence in Vemurafenib-Resistant Melanoma by Extended Inhibition of CDK4/6. Cancer Res. 2016, 76, 2990–3003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, X.; LaPak, K.M.; Hennessey, R.C.; Yu, C.Y.; Shakya, R.; Zhang, J.; Burd, C.E. Stromal Senescence By Prolonged CDK4/6 Inhibition Potentiates Tumor Growth. Mol. Cancer Res. 2017, 15, 237–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Espín, D.; Rovira, M.; Galiana, I.; Giménez, C.; Lozano-Torres, B.; Paez-Ribes, M.; Llanos, S.; Chaib, S.; Muñoz-Martín, M.; Ucero, A.C.; et al. A versatile drug delivery system targeting senescent cells. EMBO Mol. Med. 2018, 10, e9355. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, C.A.; Vargas, L.; Martinez, V.; Bravo, S.; Brown, N.E. Palbociclib-induced autophagy and senescence in gastric cancer cells. Exp. Cell Res. 2017, 360, 390–396. [Google Scholar] [CrossRef]
- Miettinen, T.P.; Peltier, J.; Härtlova, A.; Gierliński, M.; Jansen, V.M.; Trost, M.; Björklund, M. Thermal proteome profiling of breast cancer cells reveals proteasomal activation by CDK 4/6 inhibitor palbociclib. EMBO J. 2018, 37, 1–19. [Google Scholar] [CrossRef]
- Puyol, M.; Martín, A.; Dubus, P.; Mulero, F.; Pizcueta, P.; Khan, G.; Guerra, C.; Santamaría, D.; Barbacid, M. A Synthetic Lethal Interaction between K-Ras Oncogenes and Cdk4 Unveils a Therapeutic Strategy for Non-small Cell Lung Carcinoma. Cancer Cell 2010, 18, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Perez, M.; Muñoz-Galván, S.; Jiménez-García, M.P.; Marín, J.J.; Carnero, A. Efficacy of CDK4 inhibition against sarcomas depends on their levels of CDK4 and p16ink4 mRNA. Oncotarget 2015, 6, 40557–40574. [Google Scholar] [CrossRef] [Green Version]
- Torres-Guzmán, R.; Calsina, B.; Hermoso, A.; Baquero, C.; Alvarez, B.; Amat, J.; McNulty, A.M.; Gong, X.; Boehnke, K.; Du, J.; et al. Preclinical characterization of abemaciclib in hormone receptor positive breast cancer. Oncotarget 2017, 8, 69493–69507. [Google Scholar] [CrossRef] [Green Version]
- Iyengar, M.; Hayer, P.O.; Cole, A.; Sebastian, T.; Yang, K.; Coffman, L.; Buckanovich, R.J. CDK4/6 inhibition as maintenance and combination therapy for high grade serous ovarian cancer. Oncotarget 2018, 9, 15658–15672. [Google Scholar] [CrossRef] [Green Version]
- Alotaibi, M.; Sharma, K.; Saleh, T.; Povirk, L.F.; Hendrickson, E.A.; Gewirtz, D.A. Radiosensitization by PARP Inhibition in DNA Repair Proficient and Deficient Tumor Cells: Proliferative Recovery in Senescent Cells. Radiat. Res. 2016, 185, 229–245. [Google Scholar] [CrossRef] [Green Version]
- Fleury, H.; Malaquin, N.; Tu, V.; Gilbert, S.; Martinez, A.; Olivier, M.A.; Sauriol, A.; Communal, L.; Leclerc-Desaulniers, K.; Carmona, E.; et al. Exploiting interconnected synthetic lethal interactions between PARP inhibition and cancer cell reversible senescence. Nat. Commun. 2019, 10, 2556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, P.; Choudhary, G.S.; Sharma, A.; Singh, K.; Heston, W.D.; Ciezki, J.; Klein, E.A.; Almasan, A. PARP Inhibition Sensitizes to Low Dose-Rate Radiation TMPRSS2-ERG Fusion Gene-Expressing and PTEN-Deficient Prostate Cancer Cells. PLoS ONE 2013, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Morelli, M.B.; Amantini, C.; Nabissi, M.; Cardinali, C.; Santoni, M.; Bernardini, G.; Santoni, A.; Santoni, G. Axitinib induces senescence-associated cell death and necrosis in glioma cell lines: The proteasome inhibitor, bortezomib, potentiates axitinib-induced cytotoxicity in a p21(Waf/Cip1) dependent manner. Oncotarget 2017, 8, 3380–3395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrova, N.V.; Velichko, A.K.; Razin, S.V.; Kantidze, O.L. Small molecule compounds that induce cellular senescence. Aging Cell 2016, 15, 999–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weichhart, T. MTOR as Regulator of Lifespan, Aging, and Cellular Senescence: A Mini-Review. Gerontology 2018, 64, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Herranz, N.; Gallage, S.; Mellone, M.; Wuestefeld, T.; Klotz, S.; Hanley, C.J.; Raguz, S.; Acosta, J.C.; Innes, A.J.; Banito, A.; et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat. Cell Biol. 2015, 17, 1205–1217. [Google Scholar] [CrossRef] [Green Version]
- Apelo, S.I.A.; Pumper, C.P.; Baar, E.L.; Cummings, N.E.; Lamming, D.W. Intermittent administration of rapamycin extends the life span of female C57BL/6J Mice. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2016, 71, 876–881. [Google Scholar] [CrossRef]
- Wang, R.; Yu, Z.; Sunchu, B.; Shoaf, J.; Dang, I.; Zhao, S.; Caples, K.; Beaver, L.M.; Ho, E.; Christiane, V.L.; et al. Rapamycin inhibits the secretory phenotype of senescent cells by a Nrf2-independent mechanism. Aging Cell 2017, 16, 564–574. [Google Scholar] [CrossRef]
- Ghosh, H.S.; Mcburney, M.; Robbins, P.D. SIRT1 Negatively Regulates the Mammalian Target of Rapamycin. PloS ONE 2010, 5, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Huck, J.J.; Zhang, M.; Mcdonald, A.; Bowman, D.; Hoar, K.M.; Stringer, B.; Ecsedy, J.; Manfredi, M.G.; Hyer, M.L. MLN8054, an Inhibitor of Aurora A Kinase, Induces Senescence in Human Tumor Cells Both In vitro and In vivo. Mol. Cancer Res. 2010, 8, 373–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Hawkins, O.E.; Su, Y.; Vilgelm, A.E.; Sobolik, T.; Thu, Y.; Kantrow, S.; Splittgerber, R.C.; Short, S.; Amiri, K.I.; et al. Targeting aurora kinases limits tumour growth through DNA damage-mediated senescence and blockade of NF-kB impairs this drug-induced senescence. EMBO Mol. Med. 2012, 5, 149–166. [Google Scholar] [CrossRef] [PubMed]
- Cullinane, C.; Waldeck, K.L.; Binns, D.; Bogatyreva, E.; Bradley, D.P.; de Jong, R.; McArthur, G.A.; Hicks, R.J. Preclinical FLT-PET and FDG-PET imaging of tumor response to the multi-targeted Aurora B kinase inhibitor, TAK-901. Nucl. Med. Biol. 2014, 41, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Paller, C.J.; Wissing, M.D.; Mendonca, J.; Sharma, A.; Kim, E.; Kim, H.S.; Kortenhorst, M.S.Q.; Gerber, S.; Rosen, M.; Shaikh, F.; et al. Combining the pan-aurora kinase inhibitor AMG 900 with histone deacetylase inhibitors enhances antitumor activity in prostate cancer. Cancer Med. 2014, 3, 1322–1335. [Google Scholar] [CrossRef] [PubMed]
- Vilgelm, A.E.; Johnson, C.A.; Prasad, N.; Yang, J.; Chen, S.; Ayers, G.D.; Pawlikowski, J.S.; Raman, D.; Sosman, J.A.; Kelley, M.; et al. Connecting the Dots: Therapy-Induced Senescence and a Tumor-Suppressive Immune Microenvironment. J. Natl. Cancer Inst. 2016, 108, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Leite de Oliveira, R.; Wang, C.; Fernandes Neto, J.M.; Mainardi, S.; Evers, B.; Lieftink, C.; Morris, B.; Jochems, F.; Willemsen, L.; et al. High-Throughput Functional Genetic and Compound Screens Identify Targets for Senescence Induction in Cancer. Cell Rep. 2017, 21, 773–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borchert, S.; Wessolly, M.; Schmeller, J.; Mairinger, E.; Kollmeier, J.; Hager, T.; Mairinger, T.; Herold, T.; Christoph, D.C.; Walter, R.F.H.; et al. Gene expression profiling of homologous recombination repair pathway indicates susceptibility for olaparib treatment in malignant pleural mesothelioma in vitro. BMC Cancer 2019, 19, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gewirtz, D.A.; Alotaibi, M.; Yakovlev, V.A.; Povirk, L.F. Tumor Cell Recovery from Senescence Induced by Radiation with PARP Inhibition. Radiat. Res. 2016, 186, 327–332. [Google Scholar] [CrossRef] [Green Version]
- Esposito, M.T.; Zhao, L.; Fung, T.K.; Rane, J.K.; Wilson, A.; Martin, N.; Gil, J.; Leung, A.Y.; Ashworth, A.; Eric So, C.W. Synthetic lethal targeting of oncogenic transcription factors in acute leukemia by PARP inhibitors. Nat. Med. 2015, 21, 1481–1490. [Google Scholar] [CrossRef]
- Sabisz, M.; Skladanowski, A.; Sabisz, M.; Skladanowski, A. Cancer stem cells and escape from drug-induced premature senescence in human lung tumor cells: Implications for drug resistance and in vitro drug screening models. Cell Cycle 2009, 8, 3208–3217. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Wu, P.C.; Dong, D.Z.; Ivanova, I.; Chu, E.; Zeliadt, S.; Vesselle, H.; Wu, D.Y. Polyploidy road to therapy-induced cellular senescence and escape. Int. J. Cancer 2013, 132, 1505–1515. [Google Scholar] [CrossRef]
- Achuthan, S.; Santhoshkumar, T.R.; Prabhakar, J.; Nair, S.A.; Pillai, M.R. Drug-induced senescence generates chemoresistant stemlike cells with low reactive oxygen species. J. Biol. Chem. 2011, 286, 37813–37829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Fang, J.; Chen, J. Tumor cell senescence response produces aggressive variants. Cell Death Discov. 2017, 3, 17049. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; de Jong, T.V.; Melov, S.; Guryev, V.; Campisi, J.; Demaria, M. Unmasking Transcriptional Heterogeneity in Senescent Cells. Curr. Biol. 2017, 27, 2652–2660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saleh, T.; Tyutynuk-Massey, L.; Cudjoe, E.K.E.K.; Idowu, M.O.M.O.; Landry, J.W.J.W.; Gewirtz, D.A.D.A.; Tyutyunyk-Massey, L.; Cudjoe, E.K.E.K.; Idowu, M.O.M.O.; Landry, J.W.J.W.; et al. Non-Cell Autonomous Effects of the Senescence-Associated Secretory Phenotype in Cancer Therapy. Front. Oncol. 2018, 8, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Elmore, L.W.; Di, X.; Dumur, C.; Holt, S.E.; Gewirtz, D.A. Evasion of a Single-Step, Chemotherapy-Induced Senescence in Breast Cancer Cells: Implications for Treatment Response. Cancer Ther. Clin. 2005, 11, 2637–2643. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Wu, P.C.; Roberson, R.S.; Luk, B.V.; Ivanova, I.; Chu, E.; Wu, D.Y. Survivin and escaping in therapy-induced cellular senescence. Int. J. Cancer 2011, 128, 1546–1558. [Google Scholar] [CrossRef] [Green Version]
- Was, H.; Barszcz, K.; Czarnecka, J.; Kowalczyk, A.; Uzarowska, E.; Koza, P.; Klejman, A.; Kaminska, B.; Sikora, E. Bafilomycin A1 triggers proliferative potential of senescent cancer cells in vitro and in NOD/SCID mice. Oncotarget 2017, 8, 9303–9322. [Google Scholar] [CrossRef] [Green Version]
- Rajaraman, R.; Guensey, D.L.; Rajaraman, M.M.; Rajaraman, S.R. Stem cells, senescence, neosis and self-renewal in cancer. Cancer Cell Int. 2006, 8, 26. [Google Scholar]
- Erenpreisa, J.; Cragg, M.S. Three steps to the immortality of cancer cells: Senescence, polyploidy and self-renewal. Cancer Cell Int. 2013, 13, 92. [Google Scholar] [CrossRef] [Green Version]
- Puig, P.-E.E.; Guilly, M.-N.N.; Bouchot, A.A.; Droin, N.; Cathelin, D.; Bouyer, F.; Favier, L.; Ghiringhelli, F.F.; Kroemer, G.; Solary, E.; et al. Tumor cells can escape DNA-damaging cisplatin through DNA endoreduplication and reversible polyploidy. Cell Biol. Int. 2008, 32, 1031–1043. [Google Scholar] [CrossRef] [PubMed]
- Rohnalter, V.; Roth, K.; Finkernagel, F.; Adhikary, T.; Obert, J.; Dorzweiler, K.; Bensberg, M.; Müller-brüsselbach, S.; Müller, R. A multi-stage process including transient polyploidization and EMT precedes the emergence of chemoresistent ovarian carcinoma cells with a dedifferentiated and pro-inflammatory secretory phenotype. Oncotarget 2015, 6, 40005–40025. [Google Scholar] [CrossRef] [PubMed]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonnessen-Murray, C.A.; Frey, W.D.; Rao, S.G.; Shahbandi, A.; Ungerleider, N.A.; Olayiwola, J.O.; Murray, L.B.; Vinson, B.T.; Chrisey, D.B.; Lord, C.J.; et al. Chemotherapy-induced senescent cancer cells engulf other cells to enhance their survival. J. Cell Biol. 2019, 218, 3827–3844. [Google Scholar] [CrossRef] [Green Version]
- Haupt, S.; Keam, S.P.; Haupt, Y. Cannibalism in Breast Cancer: The Dangers of Overeating. Trends Cancer 2019, 5, 761–762. [Google Scholar] [CrossRef]
- Jones, K.R.; Elmore, L.W.; Jackson-Cook, C.; Demasters, G.; Povirk, L.F.; Holt, S.E.; Gewirtz, D.A. p53-Dependent accelerated senescence induced by ionizing radiation in breast tumour cells. Int. J. Radiat. Biol. 2005, 81, 445–458. [Google Scholar] [CrossRef]
- Chitikova, Z.V.; Gordeev, S.A.; Bykova, T.V.; Svetlana, G.; Pospelov, V.A.; Pospelova, T.V. Sustained activation of DNA damage response in irradiated apoptosis-resistant cells induces downregulation and expression of stem cell markers. Cell Cycle 2014, 13, 1424–1439. [Google Scholar] [CrossRef] [Green Version]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Ewald, J.A.; Desotelle, J.A.; Almassi, N.; Jarrard, D.F. Drug-induced senescence bystander proliferation in prostate cancer cells in vitro and in vivo. Br. J. Cancer 2008, 98, 1244–1249. [Google Scholar] [CrossRef]
- Ortiz-Montero, P.; Londoño-Vallejo, A.; Vernot, J.P. Senescence-associated IL-6 and IL-8 cytokines induce a self- and cross-reinforced senescence/inflammatory milieu strengthening tumorigenic capabilities in the MCF-7 breast cancer cell line. Cell Commun. Signal. 2017, 15, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Ohuchida, K.; Mizumoto, K.; Murakami, M.; Qian, L.; Sato, N.; Nagai, E.; Matsumoto, K.; Nakamura, T.; Tanaka, M. Radiation to Stromal Fibroblasts Increases Invasiveness of Pancreatic Cancer Cells through Tumor-Stromal Interactions. Cancer Res. 2004, 64, 3215–3222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohanna, M.; Cheli, Y.; Bonet, C.; Bonazzi, V.F.; Allegra, M.; Giuliano, S.; Bille, K.; Bahadoran, P.; Giacchero, D.; Lacour, J.P.; et al. Secretome from senescent melanoma engages the STAT3 pathway to favor reprogramming of naive melanoma towards a tumor-initiating cell phenotype. Oncotarget 2013, 4, 2212–2224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oubaha, M.; Miloudi, K.; Dejda, A.; Guber, V.; Mawambo, G.; Germain, M.; Bourdel, G.; Popovic, N.; Rezende, F.A.; Kaufman, R.J.; et al. Senescence-associated secretory phenotype contributes to pathological angiogenesis in retinopathy. Sci. Transl. Med. 2016, 8, 362ra144. [Google Scholar] [CrossRef] [PubMed]
- Sagiv, A.; Krizhanovsky, V. Immunosurveillance of senescent cells: The bright side of the senescence program. Biogerontology 2013, 14, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Toso, A.; Revandkar, A.; Catapano, C.V.; Alimonti, A.; Toso, A.; Revandkar, A.; Mitri, D.; Guccini, I.; Proietti, M.; Sarti, M.; et al. Enhancing Chemotherapy Efficacy in Pten -Deficient Prostate Tumors by Activating the Senescence- Associated Antitumor Immunity Article Enhancing Chemotherapy Efficacy in Pten -Deficient Prostate Tumors by Activating the Senescence-Associated Antitumor Imm. Cell Rep. 2014, 9, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.; Khan, A.A.; Yin, J.; Ferguson, T.A.; Apte, R.S. Senescence regulates macrophage activation and angiogenic fate at sites of tissue injury in mice. J. Clin. Investig. 2007, 117, 3421–3426. [Google Scholar] [CrossRef]
- Milanovic, M.; Yu, Y.; Schmitt, C.A. The Senescence–Stemness Alliance–A Cancer-Hijacked Regeneration Principle. Trends Cell Biol. 2018, 28, 1049–1061. [Google Scholar] [CrossRef]
- Soto-Gamez, A.; Quax, W.J.; Demaria, M. Regulation of Survival Networks in Senescent Cells: From Mechanisms to Interventions. J. Mol. Biol. 2019, 431, 2629–2643. [Google Scholar] [CrossRef]
- Marcotte, R.; Lacelle, C.; Wang, E. Senescent fibroblasts resist apoptosis by downregulating caspase-3. Mech. Ageing Dev. 2004, 125, 777–783. [Google Scholar] [CrossRef]
- Yeo, E.J.; Hwang, Y.C.; Kang, C.M.; Choy, H.E.; Park, S.C. Reduction of UV-induced cell death in the human senescent fibroblasts. Mol. Cells 2000, 10, 415–422. [Google Scholar]
- da Silva, P.F.L.; Ogrodnik, M.; Kucheryavenko, O.; Glibert, J.; Miwa, S.; Cameron, K.; Ishaq, A.; Saretzki, G.; Nagaraja-Grellscheid, S.; Nelson, G.; et al. The bystander effect contributes to the accumulation of senescent cells in vivo. Aging Cell 2019, 18, e12848. [Google Scholar] [CrossRef] [PubMed]
- Nelson, G.; Wordsworth, J.; Wang, C.; Jurk, D.; Lawless, C.; Martin-Ruiz, C.; von Zglinicki, T. A senescent cell bystander effect: Senescence-induced senescence. Aging Cell 2012, 11, 345–349. [Google Scholar] [CrossRef] [Green Version]
- Di, X.; Bright, A.T.; Bellott, R.; Gaskins, E.; Holt, S.; Gewirtz, D.A.; Elmore, L.W. A chemotherapy-associated senescence bystander effect in breast cancer cells. Cancer Biol. Ther. 2008, 7, 864–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkland, J.L.; Tchkonia, T. Clinical strategies and animal models for developing senolytic agents. Exp. Gerontol. 2015, 68, 19–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; Lebrasseur, N.K.; Childs, B.G.; Sluis, B.; Kirkland, J.L.; Deursen, J.M. Van Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Hara, S.P.O.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, M.; Korfei, M.; Mutze, K.; Klee, S.; Skronska-Wasek, W.; Alsafadi, H.N.; Ota, C.; Costa, R.; Schiller, H.B.; Lindner, M.; et al. Senolytic drugs target alveolar epithelial cell function and attenuate experimental lung fibrosis ex vivo. Eur. Respir. J. 2017, 50. [Google Scholar] [CrossRef] [Green Version]
- Roos, C.M.; Zhang, B.; Palmer, A.K.; Ogrodnik, M.B.; Pirtskhalava, T.; Thalji, N.M.; Hagler, M.; Jurk, D.; Smith, L.A.; Casaclang-Verzosa, G.; et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell 2016, 15, 973–977. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Miwa, S.; Tchkonia, T.; Tiniakos, D.; Wilson, C.L.; Lahat, A.; Day, C.P.; Burt, A.; Palmer, A.; Anstee, Q.M.; et al. Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun. 2017, 8, 15691. [Google Scholar] [CrossRef]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Allyson, K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Christine, M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef]
- Nath, K.A.; Brien, D.R.O.; Croatt, A.J.; Grande, J.P.; Ackerman, A.W.; Nath, M.C.; Yamada, S.; Terzic, A.; Tchkonia, T.; Kirkland, J.L.; et al. The murine dialysis fistula model exhibits a senescence phenotype: Pathobiological mechanisms and therapeutic potential. Am. J. Physiol. Ren. Physiol. 2018, 315, F1493–F1499. [Google Scholar] [CrossRef] [PubMed]
- Musi, N.; Valentine, J.M.; Sickora, K.R.; Baeuerle, E.; Thompson, C.S.; Shen, Q.; Orr, M.E. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell 2018, 17, e12840. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Kishimoto, Y.; Grammatikakis, I.; Gottimukkala, K.; Cutler, R.G.; Zhang, S.; Abdelmohsen, K.; Bohr, V.A.; Sen, J.M.; Gorospe, M.; et al. Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat. Neurosci. 2019, 22, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Cavalcante, M.B.; Saccon, T.D.; Nunes, A.D.C.; Kirkland, J.L.; Schneider, A.; Masternak, M.M. Dasatinib plus quercetin prevents uterine age-related dysfunction and fibrosis in mice. Aging 2020, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chandra, A.; Lagnado, A.B.; Farr, J.N.; Monroe, D.G.; Park, S.; Hachfeld, C.; Tchkonia, T.; Kirkland, J.L.; Khosla, S.; Passos, J.F.; et al. Targeted reduction of senescent cell burden alleviates focal radiotherapy-related bone loss. J. Bone Miner. Res. 2020, jbmr.3978. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Tchkonia, T.; Fuhrmann-Stroissnigg, H.; Dai, H.M.; Ling, Y.Y.; Stout, M.B.; Pirtskhalava, T.; Giorgadze, N.; Johnson, K.O.; Giles, C.B.; et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 2016, 15, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Li, D.; Xu, Y.; Zhang, J.; Wang, Y.; Chen, M.; Lin, S.; Huang, L.; Chung, E.J.; Citrin, D.E.; et al. Inhibition of Bcl-2/xl With ABT-263 Selectively Kills Senescent Type II Pneumocytes and Reverses Persistent Pulmonary Fibrosis Induced by Ionizing Radiation in Mice. Int. J. Radiat. Oncol. Biol. Phys. 2017, 99, 353–361. [Google Scholar] [CrossRef]
- Mosteiro, L.; Pantoja, C.; Alcazar, N.; Marión, R.M.; Chondronasiou, D.; Rovira, M.; Fernandez-Marcos, P.J.; Muñoz-Martin, M.; Blanco-Aparicio, C.; Pastor, J.; et al. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science 2016, 354, aaf4445. [Google Scholar] [CrossRef]
- Grezella, C.; Fernandez-rebollo, E.; Franzen, J.; Sofia, M.; Ferreira, V.; Beier, F.; Wagner, W. Effects of senolytic drugs on human mesenchymal stromal cells. Stem Cell Res. Ther. 2018, 9, 108. [Google Scholar] [CrossRef] [Green Version]
- Walaszczyk, A.; Dookun, E.; Redgrave, R.; Tual-Chalot, S.; Victorelli, S.; Spyridopoulos, I.; Owens, A.; Arthur, H.M.; Passos, J.F.; Richardson, G.D. Pharmacological clearance of senescent cells improves survival and recovery in aged mice following acute myocardial infarction. Aging Cell 2019, 18, e12945. [Google Scholar] [CrossRef] [PubMed]
- Aguayo-Mazzucato, C.; Andle, J.; Lee, T.B.; Midha, A.; Talemal, L.; Chipashvili, V.; Hollister-Lock, J.; van Deursen, J.; Weir, G.; Bonner-Weir, S. Acceleration of β Cell Aging Determines Diabetes and Senolysis Improves Disease Outcomes. Cell Metab. 2019, 45, 588–605. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.N.; Chang, J.; Shao, L.; Han, L.; Iyer, S.; Manolagas, S.C.; O’Brien, C.A.; Jilka, R.L.; Zhou, D.; Almeida, M. DNA damage and senescence in osteoprogenitors expressing Osx1 may cause their decrease with age. Aging Cell 2017, 16, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Xu, X.; Yin, P.; Li, Y.; Guo, H.; Kujawa, S.; Chakravarti, D.; Bulun, S.; Kim, J.J.; Wei, J.-J. Application of ex-vivo spheroid model system for the analysis of senescence and senolytic phenotypes in uterine leiomyoma. Lab. Investig. 2018, 98, 1575–1587. [Google Scholar] [CrossRef]
- Wang, Y.; Chang, J.; Liu, X.; Zhang, X.; Zhang, S.; Zhang, X.; Zhou, D.; Zheng, G. Discovery of piperlongumine as a potential novel lead for the development of senolytic agents. Aging 2016, 8, 2915–2926. [Google Scholar] [CrossRef] [Green Version]
- Yabluchanskiy, A.; Tarantini, S.; Balasubramanian, P.; Kiss, T.; Csipo, T.; Fülöp, G.A.; Lipecz, A.; Ahire, C.; DelFavero, J.; Nyul-Toth, A.; et al. Pharmacological or genetic depletion of senescent astrocytes prevents whole brain irradiation–induced impairment of neurovascular coupling responses protecting cognitive function in mice. GeroScience 2020, 1–20. [Google Scholar] [CrossRef]
- Cherif, H.; Bisson, D.; Jarzem, P.; Weber, M.; Ouellet, J.; Haglund, L. Curcumin and o-Vanillin Exhibit Evidence of Senolytic Activity in Human IVD Cells In Vitro. J. Clin. Med. 2019, 8, 433. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; He, Y.; Zhang, R.; Zheng, G.; Zhou, D. The curcumin analog EF24 is a novel senolytic agent. Aging 2019, 11, 771–782. [Google Scholar] [CrossRef]
- Yousefzadeh, M.J.; Zhu, Y.; McGowan, S.J.; Angelini, L.; Fuhrmann-Stroissnigg, H.; Xu, M.; Ling, Y.Y.; Melos, K.I.; Pirtskhalava, T.; Inman, C.L.; et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 2018, 36, 18–28. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Doornebal, E.J.; Pirtskhalava, T.; Giorgadze, N.; Wentworth, M.; Fuhrmann-Stroissnigg, H.; Niedernhofer, L.J.; Robbins, P.D.; Tchkonia, T.; Kirkland, J.L. New agents that target senescent cells: The flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging 2017, 9, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Śmieszek, A.; Stręk, Z.; Kornicka, K.; Grzesiak, J.; Weiss, C.; Marycz, K. Antioxidant and Anti-Senescence Effect of Metformin on Mouse Olfactory Ensheathing Cells (mOECs) May Be Associated with Increased Brain-Derived Neurotrophic Factor Levels—An Ex Vivo Study. Int. J. Mol. Sci. 2017, 18, 872. [Google Scholar] [CrossRef]
- Samaraweera, L.; Adomako, A.; Rodriguez-Gabin, A.; McDaid, H.M. A Novel Indication for Panobinostat as a Senolytic Drug in NSCLC and HNSCC. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Fuhrmann-Stroissnigg, H.; Ling, Y.Y.; Zhao, J.; McGowan, S.J.; Zhu, Y.; Brooks, R.W.; Grassi, D.; Gregg, S.Q.; Stripay, J.L.; Dorronsoro, A.; et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat. Commun. 2017, 8, 422. [Google Scholar] [CrossRef] [PubMed]
- Kucheryavenko, O.; Nelson, G.; von Zglinicki, T.; Korolchuk, V.I.; Carroll, B. The mTORC1-autophagy pathway is a target for senescent cell elimination. Biogerontology 2019, 20, 331–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, R.; Sharma, A.; Kumari, A.; Gulati, A.; Yogendra, P.; Sharma, R. Epigallocatechin gallate suppresses premature senescence of preadipocytes by inhibition of PI3K/Akt/mTOR pathway and induces senescent cell death by regulation of Bax/Bcl-2 pathway. Biogerontology 2018, 20, 171–189. [Google Scholar] [CrossRef] [PubMed]
- Ozsvari, B.; Nuttall, J.R.; Sotgia, F.; Lisanti, M.P. Azithromycin and Roxithromycin define a new family of “senolytic” drugs that target senescent human fibroblasts. Aging 2018, 10, 3294–3307. [Google Scholar] [CrossRef] [PubMed]
- Nogueira-Recalde, U.; Lorenzo-Gómez, I.; Blanco, F.J.; Loza, M.I.; Grassi, D.; Shirinsky, V.; Shirinsky, I.; Lotz, M.; Robbins, P.D.; Domínguez, E.; et al. Fibrates as drugs with senolytic and autophagic activity for osteoarthritis therapy. EBioMedicine 2019, 45, 588–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Triana-Martínez, F.; Picallos-Rabina, P.; Da Silva-Álvarez, S.; Pietrocola, F.; Llanos, S.; Rodilla, V.; Soprano, E.; Pedrosa, P.; Ferreirós, A.; Barradas, M.; et al. Identification and characterization of Cardiac Glycosides as senolytic compounds. Nat. Commun. 2019, 10, 4731. [Google Scholar] [CrossRef]
- Guerrero, A.; Herranz, N.; Sun, B.; Wagner, V.; Gallage, S.; Guiho, R.; Wolter, K.; Pombo, J.; Irvine, E.E.; Innes, A.J.; et al. Cardiac glycosides are broad-spectrum senolytics. Nat. Metab. 2019, 1, 1074–1088. [Google Scholar] [CrossRef]
- Izumi-Nakaseko, H.; Fujiyoshi, M.; Hagiwara-Nagasawa, M.; Goto, A.; Chiba, K.; Kambayashi, R.; Naito, A.T.; Ando, K.; Kanda, Y.; Ishii, I.; et al. Dasatinib can Impair Left Ventricular Mechanical Function But May Lack Proarrhythmic Effect: A Proposal of Non-clinical Guidance for Predicting Clinical Cardiovascular Adverse Events of Tyrosine Kinase Inhibitors. Cardiovasc. Toxicol. 2019, 20, 58–70. [Google Scholar] [CrossRef]
- Hassan, A.; moniem, S.; Abo El-Ela, F.I.; Abdel-Aziz, A.M. Investigating the potential protective effects of natural product quercetin against imidacloprid-induced biochemical toxicity and DNA damage in adults rats. Toxicol. Rep. 2019, 6, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Elkady, M.; Shalaby, S.; Fathi, F.; El-Mandouh, S. Effects of quercetin and rosuvastatin each alone or in combination on cyclophosphamide-induced premature ovarian failure in female albino mice. Hum. Exp. Toxicol. 2019, 38, 1283–1295. [Google Scholar] [CrossRef] [PubMed]
- Short, S.; Fielder, E.; Miwa, S.; von Zglinicki, T. Senolytics and senostatics as adjuvant tumour therapy. EBioMedicine 2019, 41, 683–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hickson, L.J.; Langhi, L.G.P.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L.; et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 2019, 47, 446–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacovicova, K.; Skolnaja, M.; Heinmaa, M.; Mistrik, M.; Pata, P.; Pata, I.; Bartek, J.; Vinciguerra, M. Senolytic Cocktail Dasatinib+Quercetin (D+Q) Does Not Enhance the Efficacy of Senescence-Inducing Chemotherapy in Liver Cancer. Front. Oncol. 2018, 8, 1–7. [Google Scholar] [CrossRef]
- Montero, J.; Letai, A. Why do BCL-2 inhibitors work and where should we use them in the clinic? Nat. Publ. Group 2017, 25, 56–64. [Google Scholar] [CrossRef]
- Gayle, S.S.; Sahni, J.M.; Webb, B.M.; Weber-Bonk, K.L.; Shively, M.S.; Spina, R.; Bar, E.E.; Summers, M.K.; Keri, R.A. Targeting BCL-xL improves the efficacy of bromodomain and extra-terminal protein inhibitors in triple-negative breast cancer by eliciting the death of senescent cells. J. Biol. Chem. 2019, 294, 875–886. [Google Scholar] [CrossRef] [Green Version]
- Moiseeva, O.; Deschênes-Simard, X.; St-Germain, E.; Igelmann, S.; Huot, G.; Cadar, A.E.; Bourdeau, V.; Pollak, M.N.; Ferbeyre, G. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-κB activation. Aging Cell 2013, 12, 489–498. [Google Scholar] [CrossRef]
- Goehe, R.W.; Di, X.; Sharma, K.; Bristol, M.L.; Henderson, S.C.; Valerie, K.; Rodier, F.; Davalos, A.R.; Gewirtz, D.A. The Autophagy-Senescence Connection in Chemotherapy: Must Tumor Cells (Self) Eat Before They Sleep? J. Pharmacol. Exp. Ther. 2012, 343, 763–778. [Google Scholar] [CrossRef] [Green Version]
- Gewirtz, D.A. Autophagy and senescence: A partnership in search of definition. Autophagy 2013, 9, 808–812. [Google Scholar] [CrossRef] [Green Version]
- Tai, H.; Wang, Z.; Gong, H.; Han, X.; Zhou, J.; Wang, X.; Wei, X.; Ding, Y.; Huang, N.; Qin, J.; et al. Autophagy impairment with lysosomal and mitochondrial dysfunction is an important characteristic of oxidative stress-induced senescence. Autophagy 2017, 13, 99–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, C.; Elledge, S.J. How autophagy both activates and inhibits cellular senescence. Autophagy 2016, 12, 898–899. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Goldstein, L.A.; Hou, W.; Chatterjee, S.; Burns, T.F.; Rabinowich, H. HSP90 inhibition targets autophagy and induces a CASP9-dependent resistance mechanism in NSCLC. Autophagy 2018, 14, 958–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manuscript, A.; With, P.; Failure, H. Therapeutic Ranges of Serum Digoxin Concentrations in Patients with Heart Failure. Am. J. Cardiol. 2012, 109, 1818–1821. [Google Scholar]

{kind=link}
| Drug Class | Drug Name | Model/Cell Line | Senescence Marker | Reference |
|---|---|---|---|---|
| Topoisomerase poisons/inhibitors | Doxorubicin (Adriamycin) | MCF-7, MDA-MB231 | p53, SA-β-gal | [35] |
| H460, A549 | SA-β-gal, p21Cip1, p16INK4, p53 | [36] | ||
| HCT116, HT1080 | Morphology, growth arrest, SA-β-gal | [37] | ||
| LS174T, A2780, MCF-7, patient breast cancer tissue samples | Morphology, growth arrest, SA-β-gal, p53, p16INK4a | [33] | ||
| MCF7, MDA-MB-231 | SA-β-gal | [38] | ||
| HCT116, MCF7 | SA-β-gal, SASP (IL-8, VEGF), p21Cip1, p53, low Ki67 | [39] | ||
| DU145, LNCaP | Morphology, growth arrest, polyploidy | [40] | ||
| K562 | SA-β-gal, SAHF | [41] | ||
| Rat-derived BMSCs and ADSCs | SA-β-gal | [42] | ||
| MDFs, HCA2, BJ, in vivo mouse model (p16-3MR) | SA-β-gal, p21Cip1, p16INK4a, SASP (IL-1α, IL-6, Mmp-3, Mmp-9, Cxcl-1, Cxcl-10 and Ccl20), reduced Lamin B1 | [28] | ||
| SH-SY-5Y | p21Cip1, low Ki67, growth arrest, SA-β-gal | [43] | ||
| HCT116 | p21Cip1, low Ki67, growth arrest, SA-β-gal, increased granularity, morphology, SASP (IL-8), γH2AX | [43] | ||
| MDA-MB-231 | p21Cip1, growth arrest, SA-β-gal, morphology, SASP (IL-6, IL-8, VEGF), γH2AX | [43] | ||
| MCF-7 | p21Cip1, low Ki67, growth arrest, SA-β-gal, increased granularity, morphology, γH2AX | [43] | ||
| A549 | p21Cip1, low Ki67, growth arrest, SA-β-gal, increased granularity, morphology, SASP (IL-6, IL-8), γH2AX | [43] | ||
| Daunorubicin | Jurkat cells | SA-β-gal, growth arrest | [44] | |
| Etoposide | HepG2, U2OS | SA-β-gal, p53, p21Cip1 | [45] | |
| IMR-90, MEFs, BJ | SA-β-gal, growth arrest, p16INK4, p21Cip1, p53 | [46] | ||
| BJ, MEFs, B16F10 | SA-β-gal, SASP (IL-6, IL-8, IL-1β) | [47] | ||
| NRK-52E | Morphology, SA-β-gal, growth arrest, p53, p21Cip1 | [48] | ||
| Follicular lymphoma 3D model | SA-β-gal | [49] | ||
| Mitoxantrone | Epithelial cells in human prostate cancer patients’ biopsies | SASP, SA-β-gal | [50] | |
| A549, WI38 | Growth arrest, SA-β-gal, yH2AX, morphology | [51] | ||
| Camptothecin | HCT116 | SA-β-gal, morphology, SAHF, reduced BrdU incorporation | [52] | |
| HCT116, RKO | SA-β-gal, morphology | [53] | ||
| HeLa, MCF7 | SA-β-gal, morphology | [54] | ||
| MNA, STA-NB-10, CLB-Ma mouse xenograft (MYCN-amplified neuroblastoma) | Reduced DNA synthesis, morphology, SA-β-gal, growth arrest, p21Cip1 | [55] | ||
| Irinotecan | SGC-7901, MKN-45 | SA-β-gal | [56] | |
| A549, HCT116 | p21Cip1, low Ki67, growth arrest, SA-β-gal, increased granularity, morphology, SASP (IL-8), γH2AX | |||
| SH-SY-5Y | p21Cip1, low Ki67, growth arrest, SA-β-gal, γH2AX | |||
| MDA-MB-231 | p21Cip1, low Ki67, growth arrest, SA-β-gal, morphology, SASP (IL-6, IL-8, VEGF), γH2AX | |||
| MCF-7 | p21Cip1, low Ki67, growth arrest, SA-β-gal, increased granularity, morphology, γH2AX | |||
| Topotecan | MNA, STA-NB-10, CLB-Ma mouse xenograft (MYCN-amplified neuroblastoma) | Reduced DNA synthesis, morphology, SA-β-gal, growth arrest, p21Cip1 | [55] | |
| Alkylating agents | Busulfan | Rat-derived BMSCs and ADSCs | SA-β-gal | [42] |
| WI38 | Growth arrest, SA-β-gal | [57] | ||
| U2OS, MG63 | SA-β-gal | [58] | ||
| WI38 | SA-β-gal | [59] | ||
| Murine hematopoietic cells | SA-β-gal, p16INK4, p19INK4 | [60] | ||
| Temozolomide | Patient derived glioma cells | Cell cycle arrest, polyploidy, morphology | [61] | |
| GL261 | SAHF (H3K9Me3), p53, Rb | [62] | ||
| LN229 | SA-β-gal, cell cycle arrest, SASP (IL-6, IL-8) | [63] | ||
| In vivo (p16-3MR) mouse model | p16INK4 | [28] | ||
| Carmustine | GL261 | SAHF (H3K9Me3), p53, Rb | [62] | |
| Dacarbazine | A375, B16F10 | SASP | [64] | |
| Cyclophosphamide | HSC-bcl2 lymphoma | SA-β-gal, p53, p16INK4 | [65] | |
| Melphalan | Multiple myeloma mouse model | SA-β-gal | [66] | |
| Mitomycin C | A549 | Growth arrest, SA-β-gal, yH2AX, morphology | [67] | |
| Platinum-based | Cisplatin | A375, B16F10, B16F10 xenografts | SASP, SA-β-gal | [64] |
| A2780 | SAHF (HP1-γ), morphology, SA-β-gal | [68] | ||
| CNE1 | Growth arrest, morphology, SA-β-gal | [30] | ||
| SKOV3, TOV-21G | Morphology, SA-β-gal | [69] | ||
| HepG2, SMMC-7721 | SA-β-gal, p53, p21Cip1, p16INK4 | [70] | ||
| Follicular lymphoma 3D model | SA-β-gal | [49] | ||
| In vivo mouse model (p16-3MR) | p16INK4 | [28] | ||
| Carboplatin | H1299, patients’ lung tumor samples | Cell cycle arrest, SA-β-gal, p16INK4, RB, downregulation of cyclin B1 and cyclin D1 | [71] | |
| Oxaliplatin | PROb, CT26 | SA-β-gal | [72] | |
| HepG2, SMMC-7721, patients’ colorectal tumor samples | SA-β-gal | [73] | ||
| Antimetabolites | Methotrexate | C85 | p53 | [74] |
| C85 | SA-β-gal | [75] | ||
| MCF-7 | SA-β-gal | [76] | ||
| Rat-derived BMSCs and ADSCs | SA-β-gal | [42] | ||
| A549 | p21Cip1, low Ki67, growth arrest, SA-β-gal, increased granularity, morphology, SASP (IL-6, IL-8), γH2AX | [43] | ||
| SH-SY-5Y | p21Cip1, growth arrest, SA-β-gal, γH2AX | [43] | ||
| HCT116 | p21Cip1, low Ki67, growth arrest, SA-β-gal, increased granularity, morphology, SASP (IL-8), γH2AX | [43] | ||
| MDA-MB-231 | p21Cip1, growth arrest, SA-β-gal, morphology, SASP (IL-6, IL-8, VEGF) | [43] | ||
| MCF-7 | p21Cip1, low Ki67, growth arrest, SA-β-gal, increased granularity, morphology, γH2AX | [43] | ||
| Pemetrexed | H1650, A549, H2228, H292, H226 and H1650, A549 xenografts | SA-β-gal, morphology, SASP (IL-6, IL-8, IL-1β and MCP-1) | [77] | |
| A549 | SASP, SA-β-gal | [78] | ||
| Gemcitabine | Miapaca-2 and Panc-1 | SA-β-gal | [79] | |
| AsPc1, Panc1 | SA-β-gal | [80] | ||
| Azacitidine | U2OS, MCF7 | SA-β-gal, p53, growth arrest | [81] | |
| TPC-1 | SA-β-gal | [82] | ||
| KKU100, HuCCA1, RMCCA1 | Morphology, SA-β-gal | [83] | ||
| DU145, LNCaP | Morphology, growth arrest, polyploidy | [40] | ||
| Bromodeoxyuridine | MNA, STA-NB-10, CLB-Ma mouse xenograft (MYCN-amplified neuroblastoma) | Reduced DNA synthesis, morphology, SA-β-gal, growth arrest, p21Cip1 | [55] | |
| KKU100, HuCCA1, RMCCA1 | Morphology, SA-β-gal | [83] | ||
| 5-Fluorouracil | SMMC-7721 | SA-β-gal | [84] | |
| MDA-MB-231 | SA-β-gal | [85] | ||
| Mycophenolic acid | K562 | SA-β-gal | [86] | |
| Hydroxyurea | STA-NB-9, STA-NB-10 MYCN amplified neuroblastoma | Morphology, increased granularity, telomere length, SA-β-gal | [87] | |
| MNA, STA-NB-10, CLB- primary neuroblastoma cells, mouse xenograft model for MYCN-amplified NB | Reduced DNA synthesis, morphology, SA-β-gal, cell cycle arrest, p21Cip1, DNA double-strand breaks | [55] | ||
| Actinomycin D | HDF-2, NHF-3 | SA-β-gal, P53, p21Cip1, p16INK4 | [88] | |
| Microtubule inhibitors/poisons | Paclitaxel | Human mesenchymal stem cells | Growth inhibition, SA-β-gal, yH2AX, morphology, SASP | [67] |
| MCF-7, MEFs | Growth arrest, morphology, SA-β-gal | [89] | ||
| A549 | p21Cip1, low Ki67, growth arrest, SA-β-gal, increased granularity, SASP (IL-6, IL-8), γH2AX | [43] | ||
| SH-SY-5Y | p21Cip1, low Ki67, growth arrest, SA-β-gal | [43] | ||
| HCT116 | p21Cip1, low Ki67, growth arrest, SA-β-gal, increased granularity, SASP (IL-8), γH2AX | [43] | ||
| MDA-MB-231 | p21Cip1, low Ki67, growth arrest, SASP (IL-6, IL-8, VEGF), γH2AX | [43] | ||
| MCF-7 | p21Cip1, low Ki67, growth arrest, SA-β-gal, increased granularity | [43] | ||
| Docetaxel | DU145, LNCaP | Morphology, growth arrest, polyploidy | [40] | |
| PTEN null prostate tumors | SASP | [90] | ||
| Vincristine | MCF-7 | Morphology, senescence-associated lysosomal changes | [91] | |
| Vinblastine | Patient derived glioma cells | Cell cycle arrest and nuclear morphometric changes | [61] | |
| Hormonal therapy | Tamoxifen | MCF-7, HCT116 | SA-β-gal, p53, p21Cip1 | [92] |
| SA-β-gal, p21Cip1 | [93] | |||
| Fulvestrant | MCF-7, T-47D | SA-β-gal | [94] | |
| MCF7 | SA-β-gal, morphology | [95] | ||
| Androgen Deprivation (CSS, antiandrogen, and/or castration) | LNCaP, LAPC4 | Growth arrest, p53 and p16INK4, SA-β-gal, low Ki67, cell cycle arrest | [96] | |
| LNCaP ICR castrated mice | SA-β-gal, p27Kip1 and p53, p21Cip1, SASP (IL-6 and IL-8) | [97] | ||
| LNCaP, LAPC4 | SA-β-gal, SAHF, Ki67, growth arrest, morphology, SASP, | [98] | ||
| LNCaP, LuCaP, xenografts Patient samples | SA-β-gal, decreased proliferation, increased cellular size, p27Kip1, HP1γ, low Ki67 | [99] | ||
| Kinase inhibitors | Imatinib | K562 | SA-β-gal, growth arrest, p21Cip1, p27Kip1 | [100] |
| Nilotinib | H1975 | SA-β-gal | [101] | |
| Trametinib | DMBC11, DMBC12, DMBC21, DMBC28, DMBC17 | SA-β-gal | [102] | |
| H2030, H460, A549, MSK-LX68 patient-derived xenografts | SA-β-gal, SASP | [103] | ||
| A549, H460, H1944, H2030, H358, H441, H2009, HCC441 | SA-β-gal, growth arrest, p53, p21Cip1 | [104] | ||
| Vemurafenib | DMBC11, DMBC12, DMBC21, DMBC28, DMBC17 | SA-β-gal | [102] | |
| MM034, MM070, MM074, SKMEL-28, MM050 | Growth arrest, morphology, SA-β-gal | [105] | ||
| SK-MEL-28, Mel2a, M19-Mel, SK-MEL-28, UACC-62, UACC-257, and FM88, M14, Malme 3M, Mel2a, SK-MEL-mouse xenografts | SAHF (H3K9me3), p16INK4, morphology, SA-β-gal, low Ki67, Rb | [106] | ||
| Dasatinib | H1666, Cal12T | Growth arrest, reduced BrdU incorporation, SA-β-gal | [107] | |
| A549, H1666 H661, Cal12T | SA-β-gal, γH2AX | [108] | ||
| Lapatinib | HCC1419, SKBR3, EFM-192A, MDA-MB-361 | SA-β-gal, p15INK4, p16INK4 | [109] | |
| Neratinib | HCC1419, SKBR3, EFM-192A, MDA-MB-361 | SA-β-gal, p15INK4, p16INK4 | [109] | |
| Afatinib | HCC1419, SKBR3, EFM-192A, MDA-MB-361 | SA-β-gal, p15INK4, p16INK4 | [109] | |
| Gefitinib | PC-9, EBC-2/R | Growth arrest, p53, p16INK4, p21Cip1, p27Kip1. | [110] | |
| Erlotinib | A549, A549 mouse xenografts | SA-β-gal, morphology | [111] | |
| Sorafenib | Huh7 mouse xenografts | SA-β-gal | [112] | |
| mTOR inhibitors | Rapamycin (Sirolimus) | SMMC-7721 | SA-β-gal | [84] |
| HUVECs | SA-β-gal, morphology | [113] | ||
| Monoclonal antibodies | Rituximab | EHEB, RC-K8, and SD-1 | Morphology, SA-β-gal | [114] |
| Follicular lymphoma 3D model | SA-β-gal | [49] | ||
| Obinutuzumab | Follicular lymphoma 3D model | SA-β-gal | [49] | |
| Pertuzumab | SK-BR-3 | SA-β-gal, p15INK4, p16INK4 | [115] | |
| Trastuzumab | SK-BR-3 | SA-β-gal, p15INK4, p16INK4 | [115] | |
| Bevacizumab | MIP101, RKO, SW620, SW480, MIP101 mouse xenografts | SA-β-gal | [116] | |
| Ranibizumab | Primary porcine retinal pigment epithelial cells | SA-β-gal, cathepsin D, amyloid β | [117] | |
| CDK 4/6 inhibitors | Palbociclib | U87MG, U138MG, Hs683, H4, A172, LN18, LN229, CCF-STTG1, T98G, DBTRG-05MG, DKMG, GAMG, SNB19, AM38, NMC-G1, KG-1-C, U87MG and GBM39 xenograft | SA-β-gal, morphology, growth arrest | [118] |
| HEK293, HeLa, U2OS | SA-β-gal, Rb, downregulated cyclin D1 | [119] | ||
| LS8817, LS141, LS0082 | SA-β-gal, p53, p16INK4, Rb downregulated cyclin A. | [120] | ||
| 1205Lu, 983B, 983BR | SA-β-gal, SASP (IL6, IL8, CXCL1), SAHF, DNA damage | [121] | ||
| B16-F1, B16-F10, NL212, NL216, TRIA | SA-β-gal, growth arrest, γH2AX and 53BP1, p16INK4, p65, p21Cip1, p53 | [122] | ||
| SK-MEL-103, NCI-H226, Huh7, SAOS-2, UT-SCC-42B | SA-β-gal, p21Cip1, Rb | [123] | ||
| AGS, MKN-45 | SA-β-gal | [124] | ||
| MCF7 | SA-β-gal, γH2AX, p21Cip1, morphology, reduced Ki67 | [125] | ||
| Huh7, skHep1, Huh7 mouse xenografts | SA-β-gal, morphology | [112] | ||
| Lung sections of Cdk4-deficient mice | SA-β-gal, γH2AX | [126] | ||
| Mouse-derived sarcoma cells/tissues | 53BP1, SA-β-gal, Rb | [127] | ||
| Abemaciclib | MCF7 | SA-β-gal, SAHF | [128] | |
| Ribociclib | Hey1 | SA-β-gal | [129] | |
| PARP inhibitors | Olaparib | HCT116 | Growth arrest, morphology, SA-β-gal, γH2AX | [130] |
| OV1369 (R2), OV90, OV4453, OV1946, MDA-MB-231 | Growth arrest, γH2AX, 53BP1, SA-β-gal, p21Cip1, p27Kip1, p15INK4, p16 INK4, p57, SASP (IL8) | [131] | ||
| Niraparib | HCT116 | Growth arrest, morphology, SA-β-gal, γH2AX | [130] | |
| Rucaparib | PC3, LNCaP, DU145, VCaP | SA-β-gal | [132] | |
| Proteasome inhibitors | Bortezomib | U87, T98 | SA-β-gal, morphology | [133] |
| Senolytic | Model/Cell Line | Reference |
|---|---|---|
| Dasatinib + Quercetin | - Senescent HUVEC, senescent preadipocytes in vitro - Senescent MEFs, senescent bone marrow-derived murine mesenchymal stem cells in vivo - SA-β-gal positive muscle and fat tissue of irradiated single mouse limb - Progeroid Ercc1(−/∆) mice | [186] |
| - Senescent lung fibroblasts and epithelial cells in bleomycin-induced lung injury/idiopathic pulmonary fibrosis mouse model | [18] | |
| - Senescent alveolar epithelial type (AT)II ex vivo in bleomycin-induced lung injury/idiopathic pulmonary fibrosis mouse model. | [187] | |
| - Senescent medial aortal cells of aging mice and hypercholesterolemia (atherosclerosis) mouse models | [188] | |
| - Senescent hepatocytes of dietary hepatic steatosis mouse model | [189] | |
| - Radiation-induced senescent preadipocytes in vivo - Senescent cells in freshly isolated human omental adipose tissue of obese individuals ex vivo | [190] | |
| - Arteriovenous fistula-chronic kidney disease mouse model | [191] | |
| - 20-month-old, transgenic tauNFT-Mapt0/0 mice | [192] | |
| - Aβ plaque-associated senescent oligodendrocyte progenitor cells in vivo - ZsGreen/APPPS1 p16INK4 reporter Alzheimer’s disease mouse model - Radiation-induced senescent N2a cells | [193] | |
| - Uterine fibrosis mouse model | [194] | |
| - Telomere dysfunction-induced senescent osteoblasts and osteocytes | [195] | |
| Navitoclax (ABT263) | - Radiation-induced, replication-exhausted and Ras-induced senescent WI38 fibroblasts in vitro - Radiation-induced senescent human IMR90 fibroblasts, human renal epithelial cells and mouse embryonic fibroblasts in vitro - Radiation-induced senescence in p16-3MR transgenic mouse model | [196] |
| - Radiation-induced senescent human umbilical vein epithelial cells, IMR90 human lung fibroblasts and mouse embryonic fibroblasts | [197] | |
| - Radiation-induced senescent type II alveolar epithelial cells in vitro and in vivo | [198] | |
| - Senescent pancreatic tissue of i4F mouse model | [199] | |
| - Replicative-exhausted human mesenchymal stromal cells | [200] | |
| - Aging-induced senescent cardiac myocytes - Myocardial infarction mouse model | [201] | |
| - Senescent murine pancreatic β-cells in vitro and in vivo | [202] | |
| - Aging mouse bone marrow stromal cells | [203] | |
| - WIPI1 and SLITKR4 overexpression-induced senescent uterine leiomyoma spheroid model ex vivo | [204] | |
| Piperlongumine | - Radiation-induced senescent astrocytes in vivo - Radiation-induced cognitive dysfunction mouse model | [205] |
| - SMARCB1 downregulation-induced senescent A375 melanoma cells - Therapy-induced A549 or H358 lung cancer cells | [145] | |
| - Radiation-induced, replication exhausted and Ras- induced senescent WI38 fibroblasts | [206] | |
| Curcumin | - Patient-derived senescent intervertebral disc cells | [207] |
| - Radiation-induced, oncogene-induced and replication-exhausted senescent WI38 fibroblasts | [208] | |
| Fisetin | - Replication-exhausted senescent Ercc1−/− MEFs - Therapy-induced senescent IMR90 senescent cells - Progeroid Ercc1−/∆ mice and aged C57BL/6 mouse models - Murine and human-derived senescent adipose tissue | [209] |
| - Senescent human umbilical vein endothelial cells | [210] | |
| Metformin | - Murine olfactory ensheathing cells ex vivo | [211] |
| Panobinostat | - Therapy-induced senescent A549 lung and FaDu head and neck cancer cells | [212] |
| 17-DMAG | - Oxidative-stress-induce primary Ercc1−/− - progeroid Ercc1 −/∆ mouse model | [213] |
| Torin 1 | - Murine senescent hepatocytes ex vivo | [214] |
| Epigallocatechin gallate (EGCG) | - Senescent 3T3-L1 preadipocytes | [215] |
| Bafilomycin A1 | - Therapy-induced HCT116 colorectal cancer cells | [158] |
| Azithromycin and roxithromycin | - Therapy-induced senescent MRC-5 and BJ human fibroblasts | [216] |
| Fenofibrate | - Senescent T/C28a2 human chondrocytes | [217] |
| Cardiac glycosides | - Therapy-induced senescent A549 lung cancer cells and SK-MEL-103 melanoma cells in vitro and in vivo | [218,219] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saleh, T.; Bloukh, S.; Carpenter, V.J.; Alwohoush, E.; Bakeer, J.; Darwish, S.; Azab, B.; Gewirtz, D.A. Therapy-Induced Senescence: An “Old” Friend Becomes the Enemy. Cancers 2020, 12, 822. https://doi.org/10.3390/cancers12040822
Saleh T, Bloukh S, Carpenter VJ, Alwohoush E, Bakeer J, Darwish S, Azab B, Gewirtz DA. Therapy-Induced Senescence: An “Old” Friend Becomes the Enemy. Cancers. 2020; 12(4):822. https://doi.org/10.3390/cancers12040822
Chicago/Turabian StyleSaleh, Tareq, Sarah Bloukh, Valerie J. Carpenter, Enas Alwohoush, Jomana Bakeer, Sarah Darwish, Belal Azab, and David A. Gewirtz. 2020. "Therapy-Induced Senescence: An “Old” Friend Becomes the Enemy" Cancers 12, no. 4: 822. https://doi.org/10.3390/cancers12040822
APA StyleSaleh, T., Bloukh, S., Carpenter, V. J., Alwohoush, E., Bakeer, J., Darwish, S., Azab, B., & Gewirtz, D. A. (2020). Therapy-Induced Senescence: An “Old” Friend Becomes the Enemy. Cancers, 12(4), 822. https://doi.org/10.3390/cancers12040822