Mechanisms of Resistance to NK Cell Immunotherapy

 , , and
, , and
Abstract
1. Natural Killer (NK) Cells
2. NK Cell-Based Therapies
3. Are NK Cells Suitable Targets for Immunotherapy?
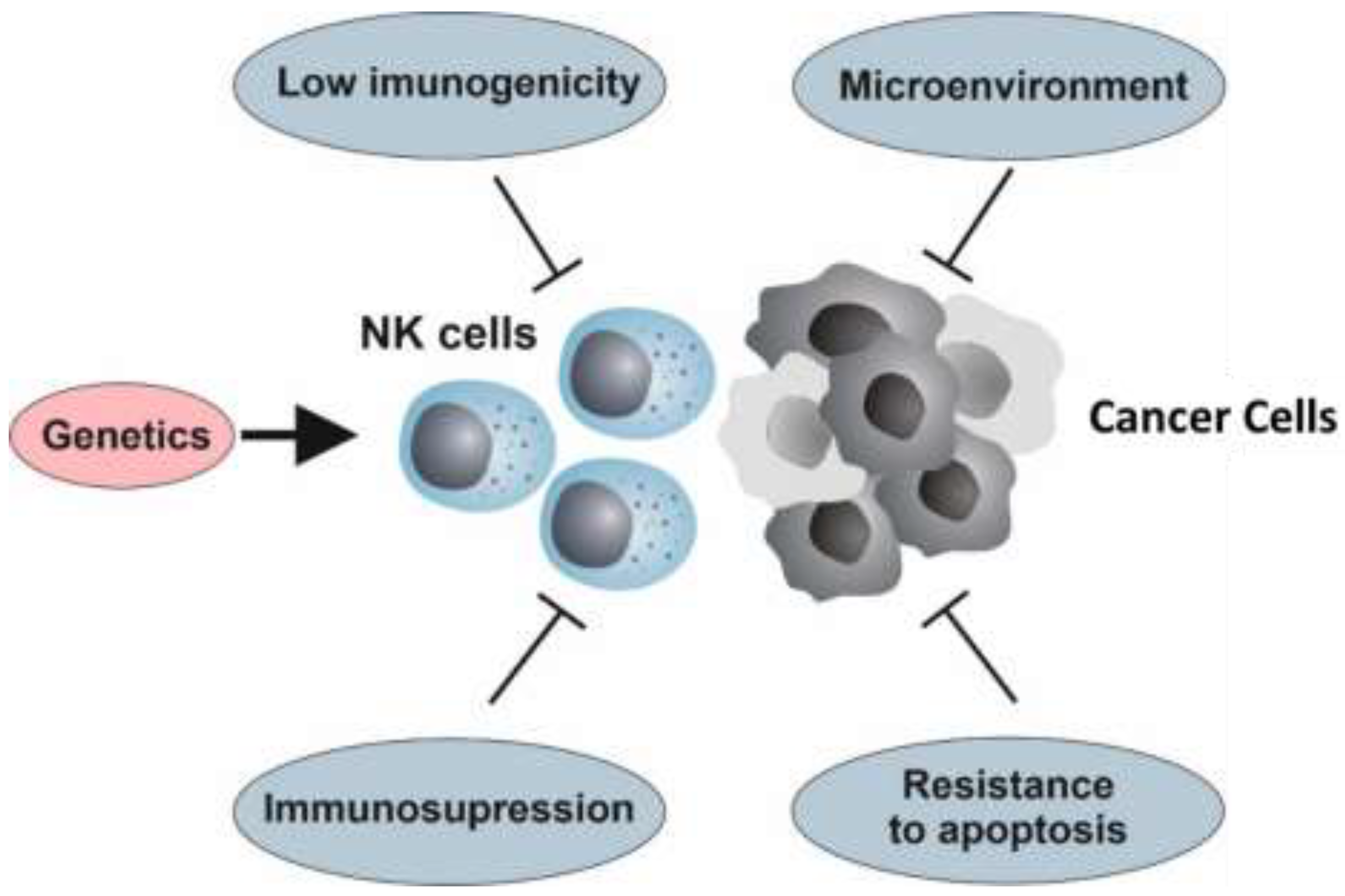
4. Resistance to NK Cell Therapy
4.1. The Genetic Background
4.2. Hallmarks of Cancer and Resistance to NK Cell Immunotherapy
4.3. Immunoediting
4.4. Immunosuppression
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cantoni, C.; Grauwet, K.; Pietra, G.; Parodi, M.; Mingari, M.C.; Maria, A.D.; Favoreel, H.; Vitale, M. Role of NK cells in immunotherapy and virotherapy of solid tumors. Immunotherapy 2015, 7, 861–882. [Google Scholar] [CrossRef] [PubMed]
- Chiossone, L.; Dumas, P.Y.; Vienne, M.; Vivier, E. Natural killer cells and other innate lymphoid cells in cancer. Nat. Rev. Immunol. 2018, 18, 671–688. [Google Scholar] [CrossRef] [PubMed]
- Karre, K.; Ljunggren, H.G.; Piontek, G.; Kiessling, R. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature 1986, 319, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.C.; Lanier, L.L. NK cell development, homeostasis and function: Parallels with CD8(+) T cells. Nat. Rev. Immunol. 2011, 11, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Moretta, L.; Locatelli, F.; Pende, D.; Sivori, S.; Falco, M.; Bottino, C.; Mingari, M.C.; Moretta, A. Human NK receptors: From the molecules to the therapy of high risk leukemias. FEBS Lett. 2011, 585, 1563–1567. [Google Scholar] [CrossRef]
- Sivori, S.; Vacca, P.; Del Zotto, G.; Munari, E.; Mingari, M.C.; Moretta, L. Human NK cells: Surface receptors, inhibitory checkpoints, and translational applications. Cell. Mol. Immunol. 2019, 16, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Muntasell, A.; Ochoa, M.C.; Cordeiro, L.; Berraondo, P.; Lopez-Diaz de Cerio, A.; Cabo, M.; Lopez-Botet, M.; Melero, I. Targeting NK-cell checkpoints for cancer immunotherapy. Curr. Opin. Immunol. 2017, 45, 73–81. [Google Scholar] [CrossRef]
- Hsu, J.; Hodgins, J.J.; Marathe, M.; Nicolai, C.J.; Bourgeois-Daigneault, M.C.; Trevino, T.N.; Azimi, C.S.; Scheer, A.K.; Randolph, H.E.; Thompson, T.W.; et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J. Clin. Investig. 2018, 128, 4654–4668. [Google Scholar] [CrossRef]
- Quatrini, L.; Wieduwild, E.; Escaliere, B.; Filtjens, J.; Chasson, L.; Laprie, C.; Vivier, E.; Ugolini, S. Endogenous glucocorticoids control host resistance to viral infection through the tissue-specific regulation of PD-1 expression on NK cells. Nat. Immunol. 2018, 19, 954–962. [Google Scholar] [CrossRef]
- Huergo-Zapico, L.; Acebes-Huerta, A.; Lopez-Soto, A.; Villa-Alvarez, M.; Gonzalez-Rodriguez, A.P.; Gonzalez, S. Molecular Bases for the Regulation of NKG2D Ligands in Cancer. Front. Immunol. 2014, 5, 106. [Google Scholar] [CrossRef]
- Lopez-Soto, A.; Huergo-Zapico, L.; Acebes-Huerta, A.; Villa-Alvarez, M.; Gonzalez, S. NKG2D signaling in cancer immunosurveillance. Int. J. Cancer 2015, 136, 1741–1750. [Google Scholar] [CrossRef]
- Moretta, A.; Bottino, C.; Vitale, M.; Pende, D.; Cantoni, C.; Mingari, M.C.; Biassoni, R.; Moretta, L. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu. Rev. Immunol. 2001, 19, 197–223. [Google Scholar] [CrossRef]
- Gasser, S.; Orsulic, S.; Brown, E.J.; Raulet, D.H. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 2005, 436, 1186–1190. [Google Scholar] [CrossRef]
- Zingoni, A.; Cecere, F.; Vulpis, E.; Fionda, C.; Molfetta, R.; Soriani, A.; Petrucci, M.T.; Ricciardi, M.R.; Fuerst, D.; Amendola, M.G.; et al. Genotoxic Stress Induces Senescence-Associated ADAM10-Dependent Release of NKG2D MIC Ligands in Multiple Myeloma Cells. J. Immunol. 2015, 195, 736–748. [Google Scholar] [CrossRef] [PubMed]
- Laurent, S.; Queirolo, P.; Boero, S.; Salvi, S.; Piccioli, P.; Boccardo, S.; Minghelli, S.; Morabito, A.; Fontana, V.; Pietra, G.; et al. The engagement of CTLA-4 on primary melanoma cell lines induces antibody-dependent cellular cytotoxicity and TNF-alpha production. J. Transl. Med. 2013, 11, 108. [Google Scholar] [CrossRef] [PubMed]
- Romano, E.; Kusio-Kobialka, M.; Foukas, P.G.; Baumgaertner, P.; Meyer, C.; Ballabeni, P.; Michielin, O.; Weide, B.; Romero, P.; Speiser, D.E. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc. Natl. Acad. Sci. USA 2015, 112, 6140–6145. [Google Scholar] [CrossRef]
- Lorenzo-Herrero, S.; Lopez-Soto, A.; Sordo-Bahamonde, C.; Gonzalez-Rodriguez, A.P.; Vitale, M.; Gonzalez, S. NK Cell-Based Immunotherapy in Cancer Metastasis. Cancers 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rodriguez, A.P.; Villa-Alvarez, M.; Sordo-Bahamonde, C.; Lorenzo-Herrero, S.; Gonzalez, S. NK Cells in the Treatment of Hematological Malignancies. J. Clin. Med. 2019, 8. [Google Scholar] [CrossRef]
- Lo Nigro, C.; Macagno, M.; Sangiolo, D.; Bertolaccini, L.; Aglietta, M.; Merlano, M.C. NK-mediated antibody-dependent cell-mediated cytotoxicity in solid tumors: Biological evidence and clinical perspectives. Ann. Transl. Med. 2019, 7, 105. [Google Scholar] [CrossRef]
- Nayyar, G.; Chu, Y.; Cairo, M.S. Overcoming Resistance to Natural Killer Cell Based Immunotherapies for Solid Tumors. Front. Oncol. 2019, 9, 51. [Google Scholar] [CrossRef]
- Giles, A.J.; Hao, S.; Padget, M.; Song, H.; Zhang, W.; Lynes, J.; Sanchez, V.; Liu, Y.; Jung, J.; Cao, X.; et al. Efficient ADCC killing of meningioma by avelumab and a high-affinity natural killer cell line, haNK. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Weiner, L.M.; Surana, R.; Wang, S. Monoclonal antibodies: Versatile platforms for cancer immunotherapy. Nat. Rev. Immunol. 2010, 10, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Pierpont, T.M.; Limper, C.B.; Richards, K.L. Past, Present, and Future of Rituximab-The World’s First Oncology Monoclonal Antibody Therapy. Front. Oncol. 2018, 8, 163. [Google Scholar] [CrossRef]
- Weiner, G.J. Rituximab: Mechanism of action. Semin. Hematol. 2010, 47, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Koch, J.; Tesar, M. Recombinant Antibodies to Arm Cytotoxic Lymphocytes in Cancer Immunotherapy. Transfus. Med. Hemotherapy 2017, 44, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Gleason, M.K.; Verneris, M.R.; Todhunter, D.A.; Zhang, B.; McCullar, V.; Zhou, S.X.; Panoskaltsis-Mortari, A.; Weiner, L.M.; Vallera, D.A.; Miller, J.S. Bispecific and trispecific killer cell engagers directly activate human NK cells through CD16 signaling and induce cytotoxicity and cytokine production. Mol. Cancer Ther. 2012, 11, 2674–2684. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.K.; Kang, S.; Youssef, Y.; Glankler, E.N.; Barrett, E.R.; Carter, A.M.; Ahmed, E.H.; Prasad, A.; Chen, L.; Zhang, J.; et al. A CS1-NKG2D Bispecific Antibody Collectively Activates Cytolytic Immune Cells against Multiple Myeloma. Cancer Immunol. Res. 2018, 6, 776–787. [Google Scholar] [CrossRef] [PubMed]
- Bottcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Reis e Sousa, C. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 2018, 172, 1022–1037. [Google Scholar] [CrossRef]
- Benson, D.M., Jr.; Bakan, C.E.; Mishra, A.; Hofmeister, C.C.; Efebera, Y.; Becknell, B.; Baiocchi, R.A.; Zhang, J.; Yu, J.; Smith, M.K.; et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: A therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood 2010, 116, 2286–2294. [Google Scholar] [CrossRef]
- Liu, Y.; Cheng, Y.; Xu, Y.; Wang, Z.; Du, X.; Li, C.; Peng, J.; Gao, L.; Liang, X.; Ma, C. Increased expression of programmed cell death protein 1 on NK cells inhibits NK-cell-mediated anti-tumor function and indicates poor prognosis in digestive cancers. Oncogene 2017, 36, 6143–6153. [Google Scholar] [CrossRef]
- Barry, K.C.; Hsu, J.; Broz, M.L.; Cueto, F.J.; Binnewies, M.; Combes, A.J.; Nelson, A.E.; Loo, K.; Kumar, R.; Rosenblum, M.D.; et al. A natural killer-dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments. Nat. Med. 2018, 24, 1178–1191. [Google Scholar] [CrossRef] [PubMed]
- Romagne, F.; Andre, P.; Spee, P.; Zahn, S.; Anfossi, N.; Gauthier, L.; Capanni, M.; Ruggeri, L.; Benson, D.M., Jr.; Blaser, B.W.; et al. Preclinical characterization of 1-7F9, a novel human anti-KIR receptor therapeutic antibody that augments natural killer-mediated killing of tumor cells. Blood 2009, 114, 2667–2677. [Google Scholar] [CrossRef] [PubMed]
- McWilliams, E.M.; Mele, J.M.; Cheney, C.; Timmerman, E.A.; Fiazuddin, F.; Strattan, E.J.; Mo, X.; Byrd, J.C.; Muthusamy, N.; Awan, F.T. Therapeutic CD94/NKG2A blockade improves natural killer cell dysfunction in chronic lymphocytic leukemia. Oncoimmunology 2016, 5, e1226720. [Google Scholar] [CrossRef] [PubMed]
- Weiden, P.L.; Flournoy, N.; Thomas, E.D.; Prentice, R.; Fefer, A.; Buckner, C.D.; Storb, R. Antileukemic effect of graft-versus-host disease in human recipients of allogeneic-marrow grafts. N. Engl. J. Med. 1979, 300, 1068–1073. [Google Scholar] [CrossRef]
- Ruggeri, L.; Mancusi, A.; Burchielli, E.; Capanni, M.; Carotti, A.; Aloisi, T.; Aversa, F.; Martelli, M.F.; Velardi, A. NK cell alloreactivity and allogeneic hematopoietic stem cell transplantation. Blood Cells Mol. Dis. 2008, 40, 84–90. [Google Scholar] [CrossRef]
- Kloess, S.; Kretschmer, A.; Stahl, L.; Fricke, S.; Koehl, U. CAR-Expressing Natural Killer Cells for Cancer Retargeting. Transfus. Med. Hemotherapy 2019, 46, 4–13. [Google Scholar] [CrossRef]
- Jiang, T.; Zhou, C.; Ren, S. Role of IL-2 in cancer immunotherapy. Oncoimmunology 2016, 5, e1163462. [Google Scholar] [CrossRef]
- Romee, R.; Leong, J.W.; Fehniger, T.A. Utilizing cytokines to function-enable human NK cells for the immunotherapy of cancer. Scientifica 2014, 2014, 205796. [Google Scholar] [CrossRef]
- Parihar, R.; Dierksheide, J.; Hu, Y.; Carson, W.E. IL-12 enhances the natural killer cell cytokine response to Ab-coated tumor cells. J. Clin. Investig. 2002, 110, 983–992. [Google Scholar] [CrossRef]
- Roda, J.M.; Parihar, R.; Lehman, A.; Mani, A.; Tridandapani, S.; Carson, W.E., 3rd. Interleukin-21 enhances NK cell activation in response to antibody-coated targets. J. Immunol. 2006, 177, 120–129. [Google Scholar] [CrossRef]
- Robinson, T.O.; Schluns, K.S. The potential and promise of IL-15 in immuno-oncogenic therapies. Immunol. Lett. 2017, 190, 159–168. [Google Scholar] [CrossRef]
- Margolin, K.; Morishima, C.; Velcheti, V.; Miller, J.S.; Lee, S.M.; Silk, A.W.; Holtan, S.G.; Lacroix, A.M.; Fling, S.P.; Kaiser, J.C.; et al. Phase I Trial of ALT-803, A Novel Recombinant IL15 Complex, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2018, 24, 5552–5561. [Google Scholar] [CrossRef]
- Pinette, A.; McMichael, E.; Courtney, N.B.; Duggan, M.; Benner, B.N.; Choueiry, F.; Yu, L.; Abood, D.; Mace, T.A.; Carson, W.E., 3rd. An IL-15-based superagonist ALT-803 enhances the NK cell response to cetuximab-treated squamous cell carcinoma of the head and neck. Cancer Immunol. Immunother. 2019, 68, 1379–1389. [Google Scholar] [CrossRef]
- Hagner, P.R.; Chiu, H.; Ortiz, M.; Apollonio, B.; Wang, M.; Couto, S.; Waldman, M.F.; Flynt, E.; Ramsay, A.G.; Trotter, M.; et al. Activity of lenalidomide in mantle cell lymphoma can be explained by NK cell-mediated cytotoxicity. Br. J. Haematol. 2017, 179, 399–409. [Google Scholar] [CrossRef]
- Gribben, J.G.; Fowler, N.; Morschhauser, F. Mechanisms of Action of Lenalidomide in B-Cell Non-Hodgkin Lymphoma. J. Clin. Oncol. 2015, 33, 2803–2811. [Google Scholar] [CrossRef]
- Acebes-Huerta, A.; Huergo-Zapico, L.; Gonzalez-Rodriguez, A.P.; Fernandez-Guizan, A.; Payer, A.R.; Lopez-Soto, A.; Gonzalez, S. Lenalidomide induces immunomodulation in chronic lymphocytic leukemia and enhances antitumor immune responses mediated by NK and CD4 T cells. Biomed Res. Int. 2014, 2014, 265840. [Google Scholar] [CrossRef]
- Gonzalez-Rodriguez, A.P.; Payer, A.R.; Acebes-Huerta, A.; Huergo-Zapico, L.; Villa-Alvarez, M.; Gonzalez-Garcia, E.; Gonzalez, S. Lenalidomide and chronic lymphocytic leukemia. Biomed Res. Int. 2013, 2013, 932010. [Google Scholar] [CrossRef][Green Version]
- Giuliani, M.; Janji, B.; Berchem, G. Activation of NK cells and disruption of PD-L1/PD-1 axis: Two different ways for lenalidomide to block myeloma progression. Oncotarget 2017, 8, 24031–24044. [Google Scholar] [CrossRef]
- Villa-Alvarez, M.; Sordo-Bahamonde, C.; Lorenzo-Herrero, S.; Gonzalez-Rodriguez, A.P.; Payer, A.R.; Gonzalez-Garcia, E.; Villa-Alvarez, M.C.; Lopez-Soto, A.; Gonzalez, S. Ig-Like Transcript 2 (ILT2) Blockade and Lenalidomide Restore NK Cell Function in Chronic Lymphocytic Leukemia. Front. Immunol. 2018, 9, 2917. [Google Scholar] [CrossRef]
- Chiu, H.; Trisal, P.; Bjorklund, C.; Carrancio, S.; Torano, E.G.; Guarinos, C.; Papazoglou, D.; Hagner, P.R.; Beldi-Ferchiou, A.; Tarte, K.; et al. Combination lenalidomide-rituximab immunotherapy activates anti-tumour immunity and induces tumour cell death by complementary mechanisms of action in follicular lymphoma. Br. J. Haematol. 2019, 185, 240–253. [Google Scholar] [CrossRef]
- Fionda, C.; Abruzzese, M.P.; Zingoni, A.; Cecere, F.; Vulpis, E.; Peruzzi, G.; Soriani, A.; Molfetta, R.; Paolini, R.; Ricciardi, M.R.; et al. The IMiDs targets IKZF-1/3 and IRF4 as novel negative regulators of NK cell-activating ligands expression in multiple myeloma. Oncotarget 2015, 6, 23609–23630. [Google Scholar] [CrossRef]
- Cifaldi, L.; Locatelli, F.; Marasco, E.; Moretta, L.; Pistoia, V. Boosting Natural Killer Cell-Based Immunotherapy with Anticancer Drugs: A Perspective. Trends Mol. Med. 2017, 23, 1156–1175. [Google Scholar] [CrossRef]
- Klingemann, H. Are natural killer cells superior CAR drivers? Oncoimmunology 2014, 3, e28147. [Google Scholar] [CrossRef]
- Ruggeri, L.; Capanni, M.; Casucci, M.; Volpi, I.; Tosti, A.; Perruccio, K.; Urbani, E.; Negrin, R.S.; Martelli, M.F.; Velardi, A. Role of natural killer cell alloreactivity in HLA-mismatched hematopoietic stem cell transplantation. Blood 1999, 94, 333–339. [Google Scholar] [CrossRef]
- Ruggeri, L.; Capanni, M.; Urbani, E.; Perruccio, K.; Shlomchik, W.D.; Tosti, A.; Posati, S.; Rogaia, D.; Frassoni, F.; Aversa, F.; et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science 2002, 295, 2097–2100. [Google Scholar] [CrossRef]
- Rezvani, K.; Rouce, R.H. The Application of Natural Killer Cell Immunotherapy for the Treatment of Cancer. Front. Immunol. 2015, 6, 578. [Google Scholar] [CrossRef]
- Stojanovic, A.; Cerwenka, A. Natural killer cells and solid tumors. J. Innate Immun. 2011, 3, 355–364. [Google Scholar] [CrossRef]
- Becker, P.S.; Suck, G.; Nowakowska, P.; Ullrich, E.; Seifried, E.; Bader, P.; Tonn, T.; Seidl, C. Selection and expansion of natural killer cells for NK cell-based immunotherapy. Cancer Immunol. Immunother. 2016, 65, 477–484. [Google Scholar] [CrossRef]
- Romee, R.; Rosario, M.; Berrien-Elliott, M.M.; Wagner, J.A.; Jewell, B.A.; Schappe, T.; Leong, J.W.; Abdel-Latif, S.; Schneider, S.E.; Willey, S.; et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci. Transl. Med. 2016, 8, 357ra123. [Google Scholar] [CrossRef]
- Schmidt-Wolf, I.G.; Negrin, R.S.; Kiem, H.P.; Blume, K.G.; Weissman, I.L. Use of a SCID mouse/human lymphoma model to evaluate cytokine-induced killer cells with potent antitumor cell activity. J. Exp. Med. 1991, 174, 139–149. [Google Scholar] [CrossRef]
- Uherek, C.; Tonn, T.; Uherek, B.; Becker, S.; Schnierle, B.; Klingemann, H.G.; Wels, W. Retargeting of natural killer-cell cytolytic activity to ErbB2-expressing cancer cells results in efficient and selective tumor cell destruction. Blood 2002, 100, 1265–1273. [Google Scholar] [CrossRef]
- Maki, G.; Klingemann, H.G.; Martinson, J.A.; Tam, Y.K. Factors regulating the cytotoxic activity of the human natural killer cell line, NK-92. J. Hematotherapy Stem Cell Res. 2001, 10, 369–383. [Google Scholar] [CrossRef]
- Cooley, S.; Xiao, F.; Pitt, M.; Gleason, M.; McCullar, V.; Bergemann, T.L.; McQueen, K.L.; Guethlein, L.A.; Parham, P.; Miller, J.S. A subpopulation of human peripheral blood NK cells that lacks inhibitory receptors for self-MHC is developmentally immature. Blood 2007, 110, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Suck, G.; Odendahl, M.; Nowakowska, P.; Seidl, C.; Wels, W.S.; Klingemann, H.G.; Tonn, T. NK-92: An ’off-the-shelf therapeutic’ for adoptive natural killer cell-based cancer immunotherapy. Cancer Immunol. Immunother. 2016, 65, 485–492. [Google Scholar] [CrossRef]
- Boissel, L.; Betancur-Boissel, M.; Lu, W.; Krause, D.S.; Van Etten, R.A.; Wels, W.S.; Klingemann, H. Retargeting NK-92 cells by means of CD19- and CD20-specific chimeric antigen receptors compares favorably with antibody-dependent cellular cytotoxicity. Oncoimmunology 2013, 2, e26527. [Google Scholar] [CrossRef] [PubMed]
- Boissel, L.; Betancur, M.; Wels, W.S.; Tuncer, H.; Klingemann, H. Transfection with mRNA for CD19 specific chimeric antigen receptor restores NK cell mediated killing of CLL cells. Leuk. Res. 2009, 33, 1255–1259. [Google Scholar] [CrossRef]
- Rezvani, A.R.; Maloney, D.G. Rituximab resistance. Best Pract. Res. Clin. Haematol. 2011, 24, 203–216. [Google Scholar] [CrossRef]
- Arribas, J.; Baselga, J.; Pedersen, K.; Parra-Palau, J.L. p95HER2 and breast cancer. Cancer Res. 2011, 71, 1515–1519. [Google Scholar] [CrossRef] [PubMed]
- Nakadate, Y.; Kodera, Y.; Kitamura, Y.; Shirasawa, S.; Tachibana, T.; Tamura, T.; Koizumi, F. KRAS mutation confers resistance to antibody-dependent cellular cytotoxicity of cetuximab against human colorectal cancer cells. Int. J. Cancer 2014, 134, 2146–2155. [Google Scholar] [CrossRef] [PubMed]
- Braig, F.; Kriegs, M.; Voigtlaender, M.; Habel, B.; Grob, T.; Biskup, K.; Blanchard, V.; Sack, M.; Thalhammer, A.; Ben Batalla, I.; et al. Cetuximab Resistance in Head and Neck Cancer Is Mediated by EGFR-K521 Polymorphism. Cancer Res. 2017, 77, 1188–1199. [Google Scholar] [CrossRef]
- Roederer, M.; Quaye, L.; Mangino, M.; Beddall, M.H.; Mahnke, Y.; Chattopadhyay, P.; Tosi, I.; Napolitano, L.; Terranova Barberio, M.; Menni, C.; et al. The genetic architecture of the human immune system: A bioresource for autoimmunity and disease pathogenesis. Cell 2015, 161, 387–403. [Google Scholar] [CrossRef]
- Moussa, P.; Marton, J.; Vidal, S.M.; Fodil-Cornu, N. Genetic dissection of NK cell responses. Front. Immunol. 2012, 3, 425. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.A.; Mangino, M.; Brumme, C.J.; Zhao, Z.Z.; Medland, S.E.; Wright, M.J.; Nyholt, D.R.; Gordon, S.; Campbell, M.; McEvoy, B.P.; et al. Quantitative trait loci for CD4:CD8 lymphocyte ratio are associated with risk of type 1 diabetes and HIV-1 immune control. Am. J. Hum. Genet. 2010, 86, 88–92. [Google Scholar] [CrossRef]
- Hall, M.A.; Ahmadi, K.R.; Norman, P.; Snieder, H.; MacGregor, A.J.; Vaughan, R.W.; Spector, T.D.; Lanchbury, J.S. Genetic influence on peripheral blood T lymphocyte levels. Genes Immun. 2000, 1, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Liu, Q.; Berger, C.T.; Keenan, B.T.; Kaliszewska, A.; Cheney, P.C.; Srivastava, G.P.; Castillo, I.W.; De Jager, P.L.; Alter, G. A 17q12 allele is associated with altered NK cell subsets and function. J. Immunol. 2012, 188, 3315–3322. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Matsuyama, S.; Miyake, S.; Suga, K.; Nakachi, K. Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: An 11-year follow-up study of a general population. Lancet 2000, 356, 1795–1799. [Google Scholar] [CrossRef]
- Klanova, M.; Oestergaard, M.Z.; Trneny, M.; Hiddemann, W.; Marcus, R.; Sehn, L.H.; Vitolo, U.; Bazeos, A.; Goede, V.; Zeuner, H.; et al. Prognostic Impact of Natural Killer Cell Count in Follicular Lymphoma and Diffuse Large B-cell Lymphoma Patients Treated with Immunochemotherapy. Clin. Cancer Res. 2019, 25, 4634–4643. [Google Scholar] [CrossRef]
- Coca, S.; Perez-Piqueras, J.; Martinez, D.; Colmenarejo, A.; Saez, M.A.; Vallejo, C.; Martos, J.A.; Moreno, M. The prognostic significance of intratumoral natural killer cells in patients with colorectal carcinoma. Cancer 1997, 79, 2320–2328. [Google Scholar] [CrossRef]
- Salgado, R.; Denkert, C.; Campbell, C.; Savas, P.; Nuciforo, P.; Aura, C.; de Azambuja, E.; Eidtmann, H.; Ellis, C.E.; Baselga, J.; et al. Tumor-Infiltrating Lymphocytes and Associations With Pathological Complete Response and Event-Free Survival in HER2-Positive Early-Stage Breast Cancer Treated With Lapatinib and Trastuzumab: A Secondary Analysis of the NeoALTTO Trial. JAMA Oncol. 2015, 1, 448–454. [Google Scholar] [CrossRef]
- Denkert, C.; von Minckwitz, G.; Darb-Esfahani, S.; Lederer, B.; Heppner, B.I.; Weber, K.E.; Budczies, J.; Huober, J.; Klauschen, F.; Furlanetto, J.; et al. Tumour-infiltrating lymphocytes and prognosis in different subtypes of breast cancer: A pooled analysis of 3771 patients treated with neoadjuvant therapy. Lancet Oncol. 2018, 19, 40–50. [Google Scholar] [CrossRef]
- Danielou-Lazareth, A.; Henry, G.; Geromin, D.; Khaznadar, Z.; Briere, J.; Tamouza, R.; Cayuela, J.M.; Thieblemont, C.; Toubert, A.; Dulphy, N. At diagnosis, diffuse large B-cell lymphoma patients show impaired rituximab-mediated NK-cell cytotoxicity. Eur. J. Immunol. 2013, 43, 1383–1388. [Google Scholar] [CrossRef]
- Hatjiharissi, E.; Xu, L.; Santos, D.D.; Hunter, Z.R.; Ciccarelli, B.T.; Verselis, S.; Modica, M.; Cao, Y.; Manning, R.J.; Leleu, X.; et al. Increased natural killer cell expression of CD16, augmented binding and ADCC activity to rituximab among individuals expressing the Fc{gamma}RIIIa-158 V/V and V/F polymorphism. Blood 2007, 110, 2561–2564. [Google Scholar] [CrossRef]
- Cartron, G.; Dacheux, L.; Salles, G.; Solal-Celigny, P.; Bardos, P.; Colombat, P.; Watier, H. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood 2002, 99, 754–758. [Google Scholar] [CrossRef] [PubMed]
- Weng, W.K.; Levy, R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J. Clin. Oncol. 2003, 21, 3940–3947. [Google Scholar] [CrossRef] [PubMed]
- Treon, S.P.; Hansen, M.; Branagan, A.R.; Verselis, S.; Emmanouilides, C.; Kimby, E.; Frankel, S.R.; Touroutoglou, N.; Turnbull, B.; Anderson, K.C.; et al. Polymorphisms in FcgammaRIIIA (CD16) receptor expression are associated with clinical response to rituximab in Waldenstrom’s macroglobulinemia. J. Clin. Oncol. 2005, 23, 474–481. [Google Scholar] [CrossRef]
- Kim, D.H.; Jung, H.D.; Kim, J.G.; Lee, J.J.; Yang, D.H.; Park, Y.H.; Do, Y.R.; Shin, H.J.; Kim, M.K.; Hyun, M.S.; et al. FCGR3A gene polymorphisms may correlate with response to frontline R-CHOP therapy for diffuse large B-cell lymphoma. Blood 2006, 108, 2720–2725. [Google Scholar] [CrossRef] [PubMed]
- Farag, S.S.; Flinn, I.W.; Modali, R.; Lehman, T.A.; Young, D.; Byrd, J.C. Fc gamma RIIIa and Fc gamma RIIa polymorphisms do not predict response to rituximab in B-cell chronic lymphocytic leukemia. Blood 2004, 103, 1472–1474. [Google Scholar] [CrossRef] [PubMed]
- Oboshi, W.; Watanabe, T.; Yukimasa, N.; Ueno, I.; Aki, K.; Tada, T.; Hosoi, E. SNPs rs4656317 and rs12071048 located within an enhancer in FCGR3A are in strong linkage disequilibrium with rs396991 and influence NK cell-mediated ADCC by transcriptional regulation. Hum. Immunol. 2016, 77, 997–1003. [Google Scholar] [CrossRef]
- Musolino, A.; Naldi, N.; Bortesi, B.; Pezzuolo, D.; Capelletti, M.; Missale, G.; Laccabue, D.; Zerbini, A.; Camisa, R.; Bisagni, G.; et al. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J. Clin. Oncol. 2008, 26, 1789–1796. [Google Scholar] [CrossRef]
- Tamura, K.; Shimizu, C.; Hojo, T.; Akashi-Tanaka, S.; Kinoshita, T.; Yonemori, K.; Kouno, T.; Katsumata, N.; Ando, M.; Aogi, K.; et al. FcγR2A and 3A polymorphisms predict clinical outcome of trastuzumab in both neoadjuvant and metastatic settings in patients with HER2-positive breast cancer. Ann. Oncol. 2010, 22, 1302–1307. [Google Scholar] [CrossRef]
- Bibeau, F.; Lopez-Crapez, E.; Di Fiore, F.; Thezenas, S.; Ychou, M.; Blanchard, F.; Lamy, A.; Penault-Llorca, F.; Frebourg, T.; Michel, P.; et al. Impact of Fc{gamma}RIIa-Fc{gamma}RIIIa polymorphisms and KRAS mutations on the clinical outcome of patients with metastatic colorectal cancer treated with cetuximab plus irinotecan. J. Clin. Oncol. 2009, 27, 1122–1129. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Albaitero, A.; Lee, S.C.; Morgan, S.; Grandis, J.R.; Gooding, W.E.; Ferrone, S.; Ferris, R.L. Role of polymorphic Fc gamma receptor IIIa and EGFR expression level in cetuximab mediated, NK cell dependent in vitro cytotoxicity of head and neck squamous cell carcinoma cells. Cancer Immunol. Immunother. 2009, 58, 1853–1864. [Google Scholar] [CrossRef] [PubMed]
- Umaña, P.; Jean–Mairet, J.; Moudry, R.; Amstutz, H.; Bailey, J.E. Engineered glycoforms of an antineuroblastoma IgG1 with optimized antibody-dependent cellular cytotoxic activity. Nat. Biotechnol. 1999, 17, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Prica, A.; Crump, M. Improving CD20 antibody therapy: Obinutuzumab in lymphoproliferative disorders. Leuk. Lymphoma 2019, 60, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Anfossi, N.; Andre, P.; Guia, S.; Falk, C.S.; Roetynck, S.; Stewart, C.A.; Breso, V.; Frassati, C.; Reviron, D.; Middleton, D.; et al. Human NK cell education by inhibitory receptors for MHC class I. Immunity 2006, 25, 331–342. [Google Scholar] [CrossRef]
- Forlenza, C.J.; Boudreau, J.E.; Zheng, J.; Le Luduec, J.B.; Chamberlain, E.; Heller, G.; Cheung, N.K.; Hsu, K.C. KIR3DL1 Allelic Polymorphism and HLA-B Epitopes Modulate Response to Anti-GD2 Monoclonal Antibody in Patients With Neuroblastoma. J. Clin. Oncol. 2016, 34, 2443–2451. [Google Scholar] [CrossRef]
- Levinson, R.D.; Yung, M.; Meguro, A.; Ashouri, E.; Yu, F.; Mizuki, N.; Ohno, S.; Rajalingam, R. KIR and HLA Genotypes Implicated in Reduced Killer Lymphocytes Immunity Are Associated with Vogt-Koyanagi-Harada Disease. PLoS ONE 2016, 11, e0160392. [Google Scholar] [CrossRef]
- Erbe, A.K.; Wang, W.; Reville, P.K.; Carmichael, L.; Kim, K.; Mendonca, E.A.; Song, Y.; Hank, J.A.; London, W.B.; Naranjo, A.; et al. HLA-Bw4-I-80 Isoform Differentially Influences Clinical Outcome As Compared to HLA-Bw4-T-80 and HLA-A-Bw4 Isoforms in Rituximab or Dinutuximab-Based Cancer Immunotherapy. Front. Immunol. 2017, 8, 675. [Google Scholar] [CrossRef]
- Du, J.; Lopez-Verges, S.; Pitcher, B.N.; Johnson, J.; Jung, S.H.; Zhou, L.; Hsu, K.; Czuczman, M.S.; Cheson, B.; Kaplan, L.; et al. CALGB 150905 (Alliance): Rituximab broadens the antilymphoma response by activating unlicensed NK cells. Cancer Immunol. Res. 2014, 2, 878–889. [Google Scholar] [CrossRef]
- Terszowski, G.; Klein, C.; Stern, M. KIR/HLA interactions negatively affect rituximab- but not GA101 (obinutuzumab)-induced antibody-dependent cellular cytotoxicity. J. Immunol. 2014, 192, 5618–5624. [Google Scholar] [CrossRef]
- Brunstein, C.G.; Wagner, J.E.; Weisdorf, D.J.; Cooley, S.; Noreen, H.; Barker, J.N.; DeFor, T.; Verneris, M.R.; Blazar, B.R.; Miller, J.S. Negative effect of KIR alloreactivity in recipients of umbilical cord blood transplant depends on transplantation conditioning intensity. Blood 2009, 113, 5628–5634. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.J.; Zhao, X.Y.; Liu, D.H.; Liu, K.Y.; Xu, L.P. Deleterious effects of KIR ligand incompatibility on clinical outcomes in haploidentical hematopoietic stem cell transplantation without in vitro T-cell depletion. Leukemia 2007, 21, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Faridi, R.M.; Kemp, T.J.; Dharmani-Khan, P.; Lewis, V.; Tripathi, G.; Rajalingam, R.; Daly, A.; Berka, N.; Storek, J.; Masood Khan, F. Donor-Recipient Matching for KIR Genotypes Reduces Chronic GVHD and Missing Inhibitory KIR Ligands Protect against Relapse after Myeloablative, HLA Matched Hematopoietic Cell Transplantation. PLoS ONE 2016, 11, e0158242. [Google Scholar] [CrossRef] [PubMed]
- Cooley, S.; Trachtenberg, E.; Bergemann, T.L.; Saeteurn, K.; Klein, J.; Le, C.T.; Marsh, S.G.; Guethlein, L.A.; Parham, P.; Miller, J.S.; et al. Donors with group B KIR haplotypes improve relapse-free survival after unrelated hematopoietic cell transplantation for acute myelogenous leukemia. Blood 2009, 113, 726–732. [Google Scholar] [CrossRef]
- Venstrom, J.M.; Pittari, G.; Gooley, T.A.; Chewning, J.H.; Spellman, S.; Haagenson, M.; Gallagher, M.M.; Malkki, M.; Petersdorf, E.; Dupont, B.; et al. HLA-C-dependent prevention of leukemia relapse by donor activating KIR2DS1. New Engl. J. Med. 2012, 367, 805–816. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Zhao, W.L.; Daneshpouy, M.E.; Mounier, N.; Briere, J.; Leboeuf, C.; Plassa, L.F.; Turpin, E.; Cayuela, J.M.; Ameisen, J.C.; Gisselbrecht, C.; et al. Prognostic significance of bcl-xL gene expression and apoptotic cell counts in follicular lymphoma. Blood 2004, 103, 695–697. [Google Scholar] [CrossRef]
- Gisselbrecht, C.; Glass, B.; Mounier, N.; Singh Gill, D.; Linch, D.C.; Trneny, M.; Bosly, A.; Ketterer, N.; Shpilberg, O.; Hagberg, H.; et al. Salvage regimens with autologous transplantation for relapsed large B-cell lymphoma in the rituximab era. J. Clin. Oncol. 2010, 28, 4184–4190. [Google Scholar] [CrossRef]
- Bannerji, R.; Kitada, S.; Flinn, I.W.; Pearson, M.; Young, D.; Reed, J.C.; Byrd, J.C. Apoptotic-regulatory and complement-protecting protein expression in chronic lymphocytic leukemia: Relationship to in vivo rituximab resistance. J. Clin. Oncol. 2003, 21, 1466–1471. [Google Scholar] [CrossRef]
- Berns, K.; Horlings, H.M.; Hennessy, B.T.; Madiredjo, M.; Hijmans, E.M.; Beelen, K.; Linn, S.C.; Gonzalez-Angulo, A.M.; Stemke-Hale, K.; Hauptmann, M.; et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell 2007, 12, 395–402. [Google Scholar] [CrossRef]
- Brand, T.M.; Lida, M.; Wheeler, D.L. Molecular mechanisms of resistance to the EGFR monoclonal antibody cetuximab. Cancer Biol. Ther. 2011, 11, 777–792. [Google Scholar] [CrossRef] [PubMed]
- Jazirehi, A.R.; Vega, M.I.; Bonavida, B. Development of rituximab-resistant lymphoma clones with altered cell signaling and cross-resistance to chemotherapy. Cancer Res. 2007, 67, 1270–1281. [Google Scholar] [CrossRef]
- Karapetis, C.S.; Khambata-Ford, S.; Jonker, D.J.; O’Callaghan, C.J.; Tu, D.; Tebbutt, N.C.; Simes, R.J.; Chalchal, H.; Shapiro, J.D.; Robitaille, S.; et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med. 2008, 359, 1757–1765. [Google Scholar] [CrossRef]
- Luque-Cabal, M.; Garcia-Teijido, P.; Fernandez-Perez, Y.; Sanchez-Lorenzo, L.; Palacio-Vazquez, I. Mechanisms Behind the Resistance to Trastuzumab in HER2-Amplified Breast Cancer and Strategies to Overcome It. Clin. Med. Insights Oncol. 2016, 10, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Olejniczak, S.H.; Hernandez-Ilizaliturri, F.J.; Clements, J.L.; Czuczman, M.S. Acquired resistance to rituximab is associated with chemotherapy resistance resulting from decreased Bax and Bak expression. Clin. Cancer Res. 2008, 14, 1550–1560. [Google Scholar] [CrossRef] [PubMed]
- Vega, M.I.; Jazirehi, A.R.; Huerta-Yepez, S.; Bonavida, B. Rituximab-induced inhibition of YY1 and Bcl-xL expression in Ramos non-Hodgkin’s lymphoma cell line via inhibition of NF-kappa B activity: Role of YY1 and Bcl-xL in Fas resistance and chemoresistance, respectively. J. Immunol. 2005, 175, 2174–2183. [Google Scholar] [CrossRef]
- Obexer, P.; Ausserlechner, M.J. X-linked inhibitor of apoptosis protein - a critical death resistance regulator and therapeutic target for personalized cancer therapy. Front. Oncol. 2014, 4, 197. [Google Scholar] [CrossRef]
- Evans, M.K.; Sauer, S.J.; Nath, S.; Robinson, T.J.; Morse, M.A.; Devi, G.R. X-linked inhibitor of apoptosis protein mediates tumor cell resistance to antibody-dependent cellular cytotoxicity. Cell Death Dis. 2016, 7, e2073. [Google Scholar] [CrossRef]
- Li, R.; Ruttinger, D.; Urba, W.; Fox, B.A.; Hu, H.M. Targeting and amplification of immune killing of tumor cells by pro-Smac. Int. J. Cancer 2004, 109, 85–94. [Google Scholar] [CrossRef]
- Pan, W.; Luo, Q.; Yan, X.; Yuan, L.; Yi, H.; Zhang, L.; Li, B.; Zhang, Y.; Sun, J.; Qiu, M.Z.; et al. A novel SMAC mimetic APG-1387 exhibits dual antitumor effect on HBV-positive hepatocellular carcinoma with high expression of cIAP2 by inducing apoptosis and enhancing innate anti-tumor immunity. Biochem. Pharmacol. 2018, 154, 127–135. [Google Scholar] [CrossRef]
- Fischer, K.; Tognarelli, S.; Roesler, S.; Boedicker, C.; Schubert, R.; Steinle, A.; Klingebiel, T.; Bader, P.; Fulda, S.; Ullrich, E. The Smac Mimetic BV6 Improves NK Cell-Mediated Killing of Rhabdomyosarcoma Cells by Simultaneously Targeting Tumor and Effector Cells. Front. Immunol. 2017, 8, 202. [Google Scholar] [CrossRef]
- Brinkmann, K.; Hombach, A.; Seeger, J.M.; Wagner-Stippich, D.; Klubertz, D.; Kronke, M.; Abken, H.; Kashkar, H. Second mitochondria-derived activator of caspase (SMAC) mimetic potentiates tumor susceptibility toward natural killer cell-mediated killing. Leuk. Lymphoma 2014, 55, 645–651. [Google Scholar] [CrossRef]
- Dougan, S.K.; Dougan, M. Regulation of innate and adaptive antitumor immunity by IAP antagonists. Immunotherapy 2018, 10, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Kearney, C.J.; Lalaoui, N.; Freeman, A.J.; Ramsbottom, K.M.; Silke, J.; Oliaro, J. PD-L1 and IAPs co-operate to protect tumors from cytotoxic lymphocyte-derived TNF. Cell Death Differ. 2017, 24, 1705–1716. [Google Scholar] [CrossRef] [PubMed]
- Runckel, K.; Barth, M.J.; Mavis, C.; Gu, J.J.; Hernandez-Ilizaliturri, F.J. The SMAC mimetic LCL-161 displays antitumor activity in preclinical models of rituximab-resistant B-cell lymphoma. Blood Adv. 2018, 2, 3516–3525. [Google Scholar] [CrossRef]
- Rettinger, E.; Glatthaar, A.; Abhari, B.A.; Oelsner, S.; Pfirrmann, V.; Huenecke, S.; Kuci, S.; Kreyenberg, H.; Willasch, A.M.; Klingebiel, T.; et al. SMAC Mimetic BV6 Enables Sensitization of Resistant Tumor Cells but also Affects Cytokine-Induced Killer (CIK) Cells: A Potential Challenge for Combination Therapy. Front. Pediatrics 2014, 2, 75. [Google Scholar] [CrossRef][Green Version]
- Beug, S.T.; Conrad, D.P.; Alain, T.; Korneluk, R.G.; Lacasse, E.C. Combinatorial cancer immunotherapy strategies with proapoptotic small-molecule IAP antagonists. Int. J. Dev. Biol. 2015, 59, 141–147. [Google Scholar] [CrossRef]
- Beug, S.T.; Beauregard, C.E.; Healy, C.; Sanda, T.; St-Jean, M.; Chabot, J.; Walker, D.E.; Mohan, A.; Earl, N.; Lun, X.; et al. Smac mimetics synergize with immune checkpoint inhibitors to promote tumour immunity against glioblastoma. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Shekhar, T.M.; Burvenich, I.J.G.; Harris, M.A.; Rigopoulos, A.; Zanker, D.; Spurling, A.; Parker, B.S.; Walkley, C.R.; Scott, A.M.; Hawkins, C.J. Smac mimetics LCL161 and GDC-0152 inhibit osteosarcoma growth and metastasis in mice. BMC Cancer 2019, 19, 924. [Google Scholar] [CrossRef] [PubMed]
- Marquez, M.E.; Hernandez-Uzcategui, O.; Cornejo, A.; Vargas, P.; Da Costa, O. Bone marrow stromal mesenchymal cells induce down regulation of CD20 expression on B-CLL: Implications for rituximab resistance in CLL. Br. J. Haematol. 2015, 169, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The three Es of cancer immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef]
- Vesely, M.D.; Kershaw, M.H.; Schreiber, R.D.; Smyth, M.J. Natural innate and adaptive immunity to cancer. Annu. Rev. Immunol. 2011, 29, 235–271. [Google Scholar] [CrossRef] [PubMed]
- Guerra, N.; Tan, Y.X.; Joncker, N.T.; Choy, A.; Gallardo, F.; Xiong, N.; Knoblaugh, S.; Cado, D.; Greenberg, N.M.; Raulet, D.H. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity 2008, 28, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Iguchi-Manaka, A.; Kai, H.; Yamashita, Y.; Shibata, K.; Tahara-Hanaoka, S.; Honda, S.; Yasui, T.; Kikutani, H.; Shibuya, K.; Shibuya, A. Accelerated tumor growth in mice deficient in DNAM-1 receptor. J. Exp. Med. 2008, 205, 2959–2964. [Google Scholar] [CrossRef] [PubMed]
- Elboim, M.; Gazit, R.; Gur, C.; Ghadially, H.; Betser-Cohen, G.; Mandelboim, O. Tumor immunoediting by NKp46. J. Immunol. 2010, 184, 5637–5644. [Google Scholar] [CrossRef]
- O’Sullivan, T.; Saddawi-Konefka, R.; Vermi, W.; Koebel, C.M.; Arthur, C.; White, J.M.; Uppaluri, R.; Andrews, D.M.; Ngiow, S.F.; Teng, M.W.; et al. Cancer immunoediting by the innate immune system in the absence of adaptive immunity. J. Exp. Med. 2012, 209, 1869–1882. [Google Scholar] [CrossRef]
- Guillerey, C.; Smyth, M.J. NK Cells and Cancer Immunoediting. Curr. Top. Microbiol. Immunol. 2016, 395, 115–145. [Google Scholar] [CrossRef]
- Holdenrieder, S.; Stieber, P.; Peterfi, A.; Nagel, D.; Steinle, A.; Salih, H.R. Soluble MICA in malignant diseases. Int. J. Cancer 2006, 118, 684–687. [Google Scholar] [CrossRef]
- Salih, H.R.; Rammensee, H.G.; Steinle, A. Cutting edge: Down-regulation of MICA on human tumors by proteolytic shedding. J. Immunol. 2002, 169, 4098–4102. [Google Scholar] [CrossRef]
- Chen, Y.; Lin, W.S.; Zhu, W.F.; Lin, J.; Zhou, Z.F.; Huang, C.Z.; Chen, G.; Shi, Y.; Guo, Z.Q.; Ye, Y.B. Tumor MICA status predicts the efficacy of immunotherapy with cytokine-induced killer cells for patients with gastric cancer. Immunol. Res. 2016, 64, 251–259. [Google Scholar] [CrossRef]
- Fan, X.Y.; Wang, P.Y.; Zhang, C.; Zhang, Y.L.; Fu, Y.; Zhang, C.; Li, Q.X.; Zhou, J.N.; Shan, B.E.; He, D.W. All-trans retinoic acid enhances cytotoxicity of CIK cells against human lung adenocarcinoma by upregulating MICA and IL-2 secretion. Sci. Rep. 2017, 7, 16481. [Google Scholar] [CrossRef]
- Rautela, J.; Baschuk, N.; Slaney, C.Y.; Jayatilleke, K.M.; Xiao, K.; Bidwell, B.N.; Lucas, E.C.; Hawkins, E.D.; Lock, P.; Wong, C.S.; et al. Loss of Host Type-I IFN Signaling Accelerates Metastasis and Impairs NK-cell Antitumor Function in Multiple Models of Breast Cancer. Cancer Immunol. Res. 2015, 3, 1207–1217. [Google Scholar] [CrossRef]
- Freeman, A.J.; Vervoort, S.J.; Ramsbottom, K.M.; Kelly, M.J.; Michie, J.; Pijpers, L.; Johnstone, R.W.; Kearney, C.J.; Oliaro, J. Natural Killer Cells Suppress T Cell-Associated Tumor Immune Evasion. Cell Rep. 2019, 28, 2784–2794. [Google Scholar] [CrossRef]
- Guillerey, C.; Huntington, N.D.; Smyth, M.J. Targeting natural killer cells in cancer immunotherapy. Nat. Immunol. 2016, 17, 1025–1036. [Google Scholar] [CrossRef]
- Grossenbacher, S.K.; Canter, R.J.; Murphy, W.J. Natural killer cell immunotherapy to target stem-like tumor cells. J. Immunother. Cancer 2016, 4, 19. [Google Scholar] [CrossRef]
- Tallerico, R.; Todaro, M.; Di Franco, S.; Maccalli, C.; Garofalo, C.; Sottile, R.; Palmieri, C.; Tirinato, L.; Pangigadde, P.N.; La Rocca, R.; et al. Human NK cells selective targeting of colon cancer-initiating cells: A role for natural cytotoxicity receptors and MHC class I molecules. J. Immunol. 2013, 190, 2381–2390. [Google Scholar] [CrossRef] [PubMed]
- Castriconi, R.; Daga, A.; Dondero, A.; Zona, G.; Poliani, P.L.; Melotti, A.; Griffero, F.; Marubbi, D.; Spaziante, R.; Bellora, F.; et al. NK cells recognize and kill human glioblastoma cells with stem cell-like properties. J. Immunol. 2009, 182, 3530–3539. [Google Scholar] [CrossRef] [PubMed]
- Pietra, G.; Manzini, C.; Vitale, M.; Balsamo, M.; Ognio, E.; Boitano, M.; Queirolo, P.; Moretta, L.; Mingari, M.C. Natural killer cells kill human melanoma cells with characteristics of cancer stem cells. Int. Immunol. 2009, 21, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.A.; Meyer, J.R.; Greco, S.J.; Corcoran, K.E.; Bryan, M.; Rameshwar, P. Mesenchymal stem cells protect breast cancer cells through regulatory T cells: Role of mesenchymal stem cell-derived TGF-beta. J. Immunol. 2010, 184, 5885–5894. [Google Scholar] [CrossRef] [PubMed]
- Kozlowska, A.K.; Tseng, H.C.; Kaur, K.; Topchyan, P.; Inagaki, A.; Bui, V.T.; Kasahara, N.; Cacalano, N.; Jewett, A. Resistance to cytotoxicity and sustained release of interleukin-6 and interleukin-8 in the presence of decreased interferon-gamma after differentiation of glioblastoma by human natural killer cells. Cancer Immunol. Immunother. 2016, 65, 1085–1097. [Google Scholar] [CrossRef] [PubMed]
- Garrido, F. MHC/HLA Class I Loss in Cancer Cells. Adv. Exp. Med. Biol. 2019, 1151, 15–78. [Google Scholar] [CrossRef]
- Zeestraten, E.C.; Reimers, M.S.; Saadatmand, S.; Goossens-Beumer, I.J.; Dekker, J.W.; Liefers, G.J.; van den Elsen, P.J.; van de Velde, C.J.; Kuppen, P.J. Combined analysis of HLA class I, HLA-E and HLA-G predicts prognosis in colon cancer patients. Br. J. Cancer 2014, 110, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, S.; Beziat, V.; Dhedin, N.; Kuentz, M.; Vernant, J.P.; Debre, P.; Vieillard, V. HLA-E upregulation on IFN-gamma-activated AML blasts impairs CD94/NKG2A-dependent NK cytolysis after haplo-mismatched hematopoietic SCT. Bone Marrow Transplant. 2009, 43, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Morandi, F.; Pistoia, V. Interactions between HLA-G and HLA-E in Physiological and Pathological Conditions. Front. Immunol. 2014, 5, 394. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, S.; Dhedin, N.; Vernant, J.P.; Kuentz, M.; Al Jijakli, A.; Rouas-Freiss, N.; Carosella, E.D.; Boudifa, A.; Debre, P.; Vieillard, V. NK-cell reconstitution after haploidentical hematopoietic stem-cell transplantations: Immaturity of NK cells and inhibitory effect of NKG2A override GvL effect. Blood 2005, 105, 4135–4142. [Google Scholar] [CrossRef] [PubMed]
- Ruggeri, L.; Urbani, E.; Andre, P.; Mancusi, A.; Tosti, A.; Topini, F.; Blery, M.; Animobono, L.; Romagne, F.; Wagtmann, N.; et al. Effects of anti-NKG2A antibody administration on leukemia and normal hematopoietic cells. Haematologica 2016, 101, 626–633. [Google Scholar] [CrossRef]
- Czuczman, M.S.; Olejniczak, S.; Gowda, A.; Kotowski, A.; Binder, A.; Kaur, H.; Knight, J.; Starostik, P.; Deans, J.; Hernandez-Ilizaliturri, F.J. Acquirement of rituximab resistance in lymphoma cell lines is associated with both global CD20 gene and protein down-regulation regulated at the pretranscriptional and posttranscriptional levels. Clin. Cancer Res. 2008, 14, 1561–1570. [Google Scholar] [CrossRef]
- Hiraga, J.; Tomita, A.; Sugimoto, T.; Shimada, K.; Ito, M.; Nakamura, S.; Kiyoi, H.; Kinoshita, T.; Naoe, T. Down-regulation of CD20 expression in B-cell lymphoma cells after treatment with rituximab-containing combination chemotherapies: Its prevalence and clinical significance. Blood 2009, 113, 4885–4893. [Google Scholar] [CrossRef]
- Terui, Y.; Mishima, Y.; Sugimura, N.; Kojima, K.; Sakurai, T.; Mishima, Y.; Kuniyoshi, R.; Taniyama, A.; Yokoyama, M.; Sakajiri, S.; et al. Identification of CD20 C-terminal deletion mutations associated with loss of CD20 expression in non-Hodgkin’s lymphoma. Clin. Cancer Res. 2009, 15, 2523–2530. [Google Scholar] [CrossRef]
- Beers, S.A.; French, R.R.; Chan, H.T.; Lim, S.H.; Jarrett, T.C.; Vidal, R.M.; Wijayaweera, S.S.; Dixon, S.V.; Kim, H.; Cox, K.L.; et al. Antigenic modulation limits the efficacy of anti-CD20 antibodies: Implications for antibody selection. Blood 2010, 115, 5191–5201. [Google Scholar] [CrossRef]
- Winiarska, M.; Bil, J.; Wilczek, E.; Wilczynski, G.M.; Lekka, M.; Engelberts, P.J.; Mackus, W.J.; Gorska, E.; Bojarski, L.; Stoklosa, T.; et al. Statins impair antitumor effects of rituximab by inducing conformational changes of CD20. PLoS Med. 2008, 5, e64. [Google Scholar] [CrossRef]
- Battke, C.; Ruiss, R.; Welsch, U.; Wimberger, P.; Lang, S.; Jochum, S.; Zeidler, R. Tumour exosomes inhibit binding of tumour-reactive antibodies to tumour cells and reduce ADCC. Cancer Immunol. Immunother. 2011, 60, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, A.E.; Jungersen, M.B.; Pedersen, C.D. Monocytes mediate shaving of B-cell-bound anti-CD20 antibodies. Immunology 2011, 133, 239–245. [Google Scholar] [CrossRef]
- Sperinde, J.; Jin, X.; Banerjee, J.; Penuel, E.; Saha, A.; Diedrich, G.; Huang, W.; Leitzel, K.; Weidler, J.; Ali, S.M.; et al. Quantitation of p95HER2 in paraffin sections by using a p95-specific antibody and correlation with outcome in a cohort of trastuzumab-treated breast cancer patients. Clin. Cancer Res. 2010, 16, 4226–4235. [Google Scholar] [CrossRef] [PubMed]
- Scaltriti, M.; Rojo, F.; Ocana, A.; Anido, J.; Guzman, M.; Cortes, J.; Di Cosimo, S.; Matias-Guiu, X.; Ramon y Cajal, S.; Arribas, J.; et al. Expression of p95HER2, a truncated form of the HER2 receptor, and response to anti-HER2 therapies in breast cancer. J. Natl. Cancer Inst. 2007, 99, 628–638. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Mamessier, E.; Sylvain, A.; Thibult, M.L.; Houvenaeghel, G.; Jacquemier, J.; Castellano, R.; Goncalves, A.; Andre, P.; Romagne, F.; Thibault, G.; et al. Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J. Clin. Investig. 2011, 121, 3609–3622. [Google Scholar] [CrossRef]
- Platonova, S.; Cherfils-Vicini, J.; Damotte, D.; Crozet, L.; Vieillard, V.; Validire, P.; Andre, P.; Dieu-Nosjean, M.C.; Alifano, M.; Regnard, J.F.; et al. Profound coordinated alterations of intratumoral NK cell phenotype and function in lung carcinoma. Cancer Res. 2011, 71, 5412–5422. [Google Scholar] [CrossRef]
- Halama, N.; Braun, M.; Kahlert, C.; Spille, A.; Quack, C.; Rahbari, N.; Koch, M.; Weitz, J.; Kloor, M.; Zoernig, I.; et al. Natural killer cells are scarce in colorectal carcinoma tissue despite high levels of chemokines and cytokines. Clin. Cancer Res. 2011, 17, 678–689. [Google Scholar] [CrossRef]
- Pesce, S.; Tabellini, G.; Cantoni, C.; Patrizi, O.; Coltrini, D.; Rampinelli, F.; Matta, J.; Vivier, E.; Moretta, A.; Parolini, S.; et al. B7-H6-mediated downregulation of NKp30 in NK cells contributes to ovarian carcinoma immune escape. Oncoimmunology 2015, 4, e1001224. [Google Scholar] [CrossRef]
- Han, B.; Mao, F.Y.; Zhao, Y.L.; Lv, Y.P.; Teng, Y.S.; Duan, M.; Chen, W.; Cheng, P.; Wang, T.T.; Liang, Z.Y.; et al. Altered NKp30, NKp46, NKG2D, and DNAM-1 Expression on Circulating NK Cells Is Associated with Tumor Progression in Human Gastric Cancer. J. Immunol. Res. 2018, 2018, 6248590. [Google Scholar] [CrossRef]
- Sanchez, C.J.; Le Treut, T.; Boehrer, A.; Knoblauch, B.; Imbert, J.; Olive, D.; Costello, R.T. Natural killer cells and malignant haemopathies: A model for the interaction of cancer with innate immunity. Cancer Immunol. Immunother. 2011, 60, 1–13. [Google Scholar] [CrossRef]
- Garcia-Iglesias, T.; Del Toro-Arreola, A.; Albarran-Somoza, B.; Del Toro-Arreola, S.; Sanchez-Hernandez, P.E.; Ramirez-Duenas, M.G.; Balderas-Pena, L.M.; Bravo-Cuellar, A.; Ortiz-Lazareno, P.C.; Daneri-Navarro, A. Low NKp30, NKp46 and NKG2D expression and reduced cytotoxic activity on NK cells in cervical cancer and precursor lesions. BMC Cancer 2009, 9, 186. [Google Scholar] [CrossRef]
- Fregni, G.; Perier, A.; Pittari, G.; Jacobelli, S.; Sastre, X.; Gervois, N.; Allard, M.; Bercovici, N.; Avril, M.F.; Caignard, A. Unique functional status of natural killer cells in metastatic stage IV melanoma patients and its modulation by chemotherapy. Clin. Cancer Res. 2011, 17, 2628–2637. [Google Scholar] [CrossRef]
- Zaiatz-Bittencourt, V.; Finlay, D.K.; Gardiner, C.M. Canonical TGF-beta Signaling Pathway Represses Human NK Cell Metabolism. J. Immunol. 2018, 200, 3934–3941. [Google Scholar] [CrossRef] [PubMed]
- Castriconi, R.; Cantoni, C.; Della Chiesa, M.; Vitale, M.; Marcenaro, E.; Conte, R.; Biassoni, R.; Bottino, C.; Moretta, L.; Moretta, A. Transforming growth factor beta 1 inhibits expression of NKp30 and NKG2D receptors: Consequences for the NK-mediated killing of dendritic cells. Proc. Natl. Acad. Sci. USA 2003, 100, 4120–4125. [Google Scholar] [CrossRef] [PubMed]
- Park, A.; Lee, Y.; Kim, M.S.; Kang, Y.J.; Park, Y.J.; Jung, H.; Kim, T.D.; Lee, H.G.; Choi, I.; Yoon, S.R. Prostaglandin E2 Secreted by Thyroid Cancer Cells Contributes to Immune Escape Through the Suppression of Natural Killer (NK) Cell Cytotoxicity and NK Cell Differentiation. Front. Immunol. 2018, 9, 1859. [Google Scholar] [CrossRef] [PubMed]
- Balsamo, M.; Scordamaglia, F.; Pietra, G.; Manzini, C.; Cantoni, C.; Boitano, M.; Queirolo, P.; Vermi, W.; Facchetti, F.; Moretta, A.; et al. Melanoma-associated fibroblasts modulate NK cell phenotype and antitumor cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 20847–20852. [Google Scholar] [CrossRef]
- Della Chiesa, M.; Carlomagno, S.; Frumento, G.; Balsamo, M.; Cantoni, C.; Conte, R.; Moretta, L.; Moretta, A.; Vitale, M. The tryptophan catabolite L-kynurenine inhibits the surface expression of NKp46- and NKG2D-activating receptors and regulates NK-cell function. Blood 2006, 108, 4118–4125. [Google Scholar] [CrossRef]
- Gaggero, S.; Bruschi, M.; Petretto, A.; Parodi, M.; Del Zotto, G.; Lavarello, C.; Prato, C.; Santucci, L.; Barbuto, A.; Bottino, C.; et al. Nidogen-1 is a novel extracellular ligand for the NKp44 activating receptor. Oncoimmunology 2018, 7, e1470730. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Guo, H.; Geng, J.; Zheng, X.; Wei, H.; Sun, R.; Tian, Z. Tumor-released Galectin-3, a soluble inhibitory ligand of human NKp30, plays an important role in tumor escape from NK cell attack. J. Biol. Chem. 2014, 289, 33311–33319. [Google Scholar] [CrossRef] [PubMed]
- de Andrade, L.F.; Tay, R.E.; Pan, D.; Luoma, A.M.; Ito, Y.; Badrinath, S.; Tsoucas, D.; Franz, B.; May, K.F., Jr.; Harvey, C.J.; et al. Antibody-mediated inhibition of MICA and MICB shedding promotes NK cell-driven tumor immunity. Science 2018, 359, 1537–1542. [Google Scholar] [CrossRef]
- Schlecker, E.; Fiegler, N.; Arnold, A.; Altevogt, P.; Rose-John, S.; Moldenhauer, G.; Sucker, A.; Paschen, A.; von Strandmann, E.P.; Textor, S.; et al. Metalloprotease-mediated tumor cell shedding of B7-H6, the ligand of the natural killer cell-activating receptor NKp30. Cancer Res. 2014, 74, 3429–3440. [Google Scholar] [CrossRef]
- MacFarlane, A.W.t.; Jillab, M.; Plimack, E.R.; Hudes, G.R.; Uzzo, R.G.; Litwin, S.; Dulaimi, E.; Al-Saleem, T.; Campbell, K.S. PD-1 expression on peripheral blood cells increases with stage in renal cell carcinoma patients and is rapidly reduced after surgical tumor resection. Cancer Immunol. Res. 2014, 2, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Pesce, S.; Greppi, M.; Tabellini, G.; Rampinelli, F.; Parolini, S.; Olive, D.; Moretta, L.; Moretta, A.; Marcenaro, E. Identification of a subset of human natural killer cells expressing high levels of programmed death 1: A phenotypic and functional characterization. J. Allergy Clin. Immunol. 2017, 139, 335–346. [Google Scholar] [CrossRef]
- Merino, A.; Zhang, B.; Dougherty, P.; Luo, X.; Wang, J.; Blazar, B.R.; Miller, J.S.; Cichocki, F. Chronic stimulation drives human NK cell dysfunction and epigenetic reprograming. J. Clin. Investig. 2019, 130, 3770–3785. [Google Scholar] [CrossRef]
- da Silva, I.P.; Gallois, A.; Jimenez-Baranda, S.; Khan, S.; Anderson, A.C.; Kuchroo, V.K.; Osman, I.; Bhardwaj, N. Reversal of NK-cell exhaustion in advanced melanoma by Tim-3 blockade. Cancer Immunol. Res. 2014, 2, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bi, J.; Zheng, X.; Chen, Y.; Wang, H.; Wu, W.; Wang, Z.; Wu, Q.; Peng, H.; Wei, H.; et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 2018, 19, 723–732. [Google Scholar] [CrossRef]
- Putz, E.M.; Guillerey, C.; Kos, K.; Stannard, K.; Miles, K.; Delconte, R.B.; Takeda, K.; Nicholson, S.E.; Huntington, N.D.; Smyth, M.J. Targeting cytokine signaling checkpoint CIS activates NK cells to protect from tumor initiation and metastasis. Oncoimmunology 2017, 6, e1267892. [Google Scholar] [CrossRef]
- Molgora, M.; Bonavita, E.; Ponzetta, A.; Riva, F.; Barbagallo, M.; Jaillon, S.; Popovic, B.; Bernardini, G.; Magrini, E.; Gianni, F.; et al. IL-1R8 is a checkpoint in NK cells regulating anti-tumour and anti-viral activity. Nature 2017, 551, 110–114. [Google Scholar] [CrossRef]
- Vitale, M.; Cantoni, C.; Pietra, G.; Mingari, M.C.; Moretta, L. Effect of tumor cells and tumor microenvironment on NK-cell function. Eur. J. Immunol. 2014, 44, 1582–1592. [Google Scholar] [CrossRef]
- Terren, I.; Orrantia, A.; Vitalle, J.; Zenarruzabeitia, O.; Borrego, F. NK Cell Metabolism and Tumor Microenvironment. Front. Immunol. 2019, 10, 2278. [Google Scholar] [CrossRef]
- Velasquez, S.Y.; Killian, D.; Schulte, J.; Sticht, C.; Thiel, M.; Lindner, H.A. Short Term Hypoxia Synergizes with Interleukin 15 Priming in Driving Glycolytic Gene Transcription and Supports Human Natural Killer Cell Activities. J. Biol. Chem. 2016, 291, 12960–12977. [Google Scholar] [CrossRef]
- Balsamo, M.; Manzini, C.; Pietra, G.; Raggi, F.; Blengio, F.; Mingari, M.C.; Varesio, L.; Moretta, L.; Bosco, M.C.; Vitale, M. Hypoxia downregulates the expression of activating receptors involved in NK-cell-mediated target cell killing without affecting ADCC. Eur. J. Immunol. 2013, 43, 2756–2764. [Google Scholar] [CrossRef]
- Parodi, M.; Raggi, F.; Cangelosi, D.; Manzini, C.; Balsamo, M.; Blengio, F.; Eva, A.; Varesio, L.; Pietra, G.; Moretta, L.; et al. Hypoxia Modifies the Transcriptome of Human NK Cells, Modulates Their Immunoregulatory Profile, and Influences NK Cell Subset Migration. Front. Immunol. 2018, 9, 2358. [Google Scholar] [CrossRef] [PubMed]
- Terry, S.; Abdou, A.; Engelsen, A.S.T.; Buart, S.; Dessen, P.; Corgnac, S.; Collares, D.; Meurice, G.; Gausdal, G.; Baud, V.; et al. AXL Targeting Overcomes Human Lung Cancer Cell Resistance to NK- and CTL-Mediated Cytotoxicity. Cancer Immunol. Res. 2019, 7, 1789–1802. [Google Scholar] [CrossRef]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef]
- Noman, M.Z.; Chouaib, S. Targeting hypoxia at the forefront of anticancer immune responses. Oncoimmunology 2014, 3, e954463. [Google Scholar] [CrossRef] [PubMed]
- Krzywinska, E.; Kantari-Mimoun, C.; Kerdiles, Y.; Sobecki, M.; Isagawa, T.; Gotthardt, D.; Castells, M.; Haubold, J.; Millien, C.; Viel, T.; et al. Loss of HIF-1alpha in natural killer cells inhibits tumour growth by stimulating non-productive angiogenesis. Nat. Commun. 2017, 8, 1597. [Google Scholar] [CrossRef] [PubMed]
- Muz, B.; de la Puente, P.; Azab, F.; Luderer, M.; Azab, A.K. The role of hypoxia and exploitation of the hypoxic environment in hematologic malignancies. Mol. Cancer Res. 2014, 12, 1347–1354. [Google Scholar] [CrossRef]
- Cantoni, C.; Huergo-Zapico, L.; Parodi, M.; Pedrazzi, M.; Mingari, M.C.; Moretta, A.; Sparatore, B.; Gonzalez, S.; Olive, D.; Bottino, C.; et al. NK Cells, Tumor Cell Transition, and Tumor Progression in Solid Malignancies: New Hints for NK-Based Immunotherapy? J. Immunol. Res. 2016, 2016, 4684268. [Google Scholar] [CrossRef] [PubMed]
- Carrega, P.; Bonaccorsi, I.; Di Carlo, E.; Morandi, B.; Paul, P.; Rizzello, V.; Cipollone, G.; Navarra, G.; Mingari, M.C.; Moretta, L.; et al. CD56(bright)perforin(low) noncytotoxic human NK cells are abundant in both healthy and neoplastic solid tissues and recirculate to secondary lymphoid organs via afferent lymph. J. Immunol. 2014, 192, 3805–3815. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.J.; Qin, S.; Unutmaz, D.; Soler, D.; Murphy, K.E.; Hodge, M.R.; Wu, L.; Butcher, E.C. Unique subpopulations of CD56+ NK and NK-T peripheral blood lymphocytes identified by chemokine receptor expression repertoire. J. Immunol. 2001, 166, 6477–6482. [Google Scholar] [CrossRef] [PubMed]
- Meng, W.; Xue, S.; Chen, Y. The role of CXCL12 in tumor microenvironment. Gene 2018, 641, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Tutunea-Fatan, E.; Majumder, M.; Xin, X.; Lala, P.K. The role of CCL21/CCR7 chemokine axis in breast cancer-induced lymphangiogenesis. Mol. Cancer 2015, 14, 35. [Google Scholar] [CrossRef] [PubMed]
- Sand, L.G.; Berghuis, D.; Szuhai, K.; Hogendoorn, P.C. Expression of CCL21 in Ewing sarcoma shows an inverse correlation with metastases and is a candidate target for immunotherapy. Cancer Immunol. Immunother. 2016, 65, 995–1002. [Google Scholar] [CrossRef]
- Castriconi, R.; Dondero, A.; Bellora, F.; Moretta, L.; Castellano, A.; Locatelli, F.; Corrias, M.V.; Moretta, A.; Bottino, C. Neuroblastoma-derived TGF-beta1 modulates the chemokine receptor repertoire of human resting NK cells. J. Immunol. 2013, 190, 5321–5328. [Google Scholar] [CrossRef]
- Costa, D.; Vene, R.; Benelli, R.; Romairone, E.; Scabini, S.; Catellani, S.; Rebesco, B.; Mastracci, L.; Grillo, F.; Minghelli, S.; et al. Targeting the Epidermal Growth Factor Receptor Can Counteract the Inhibition of Natural Killer Cell Function Exerted by Colorectal Tumor-Associated Fibroblasts. Front. Immunol. 2018, 9, 1150. [Google Scholar] [CrossRef]
- Trotta, R.; Dal Col, J.; Yu, J.; Ciarlariello, D.; Thomas, B.; Zhang, X.; Allard, J., 2nd; Wei, M.; Mao, H.; Byrd, J.C.; et al. TGF-beta utilizes SMAD3 to inhibit CD16-mediated IFN-gamma production and antibody-dependent cellular cytotoxicity in human NK cells. J. Immunol. 2008, 181, 3784–3792. [Google Scholar] [CrossRef]
- Watanabe, M.; Kono, K.; Kawaguchi, Y.; Mizukami, Y.; Mimura, K.; Maruyama, T.; Izawa, S.; Fujii, H. NK cell dysfunction with down-regulated CD16 and up-regulated CD56 molecules in patients with esophageal squamous cell carcinoma. Dis. Esophagus 2010, 23, 675–681. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Kono, K.; Mimura, K.; Sugai, H.; Akaike, H.; Fujii, H. Cetuximab induce antibody-dependent cellular cytotoxicity against EGFR-expressing esophageal squamous cell carcinoma. Int. J. Cancer 2007, 120, 781–787. [Google Scholar] [CrossRef]
- Bedi, A.; Chang, X.; Noonan, K.; Pham, V.; Bedi, R.; Fertig, E.J.; Considine, M.; Califano, J.A.; Borrello, I.; Chung, C.H.; et al. Inhibition of TGF-beta enhances the in vivo antitumor efficacy of EGF receptor-targeted therapy. Mol. Cancer Ther. 2012, 11, 2429–2439. [Google Scholar] [CrossRef]
- Wang, S.Y.; Veeramani, S.; Racila, E.; Cagley, J.; Fritzinger, D.C.; Vogel, C.W.; St John, W.; Weiner, G.J. Depletion of the C3 component of complement enhances the ability of rituximab-coated target cells to activate human NK cells and improves the efficacy of monoclonal antibody therapy in an in vivo model. Blood 2009, 114, 5322–5330. [Google Scholar] [CrossRef] [PubMed]
- Battella, S.; Cox, M.C.; Santoni, A.; Palmieri, G. Natural killer (NK) cells and anti-tumor therapeutic mAb: Unexplored interactions. J. Leukoc. Biol. 2016, 99, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Mishra, H.K.; Walcheck, B. Role of ADAM17 as a regulatory checkpoint of CD16A in NK cells and as a potential target for cancer immunotherapy. J. Leukoc. Biol. 2019, 105, 1297–1303. [Google Scholar] [CrossRef]
- Balsamo, M.; Vermi, W.; Parodi, M.; Pietra, G.; Manzini, C.; Queirolo, P.; Lonardi, S.; Augugliaro, R.; Moretta, A.; Facchetti, F.; et al. Melanoma cells become resistant to NK-cell-mediated killing when exposed to NK-cell numbers compatible with NK-cell infiltration in the tumor. Eur. J. Immunol. 2012, 42, 1833–1842. [Google Scholar] [CrossRef] [PubMed]
- Andre, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Blery, M.; Bonnafous, C.; Gauthier, L.; Morel, A.; et al. Anti-NKG2A mAb Is a Checkpoint Inhibitor that Promotes Anti-tumor Immunity by Unleashing Both T and NK Cells. Cell 2018, 175, 1731–1743. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, L.; Morel, A.; Anceriz, N.; Rossi, B.; Blanchard-Alvarez, A.; Grondin, G.; Trichard, S.; Cesari, C.; Sapet, M.; Bosco, F.; et al. Multifunctional Natural Killer Cell Engagers Targeting NKp46 Trigger Protective Tumor Immunity. Cell 2019, 177, 1701–1713. [Google Scholar] [CrossRef]
- Sarhan, D.; Brandt, L.; Felices, M.; Guldevall, K.; Lenvik, T.; Hinderlie, P.; Curtsinger, J.; Warlick, E.; Spellman, S.R.; Blazar, B.R.; et al. 161533 TriKE stimulates NK-cell function to overcome myeloid-derived suppressor cells in MDS. Blood Adv. 2018, 2, 1459–1469. [Google Scholar] [CrossRef]
- Tumino, N.; Besi, F.; Di Pace, A.L.; Mariotti, F.R.; Merli, P.; Li Pira, G.; Galaverna, F.; Pitisci, A.; Ingegnere, T.; Pelosi, A.; et al. PMN-MDSC are a new target to rescue graft-versus-leukemia activity of NK cells in haplo-HSC transplantation. Leukemia 2019. [Google Scholar] [CrossRef]
- Khan, K.D.; Emmanouilides, C.; Benson, D.M., Jr.; Hurst, D.; Garcia, P.; Michelson, G.; Milan, S.; Ferketich, A.K.; Piro, L.; Leonard, J.P.; et al. A phase 2 study of rituximab in combination with recombinant interleukin-2 for rituximab-refractory indolent non-Hodgkin’s lymphoma. Clin. Cancer Res. 2006, 12, 7046–7053. [Google Scholar] [CrossRef] [PubMed]
- Ahmadzadeh, M.; Rosenberg, S.A. IL-2 administration increases CD4+ CD25(hi) Foxp3+ regulatory T cells in cancer patients. Blood 2006, 107, 2409–2414. [Google Scholar] [CrossRef] [PubMed]
- Jie, H.B.; Schuler, P.J.; Lee, S.C.; Srivastava, R.M.; Argiris, A.; Ferrone, S.; Whiteside, T.L.; Ferris, R.L. CTLA-4(+) Regulatory T Cells Increased in Cetuximab-Treated Head and Neck Cancer Patients Suppress NK Cell Cytotoxicity and Correlate with Poor Prognosis. Cancer Res. 2015, 75, 2200–2210. [Google Scholar] [CrossRef]
- Julia, E.P.; Amante, A.; Pampena, M.B.; Mordoh, J.; Levy, E.M. Avelumab, an IgG1 anti-PD-L1 Immune Checkpoint Inhibitor, Triggers NK Cell-Mediated Cytotoxicity and Cytokine Production Against Triple Negative Breast Cancer Cells. Front. Immunol. 2018, 9, 2140. [Google Scholar] [CrossRef]
- Datar, I.; Sanmamed, M.F.; Wang, J.; Henick, B.S.; Choi, J.; Badri, T.; Dong, W.; Mani, N.; Toki, M.; Mejias, L.D.; et al. Expression Analysis and Significance of PD-1, LAG-3, and TIM-3 in Human Non-Small Cell Lung Cancer Using Spatially Resolved and Multiparametric Single-Cell Analysis. Clin. Cancer Res. 2019, 25, 4663–4673. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Wu, X.; Ma, S.; Wang, Y.; Nalin, A.P.; Zhu, Z.; Zhang, J.; Benson, D.M.; He, K.; Caligiuri, M.A.; et al. The Mechanism of Anti-PD-L1 Antibody Efficacy against PD-L1-Negative Tumors Identifies NK Cells Expressing PD-L1 as a Cytolytic Effector. Cancer Discov. 2019, 9, 1422–1437. [Google Scholar] [CrossRef]
- Hwang, W.L.; Pike, L.R.G.; Royce, T.J.; Mahal, B.A.; Loeffler, J.S. Safety of combining radiotherapy with immune-checkpoint inhibition. Nat. Rev. Clin. Oncol. 2018, 15, 477–494. [Google Scholar] [CrossRef]
- Roger, A.; Finet, A.; Boru, B.; Beauchet, A.; Mazeron, J.J.; Otzmeguine, Y.; Blom, A.; Longvert, C.; de Maleissye, M.F.; Fort, M.; et al. Efficacy of combined hypo-fractionated radiotherapy and anti-PD-1 monotherapy in difficult-to-treat advanced melanoma patients. Oncoimmunology 2018, 7, e1442166. [Google Scholar] [CrossRef]
- Formenti, S.C.; Lee, P.; Adams, S.; Goldberg, J.D.; Li, X.; Xie, M.W.; Ratikan, J.A.; Felix, C.; Hwang, L.; Faull, K.F.; et al. Focal Irradiation and Systemic TGFbeta Blockade in Metastatic Breast Cancer. Clin. Cancer Res. 2018, 24, 2493–2504. [Google Scholar] [CrossRef]
- Aboudaram, A.; Modesto, A.; Chaltiel, L.; Gomez-Roca, C.; Boulinguez, S.; Sibaud, V.; Delord, J.P.; Chira, C.; Delannes, M.; Moyal, E.; et al. Concurrent radiotherapy for patients with metastatic melanoma and receiving anti-programmed-death 1 therapy: A safe and effective combination. Melanoma Res. 2017, 27, 485–491. [Google Scholar] [CrossRef]
- Sivori, S.; Meazza, R.; Quintarelli, C.; Carlomagno, S.; Della Chiesa, M.; Falco, M.; Moretta, L.; Locatelli, F.; Pende, D. NK Cell-Based Immunotherapy for Hematological Malignancies. J. Clin. Med. 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Souza-Fonseca-Guimaraes, F.; Cursons, J.; Huntington, N.D. The Emergence of Natural Killer Cells as a Major Target in Cancer Immunotherapy. Trends Immunol. 2019, 40, 142–158. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Mechanisms | Therapy | Main Resistance Mechanisms | Overcoming Resistance |
|---|---|---|---|
| ADCC | MAbs | - Low NK cell numbers or activity - FCGR3A polymorphisms - Shedding or low expression of CD16 - Loss or modulation of expression of the target antigen - Expression of anti-apoptotic proteins by cancer cells - Tumor microenvironment interactions - Immunosuppressive cytokines and microenvironment - Expression of checkpoint proteins and inhibitory receptors | - Improving mAbs with increased affinity - Combination with immunomodulatory drugs and cytokines - Using multitarget BiKEs and TRiKEs - Metalloproteinase inhibitors to avoid CD16 shedding - Using pro-apoptotic drugs - Targeting inhibitory and immunosuppressive proteins |
| Missing-self recognition | HSTC allogenic NK cell transfer | - KIR/HLA repertoire - Presence of specific inhibitory KIR genes - Increased expression of non-classical MHC class I molecules - Short lifespan (adoptive transfer) - Poor persistence and trafficking (adoptive transfer) | - Activation and expansion of NK cells - Using allogenic HSTC - Using anti-KIR antibodies |
| Activating receptors | NK cell transfer Immuno-modulatory drugs and cytokines | - Downregulation of activating receptors or ligands - Increased expression of checkpoint proteins and inhibitory proteins - Expression of anti-apoptotic proteins by cancer cells - Tumor microenvironment interactions - Immunosuppressive cytokines and microenvironment - Short lifespan (adoptive transfer) - Poor persistence and trafficking (adoptive transfer) | - Activation and expansion of NK cells - Induction of NKG2D ligand expression - Using pro-apoptotic drugs - Targeting inhibitory and immunosuppressive proteins - Using NK cell lines (i.e., NK-92) - Therapy with NKG2D CAR-NK cells |
| Chimeric antigenic receptors | CAR-NK cells | - Short lifespan - Poor activation, persistence and trafficking | - IL-15-expressing CAR-NK cells - Using NK-92 cells as carriers - Combination with mAbs |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sordo-Bahamonde, C.; Vitale, M.; Lorenzo-Herrero, S.; López-Soto, A.; Gonzalez, S. Mechanisms of Resistance to NK Cell Immunotherapy. Cancers 2020, 12, 893. https://doi.org/10.3390/cancers12040893
Sordo-Bahamonde C, Vitale M, Lorenzo-Herrero S, López-Soto A, Gonzalez S. Mechanisms of Resistance to NK Cell Immunotherapy. Cancers. 2020; 12(4):893. https://doi.org/10.3390/cancers12040893
Chicago/Turabian StyleSordo-Bahamonde, Christian, Massimo Vitale, Seila Lorenzo-Herrero, Alejandro López-Soto, and Segundo Gonzalez. 2020. "Mechanisms of Resistance to NK Cell Immunotherapy" Cancers 12, no. 4: 893. https://doi.org/10.3390/cancers12040893
APA StyleSordo-Bahamonde, C., Vitale, M., Lorenzo-Herrero, S., López-Soto, A., & Gonzalez, S. (2020). Mechanisms of Resistance to NK Cell Immunotherapy. Cancers, 12(4), 893. https://doi.org/10.3390/cancers12040893