Preclinical Study Using ABT263 to Increase Enzalutamide Sensitivity to Suppress Prostate Cancer Progression Via Targeting BCL2/ROS/USP26 Axis Through Altering ARv7 Protein Degradation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. Cell Proliferation Assays
2.3. Drug Synergy Assay
2.4. Lentivirus Packaging and Cell Transfection
2.5. RNA Extraction and Quantitative Real-Time PCR (PCR) Analysis
2.6. Western Blot and Protein Stability Analysis
2.7. Immunoprecipitation (IP)
2.8. Reactive oxygen species (ROS) Measurement
2.9. In Vitro Deubiquitinating Enzymes (DUB) Activity Assay
2.10. In Vivo Studies
2.11. Immunohistochemistry (IHC) Staining
2.12. Statistics
3. Results
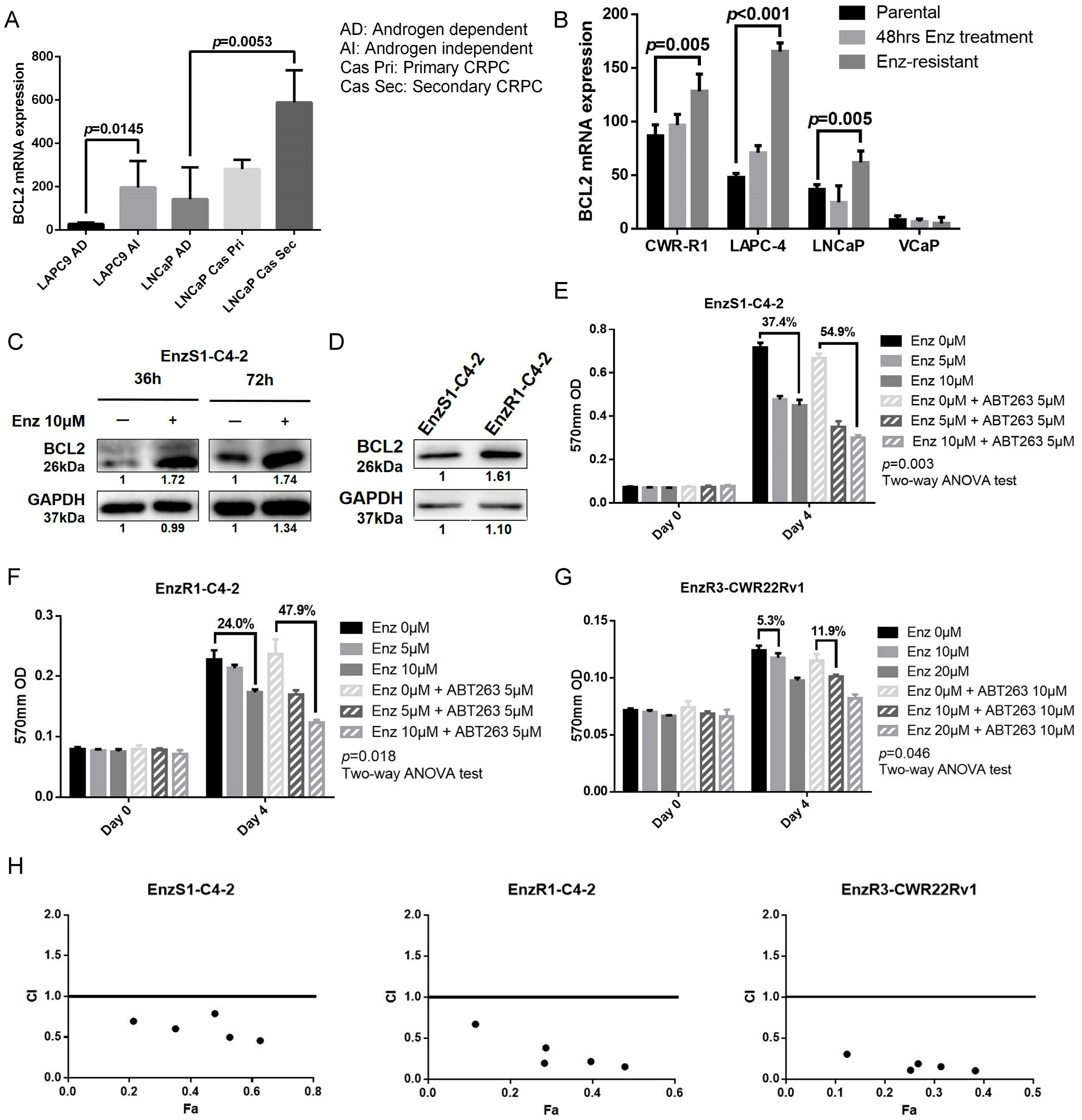
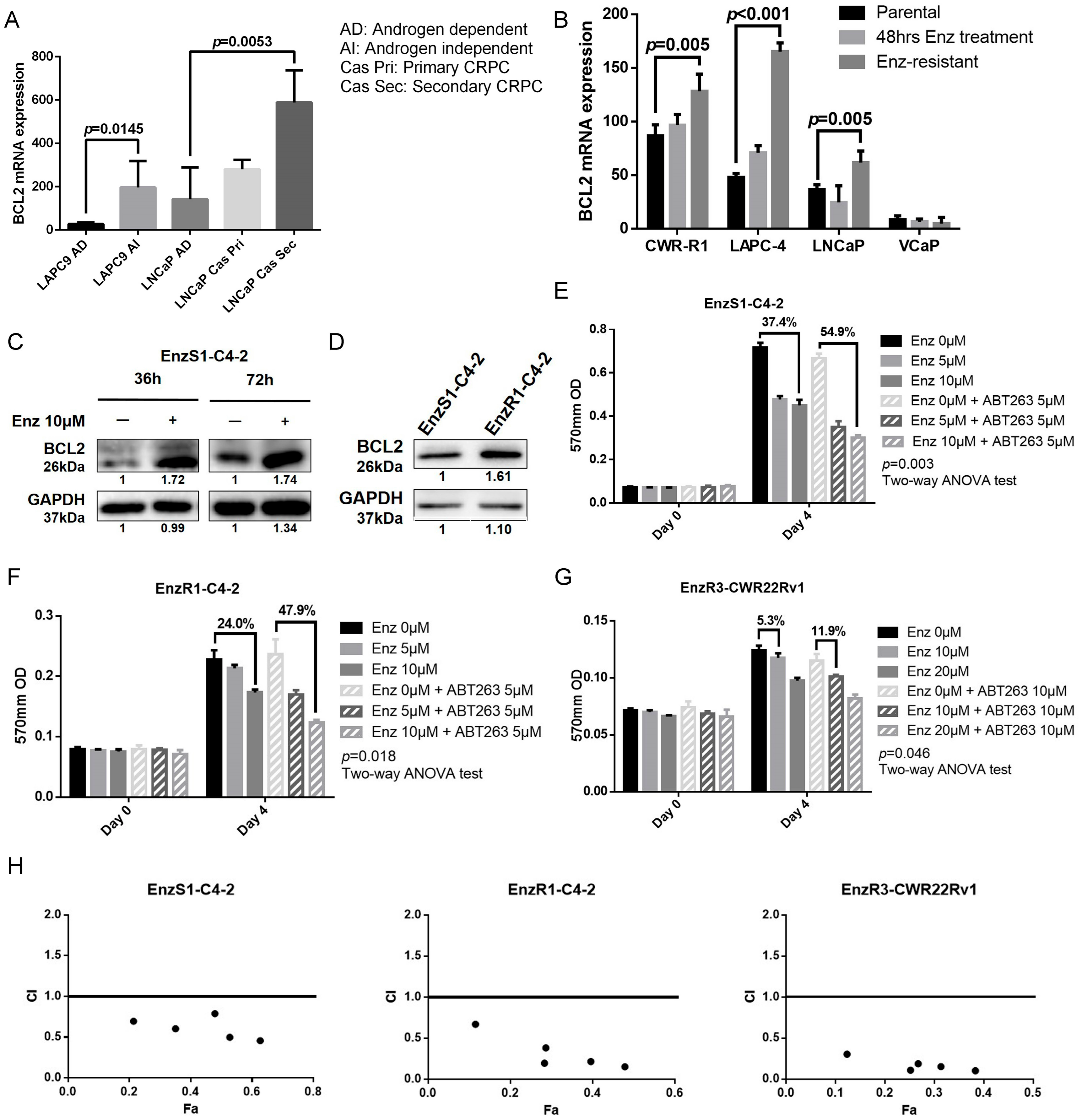
3.1. BCL2 Expression Increased in Enz-Resistant Prostate Cancer (PCa) Cells
3.2. BCL2 Targeting by ABT263 Impedes Cell Growth by Enhancing Enz Potency in Both Enz-Sensitive and Enz-Resistant PCa Cells
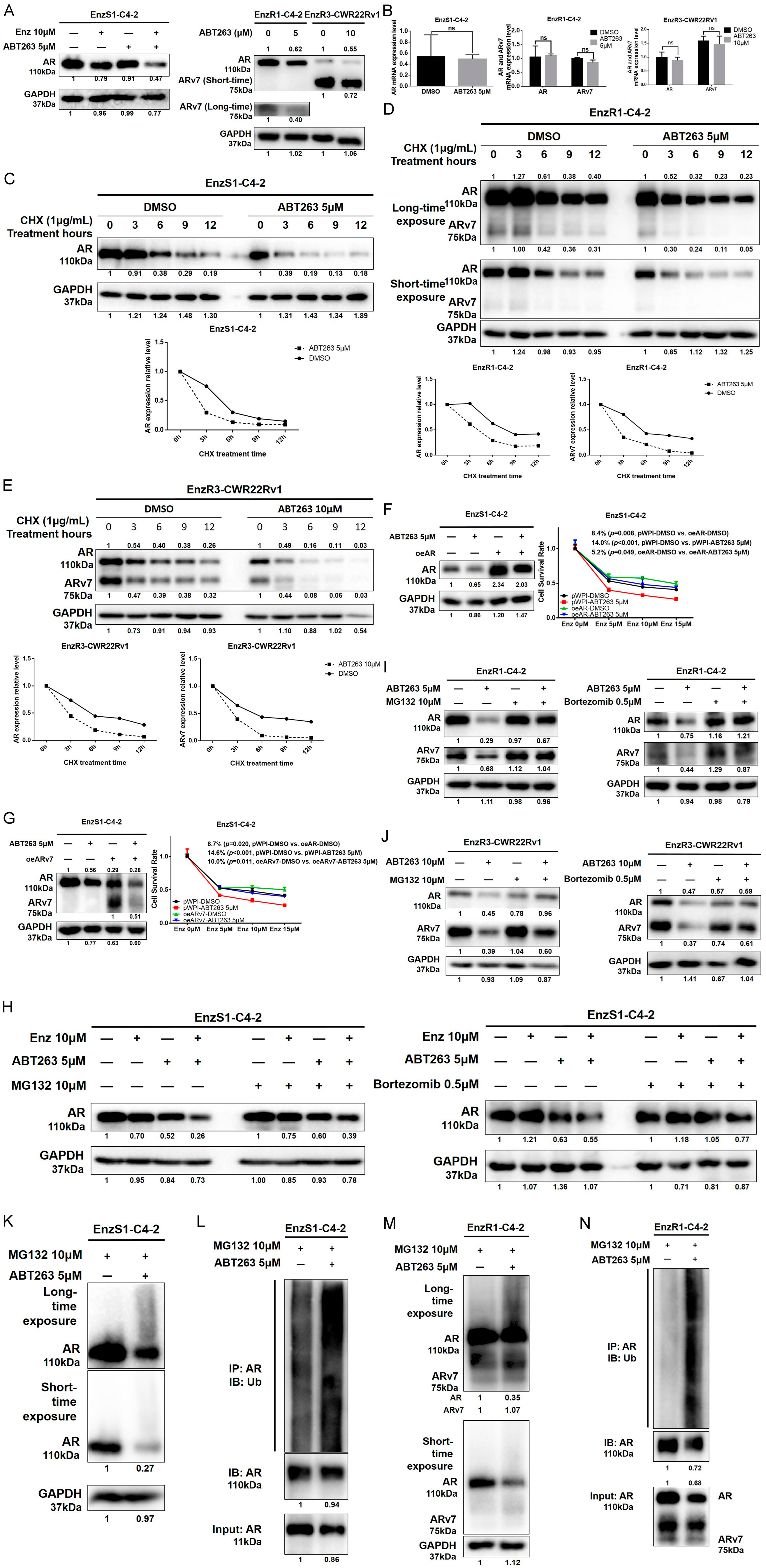
3.3. ABT263 Mechanistically Increases Enz Sensitivity by Enhancing Proteasome-Dependent Degradation of AR and ARv7
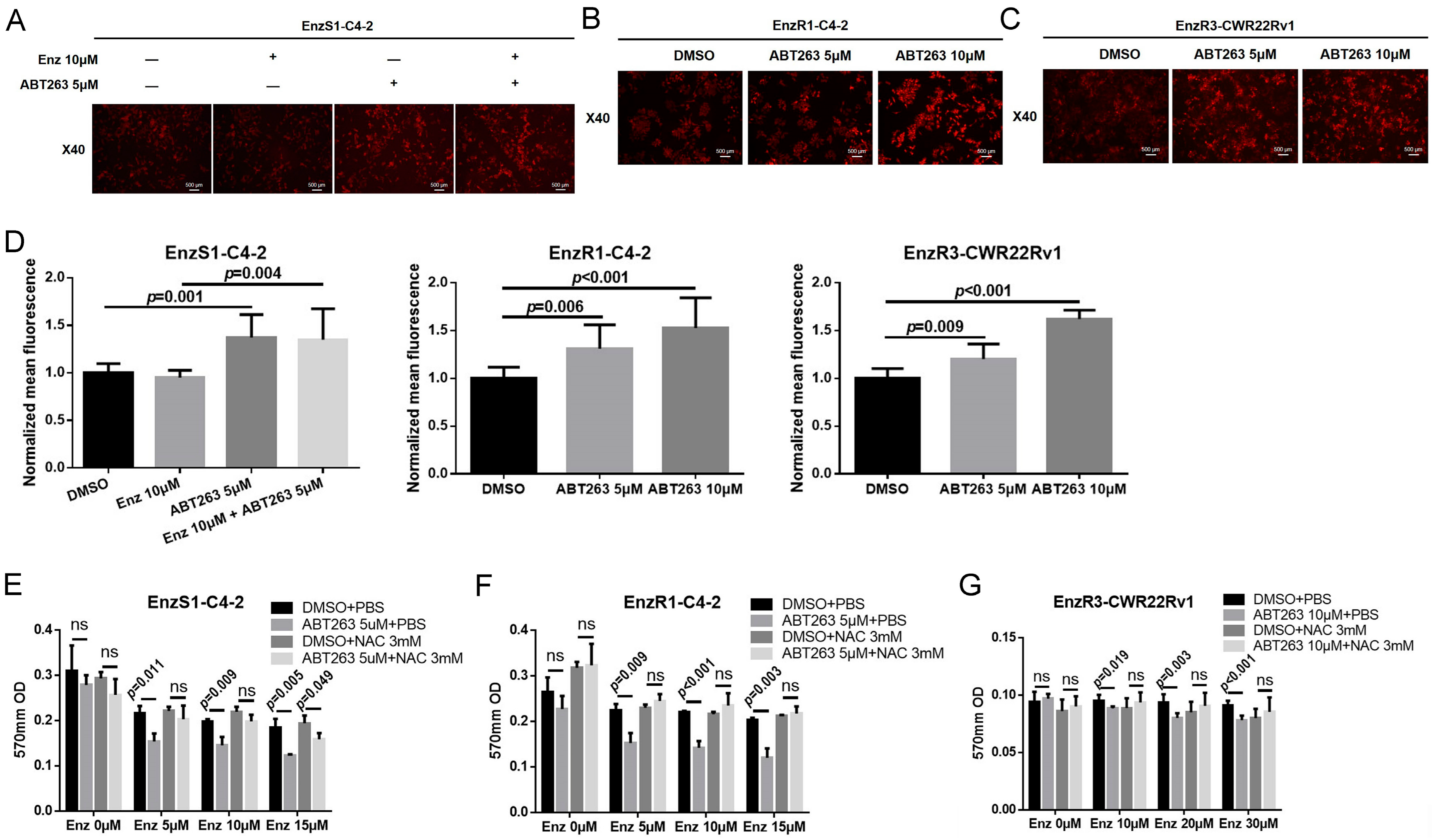
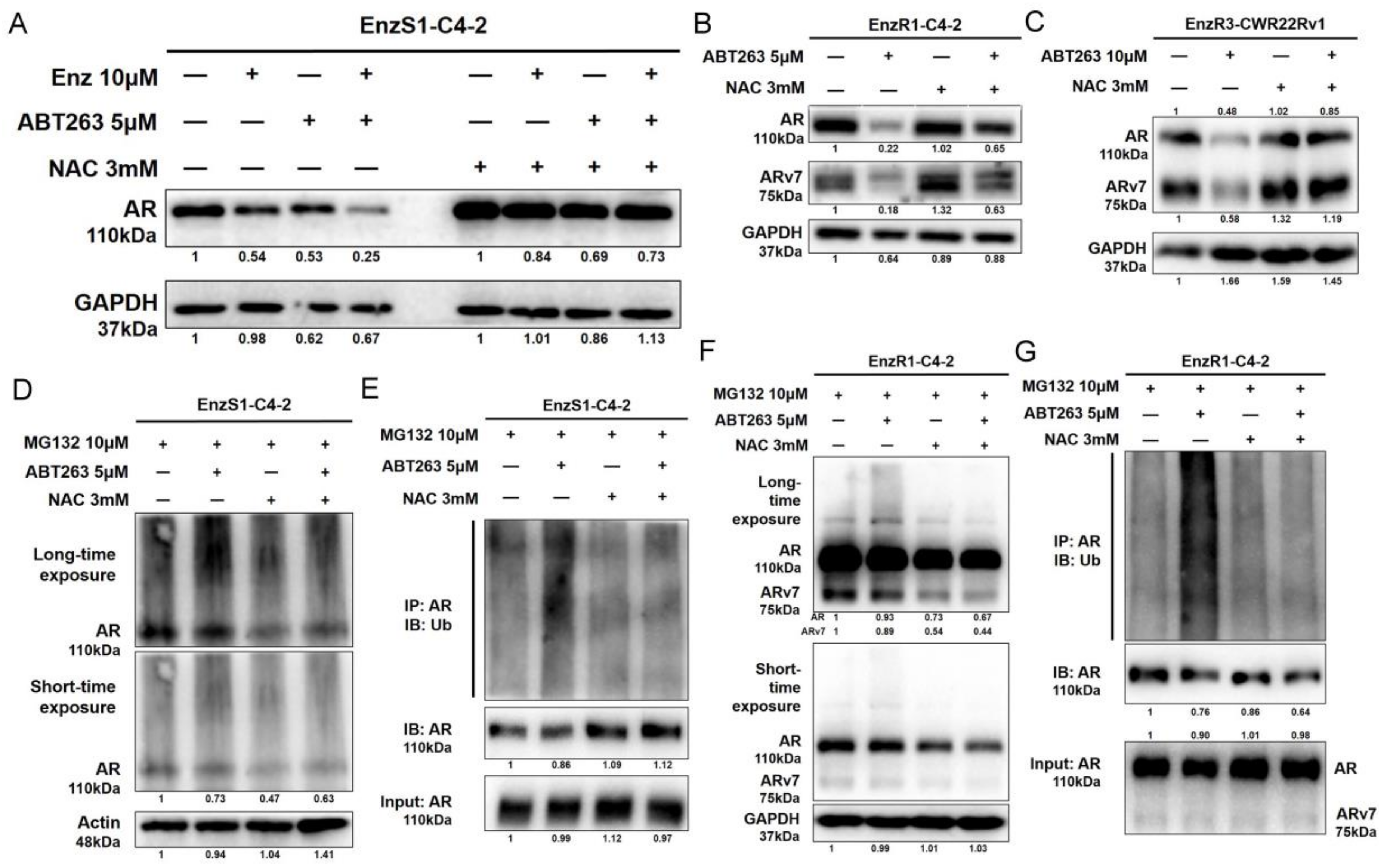
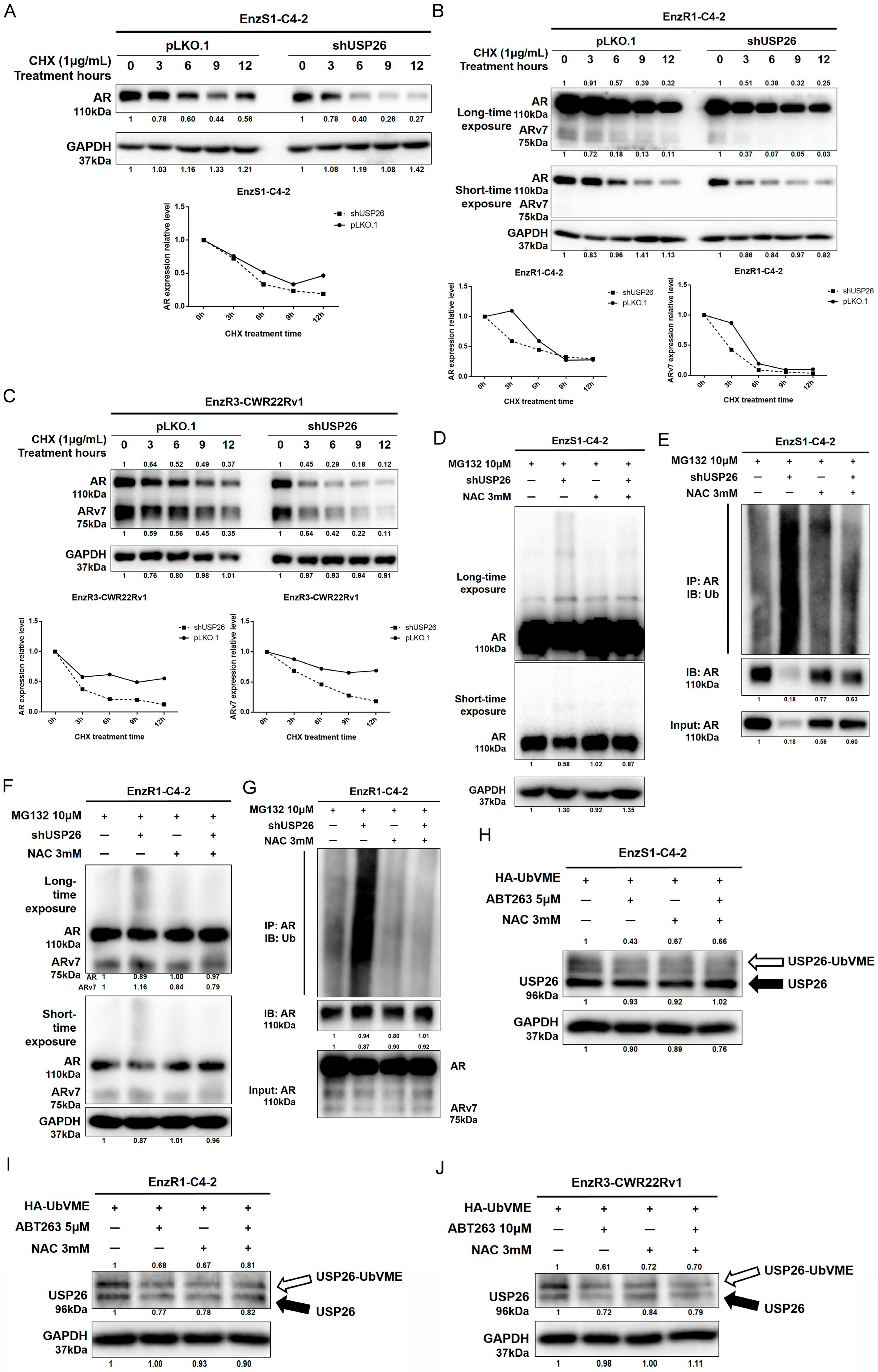
3.4. ABT263 Mechanistically Increases AR and ARv7 Degradation by Inducing Cellular ROS Level
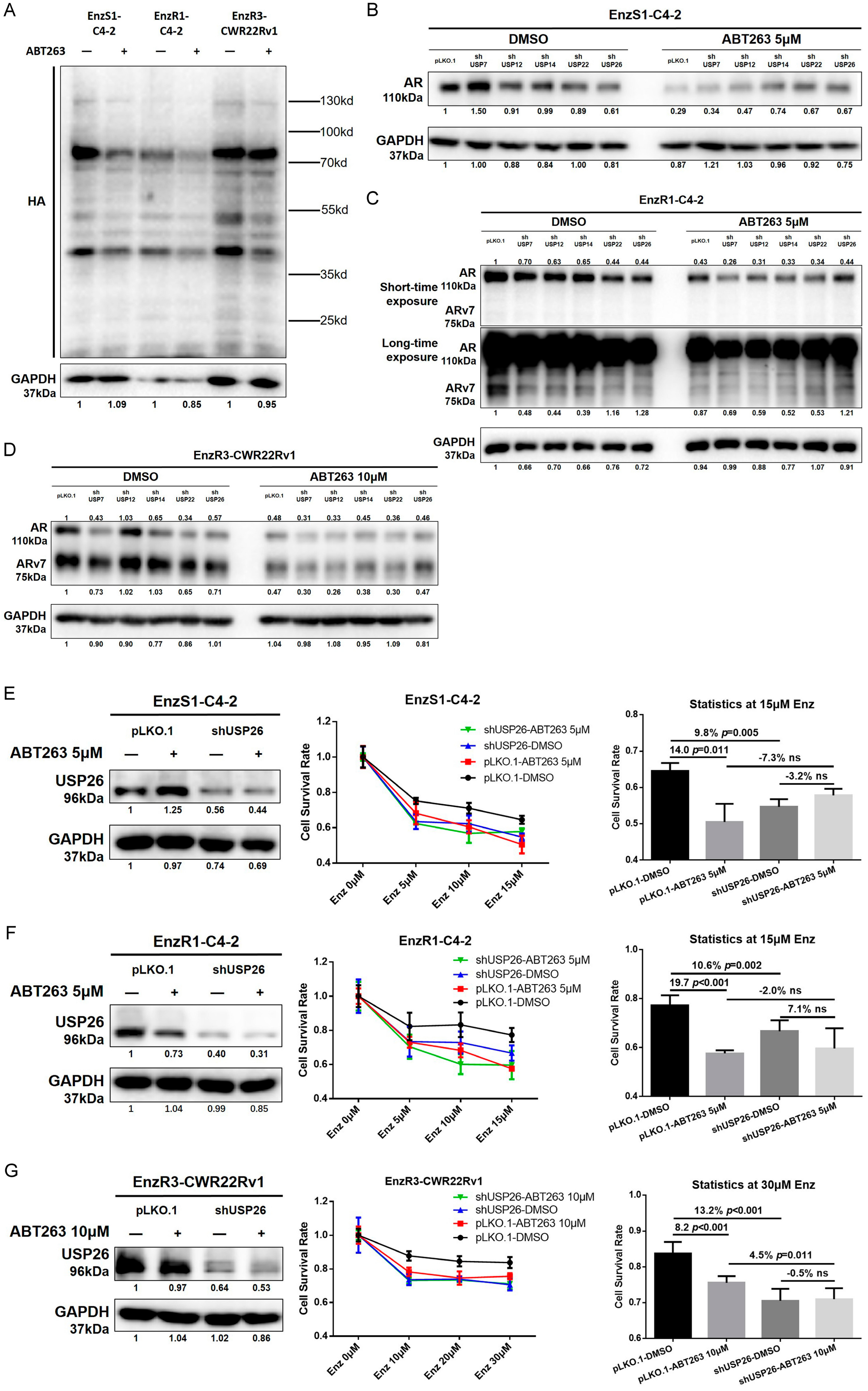
3.5. ABT263 Increases Enz Sensitivity and AR/ARv7 Degradation via Inhibiting USP26 Activity
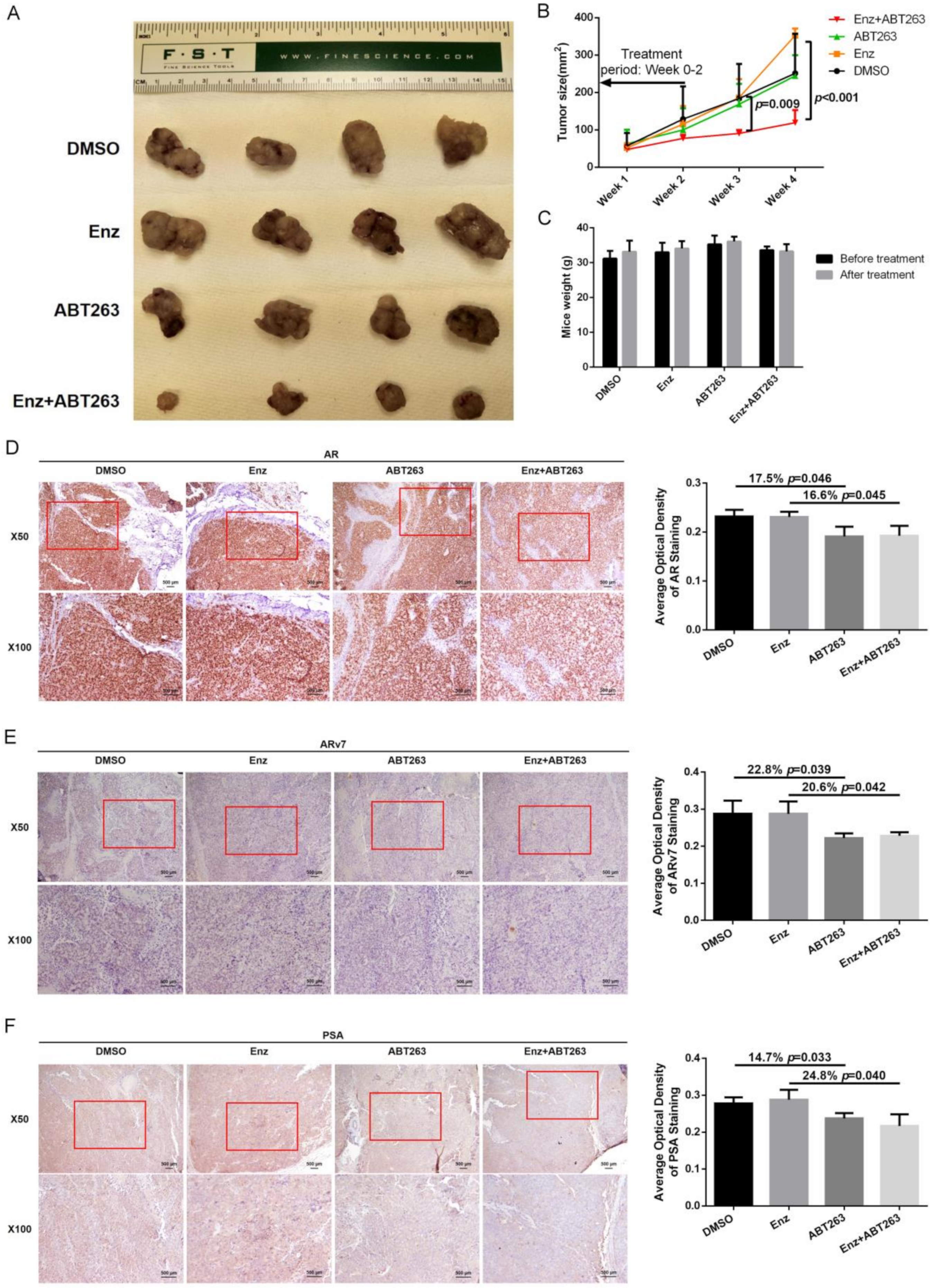
3.6. Preclinical Study Using in Vivo Mouse Model to Demonstrate ABT263 Can Increase Enz Sensitivity in Enz-Resistant PCa Cells and Better Suppress of PCa Progression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA A Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zheng, R.; Baade, P.D.; Zhang, S.; Zeng, H.; Bray, F.; Jemal, A.; Yu, X.Q.; He, J. Cancer statistics in China, 2015. CA A Cancer J. Clin. 2016, 66, 115–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loblaw, D.A.; Virgo, K.S.; Nam, R.; Somerfield, M.R.; Ben-Josef, E.; Mendelson, D.S.; Middleton, R.; Sharp, S.A.; Smith, T.J.; Talcott, J.; et al. Initial hormonal management of androgen-sensitive metastatic, recurrent, or progressive prostate cancer: 2006 update of an american society of clinical oncology practice guideline. J. Clin. Oncol. 2007, 25, 1596–1605. [Google Scholar] [CrossRef]
- Smith, M.R.; Kabbinavar, F.; Saad, F.; Hussain, A.; Gittelman, M.C.; Bilhartz, D.L.; Wynne, C.; Murray, R.; Zinner, N.R.; Schulman, C.; et al. Natural history of rising serum prostate-specific antigen in men with castrate nonmetastatic prostate cancer. J. Clin. Oncol. 2005, 23, 2918–2925. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berruti, A.; Pia, A.; Terzolo, M. Abiraterone and increased survival in metastatic prostate cancer. N. Engl. J. Med. 2011, 365, 767–768. [Google Scholar]
- Miyamoto, H.; Messing, E.M.; Chang, C. Does androgen deprivation improve treatment outcomes in patients with low-risk and intermediate-risk prostate cancer? Nat. Clin. Pract. Oncol. 2005, 2, 236–237. [Google Scholar] [CrossRef]
- Li, Y.; Chan, S.C.; Brand, L.J.; Hwang, T.H.; Silverstein, K.A.; Dehm, S.M. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2013, 73, 483–489. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.D.; Welsbie, D.S.; Tran, C.; Baek, S.H.; Chen, R.; Vessella, R.; Rosenfeld, M.G.; Sawyers, C.L. Molecular determinants of resistance to antiandrogen therapy. Nat. Med. 2004, 10, 33–39. [Google Scholar] [CrossRef]
- Hoefer, J.; Akbor, M.; Handle, F.; Ofer, P.; Puhr, M.; Parson, W.; Culig, Z.; Klocker, H.; Heidegger, I. Critical role of androgen receptor level in prostate cancer cell resistance to new generation antiandrogen enzalutamide. Oncotarget 2016, 7, 59781–59794. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.K.; Altuwaijri, S.; Lin, W.J.; Kan, P.Y.; Collins, L.L.; Chang, C. Proteasome activity is required for androgen receptor transcriptional activity via regulation of androgen receptor nuclear translocation and interaction with coregulators in prostate cancer cells. J. Biol. Chem. 2002, 277, 36570–36576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes-Turcu, F.E.; Ventii, K.H.; Wilkinson, K.D. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Ann. Rev. Biochem. 2009, 78, 363–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dirac, A.M.; Bernards, R. The deubiquitinating enzyme usp26 is a regulator of androgen receptor signaling. Mol. Cancer Res. MCR 2010, 8, 844–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burska, U.L.; Harle, V.J.; Coffey, K.; Darby, S.; Ramsey, H.; O’Neill, D.; Logan, I.R.; Gaughan, L.; Robson, C.N. Deubiquitinating enzyme usp12 is a novel co-activator of the androgen receptor. J. Biol. Chem. 2013, 288, 32641–32650. [Google Scholar] [CrossRef] [Green Version]
- Schrecengost, R.S.; Dean, J.L.; Goodwin, J.F.; Schiewer, M.J.; Urban, M.W.; Stanek, T.J.; Sussman, R.T.; Hicks, J.L.; Birbe, R.C.; Draganova-Tacheva, R.A.; et al. Usp22 regulates oncogenic signaling pathways to drive lethal cancer progression. Cancer Res. 2014, 74, 272–286. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.T.; Okada, M.; Nakato, R.; Izumi, K.; Bando, M.; Shirahige, K. The deubiquitinating enzyme usp7 regulates androgen receptor activity by modulating its binding to chromatin. J. Biol. Chem. 2015, 290, 21713–21723. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Liu, N.; Hua, X.; Cai, J.; Xia, X.; Wang, X.; Huang, H.; Liu, J. Proteasome-associated deubiquitinase ubiquitin-specific protease 14 regulates prostate cancer proliferation by deubiquitinating and stabilizing androgen receptor. Cell Death Dis. 2017, 8, e2585. [Google Scholar] [CrossRef]
- Tse, C.; Shoemaker, A.R.; Adickes, J.; Anderson, M.G.; Chen, J.; Jin, S.; Johnson, E.F.; Marsh, K.C.; Mitten, M.J.; Nimmer, P.; et al. Abt-263: A potent and orally bioavailable bcl-2 family inhibitor. Cancer Res. 2008, 68, 3421–3428. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. Nih image to imagej: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Crowley, L.C.; Christensen, M.E.; Waterhouse, N.J. Measuring survival of adherent cells with the colony-forming assay. Cold Spring Harb. Protoc. 2016, 8, 721–724. [Google Scholar] [CrossRef] [Green Version]
- Chou, T.C. Drug combination studies and their synergy quantification using the chou-talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borodovsky, A.; Ovaa, H.; Kolli, N.; Gan-Erdene, T.; Wilkinson, K.D.; Ploegh, H.L.; Kessler, B.M. Chemistry-based functional proteomics reveals novel members of the deubiquitinating enzyme family. Chem. Biol. 2002, 9, 1149–1159. [Google Scholar] [CrossRef] [Green Version]
- Reed, J.C. Dysregulation of apoptosis in cancer. J. Clin. Oncol. 1999, 17, 2941–2953. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Deng, Q.; Chao, H.P.; Liu, X.; Lu, Y.; Lin, K.; Liu, B.; Tang, G.W.; Zhang, D.; Tracz, A.; et al. Linking prostate cancer cell ar heterogeneity to distinct castration and enzalutamide responses. Nat. Commun. 2018, 9, 3600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kregel, S.; Chen, J.L.; Tom, W.; Krishnan, V.; Kach, J.; Brechka, H.; Fessenden, T.B.; Isikbay, M.; Paner, G.P.; Szmulewitz, R.Z.; et al. Acquired resistance to the second-generation androgen receptor antagonist enzalutamide in castration-resistant prostate cancer. Oncotarget 2016, 7, 26259–26274. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. Ar-v7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [Green Version]
- Sun, F.; Indran, I.R.; Zhang, Z.W.; Tan, M.H.; Li, Y.; Lim, Z.L.; Hua, R.; Yang, C.; Soon, F.F.; Li, J.; et al. A novel prostate cancer therapeutic strategy using icaritin-activated arylhydrocarbon-receptor to co-target androgen receptor and its splice variants. Carcinogenesis 2015, 36, 757–768. [Google Scholar] [CrossRef] [Green Version]
- Deng, X.; Gao, F.; May, W.S., Jr. Bcl2 retards g1/s cell cycle transition by regulating intracellular ros. Blood 2003, 102, 3179–3185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dikalov, S.I.; Harrison, D.G. Methods for detection of mitochondrial and cellular reactive oxygen species. Antioxid. Redox Signal. 2014, 20, 372–382. [Google Scholar] [CrossRef] [Green Version]
- Kerksick, C.; Willoughby, D. The antioxidant role of glutathione and n-acetyl-cysteine supplements and exercise-induced oxidative stress. J. Int. Soc. Sports Nutr. 2005, 2, 38–44. [Google Scholar] [CrossRef] [Green Version]
- McDonnell, T.J.; Troncoso, P.; Brisbay, S.M.; Logothetis, C.; Chung, L.W.; Hsieh, J.T.; Tu, S.M.; Campbell, M.L. Expression of the protooncogene bcl-2 in the prostate and its association with emergence of androgen-independent prostate cancer. Cancer Res. 1992, 52, 6940–6944. [Google Scholar] [PubMed]
- Raffo, A.J.; Perlman, H.; Chen, M.W.; Day, M.L.; Streitman, J.S.; Buttyan, R. Overexpression of bcl-2 protects prostate cancer cells from apoptosis in vitro and confers resistance to androgen depletion in vivo. Cancer Res. 1995, 55, 4438–4445. [Google Scholar] [PubMed]
- Kajiwara, T.; Takeuchi, T.; Ueki, T.; Moriyama, N.; Ueki, K.; Kakizoe, T.; Kawabe, K. Effect of bcl-2 overexpression in human prostate cancer cells in vitro and in vivo. Int. J. Urol. Off. J. Jpn. Urol. Assoc. 1999, 6, 520–525. [Google Scholar]
- Thoma, C. Prostate cancer: Targeting apoptosis resistance in crpc. Nat. Rev. Urol. 2016, 13, 631. [Google Scholar] [CrossRef]
- Gandhi, L.; Camidge, D.R.; Ribeiro de Oliveira, M.; Bonomi, P.; Gandara, D.; Khaira, D.; Hann, C.L.; McKeegan, E.M.; Litvinovich, E.; Hemken, P.M.; et al. Phase i study of navitoclax (abt-263), a novel bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. J. Clin. Oncol. 2011, 29, 909–916. [Google Scholar] [CrossRef] [Green Version]
- Kipps, T.J.; Eradat, H.; Grosicki, S.; Catalano, J.; Cosolo, W.; Dyagil, I.S.; Yalamanchili, S.; Chai, A.; Sahasranaman, S.; Punnoose, E.; et al. A phase 2 study of the bh3 mimetic bcl2 inhibitor navitoclax (abt-263) with or without rituximab, in previously untreated b-cell chronic lymphocytic leukemia. Leuk. Lymphoma 2015, 56, 2826–2833. [Google Scholar] [CrossRef] [Green Version]
- Vogler, M.; Dinsdale, D.; Dyer, M.J.; Cohen, G.M. Bcl-2 inhibitors: Small molecules with a big impact on cancer therapy. Cell Death Diff. 2009, 16, 360–367. [Google Scholar] [CrossRef] [Green Version]
- Ogura, T.; Tanaka, Y.; Tamaki, H.; Harada, M. Docetaxel induces bcl-2- and pro-apoptotic caspase-independent death of human prostate cancer du145 cells. Int. J. Oncol. 2016, 48, 2330–2338. [Google Scholar] [CrossRef] [Green Version]
- Karnak, D.; Xu, L. Chemosensitization of prostate cancer by modulating bcl-2 family proteins. Curr. Drug Targets 2010, 11, 699–707. [Google Scholar] [CrossRef] [Green Version]
- Tamaki, H.; Harashima, N.; Hiraki, M.; Arichi, N.; Nishimura, N.; Shiina, H.; Naora, K.; Harada, M. Bcl-2 family inhibition sensitizes human prostate cancer cells to docetaxel and promotes unexpected apoptosis under caspase-9 inhibition. Oncotarget 2014, 5, 11399–11412. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Huang, S.B.; Yang, M.C.; Lin, Y.T.; Chu, I.H.; Shen, Y.N.; Chiu, Y.H.; Hung, S.H.; Kang, L.; Hong, Y.R.; et al. Combining paclitaxel with abt-263 has a synergistic effect on paclitaxel resistant prostate cancer cells. PLoS ONE 2015, 10, e0120913. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Chervin, A.S.; Nie, A.; Ducoff, H.S.; Huang, Z. Overcoming the radioresistance of prostate cancer cells with a novel bcl-2 inhibitor. Oncogene 2007, 26, 652–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.C.; Wang, L.J.; Huang, C.H.; Shi, Y.J.; Chang, L.S. Abt-263-induced mcl1 upregulation depends on autophagy-mediated 4ebp1 downregulation in human leukemia cells. Cancer Lett. 2018, 432, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, Y.; Wan, J.; Liu, X.; Yu, C.; Li, W. Abt-263 enhances sorafenib-induced apoptosis associated with akt activity and the expression of bax and p21((cip1/waf1)) in human cancer cells. Br. J. Pharmacol. 2014, 171, 3182–3195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, A.T.; Wang, Y.; Chiu, J.F. Reactive oxygen species: Current knowledge and applications in cancer research and therapeutic. J. Cell. Biochem. 2008, 104, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Dharmaraja, A.T. Role of reactive oxygen species (ros) in therapeutics and drug resistance in cancer and bacteria. J. Med.Chem. 2017, 60, 3221–3240. [Google Scholar] [CrossRef]
- Halliwell, B. Reactive oxygen species in living systems: Source, biochemistry, and role in human disease. Am. J. Med. 1991, 91, 14S–22S. [Google Scholar] [CrossRef]
- Guterman, A.; Glickman, M.H. Deubiquitinating enzymes are in/(trinsic to proteasome function). Curr. Protein Pept. Sci. 2004, 5, 201–211. [Google Scholar] [CrossRef]
- Cotto-Rios, X.M.; Bekes, M.; Chapman, J.; Ueberheide, B.; Huang, T.T. Deubiquitinases as a signaling target of oxidative stress. Cell Rep. 2012, 2, 1475–1484. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, D.; Tian, Z.; Nicholson, B.; Kumar, K.G.; Zhou, B.; Carrasco, R.; McDermott, J.L.; Leach, C.A.; Fulcinniti, M.; Kodrasov, M.P.; et al. A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell 2012, 22, 345–358. [Google Scholar] [CrossRef] [Green Version]
- Reverdy, C.; Conrath, S.; Lopez, R.; Planquette, C.; Atmanene, C.; Collura, V.; Harpon, J.; Battaglia, V.; Vivat, V.; Sippl, W.; et al. Discovery of specific inhibitors of human usp7/hausp deubiquitinating enzyme. Chem. Biol. 2012, 19, 467–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, B.H.; Lee, M.J.; Park, S.; Oh, D.C.; Elsasser, S.; Chen, P.C.; Gartner, C.; Dimova, N.; Hanna, J.; Gygi, S.P.; et al. Enhancement of proteasome activity by a small-molecule inhibitor of usp14. Nature 2010, 467, 179–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byun, S.; Lee, S.Y.; Lee, J.; Jeong, C.H.; Farrand, L.; Lim, S.; Reddy, K.; Kim, J.Y.; Lee, M.H.; Lee, H.J.; et al. Usp8 is a novel target for overcoming gefitinib resistance in lung cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 3894–3904. [Google Scholar] [CrossRef] [PubMed] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, H.; Sun, Y.; Huang, C.-P.; You, B.; Ye, D.; Chang, C. Preclinical Study Using ABT263 to Increase Enzalutamide Sensitivity to Suppress Prostate Cancer Progression Via Targeting BCL2/ROS/USP26 Axis Through Altering ARv7 Protein Degradation. Cancers 2020, 12, 831. https://doi.org/10.3390/cancers12040831
Xu H, Sun Y, Huang C-P, You B, Ye D, Chang C. Preclinical Study Using ABT263 to Increase Enzalutamide Sensitivity to Suppress Prostate Cancer Progression Via Targeting BCL2/ROS/USP26 Axis Through Altering ARv7 Protein Degradation. Cancers. 2020; 12(4):831. https://doi.org/10.3390/cancers12040831
Chicago/Turabian StyleXu, Hua, Yin Sun, Chi-Ping Huang, Bosen You, Dingwei Ye, and Chawnshang Chang. 2020. "Preclinical Study Using ABT263 to Increase Enzalutamide Sensitivity to Suppress Prostate Cancer Progression Via Targeting BCL2/ROS/USP26 Axis Through Altering ARv7 Protein Degradation" Cancers 12, no. 4: 831. https://doi.org/10.3390/cancers12040831
APA StyleXu, H., Sun, Y., Huang, C.-P., You, B., Ye, D., & Chang, C. (2020). Preclinical Study Using ABT263 to Increase Enzalutamide Sensitivity to Suppress Prostate Cancer Progression Via Targeting BCL2/ROS/USP26 Axis Through Altering ARv7 Protein Degradation. Cancers, 12(4), 831. https://doi.org/10.3390/cancers12040831