Secondary Resistant Mutations to Small Molecule Inhibitors in Cancer Cells

Abstract
:1. Introduction
2. Data Acquisition and Manipulation
3. Overview of Resistance Mutations
4. Resistant Mutations Appear in Several Cancers in Response to Small Molecule Inhibitors
4.1. Non-Small Cell Lung Cancer
4.1.1. Classification
4.1.2. Epidermal Growth Factor Receptor (EGFR)
4.1.3. Hepatocyte Growth Factor Receptor (HFGR, c-Met)
4.1.4. Anaplastic Lymphoma Kinase (ALK)
4.2. Hematopoietic and Lymphoid Tissue
4.2.1. Classification
4.2.2. Abelson Murine Leukemia Viral Oncogene Homolog 1 (ABL1)
4.2.3. Bruton’s Tyrosine Kinase (BTK)
4.2.4. FMS-like Tyrosine Kinase 3 (FLT3)
4.3. Gastrointestinal Stromal Tumors (GISTs)
4.3.1. Tyrosine-Protein Kinase KIT
4.3.2. Platelet Derived Growth Factor Receptor Alpha (PDGFRA)
4.4. Melanoma
Classification
4.5. ESR1 in Breast Cancer
Classification
4.6. Androgen Receptor in Prostate Cancer
4.7. Other Resistant Mutations
5. Conclusions and Future Directions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- DeVita, V.T., Jr.; Chu, E. A history of cancer chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farber, S. Chemotherapy in the treatment of leukemia and Wilms’ tumor. JAMA 1966, 198, 826–836. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D. Sidney Farber and the treatment of childhood acute lymphoblastic leukemia with a chemotherapeutic agent. Pediatr. Hematol. Oncol. 2012, 29, 299–302. [Google Scholar] [CrossRef] [PubMed]
- Farber, S.; Diamond, L.K. Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid. N. Engl. J. Med. 1948, 238, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Grever, M.R.; Schepartz, S.A.; Chabner, B.A. The National Cancer Institute: Cancer drug discovery and development program. Semin. Oncol. 1992, 19, 622–638. [Google Scholar] [PubMed]
- Mukherjee, S. The Emperor of all Maladies: A Biography of Cancer; Scribner: New York, NY, USA, 2010. [Google Scholar]
- Meisner, N.C.; Hintersteiner, M.; Uhl, V.; Weidemann, T.; Schmied, M.; Gstach, H.; Auer, M. The chemical hunt for the identification of drugable targets. Curr. Opin. Chem. Biol. 2004, 8, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Piggott, A.M.; Karuso, P. Quality, not quantity: The role of natural products and chemical proteomics in modern drug discovery. Comb. Chem. High. Throughput Scr. 2004, 7, 607–630. [Google Scholar] [CrossRef]
- Sioud, M.; Leirdal, M. Druggable signaling proteins. Methods Mol. Biol. 2007, 361, 1–24. [Google Scholar] [CrossRef]
- Yuan, Y.X.; Pei, J.F.; Lai, L.H. Binding site detection and druggability prediction of protein targets for structure-based drug design. Curr. Pharm. Des. 2013, 19, 2326–2333. [Google Scholar] [CrossRef]
- Perola, E.; Herman, L.; Weiss, J. Development of a rule-based method for the assessment of protein druggability. J. Chem. Inf. Modeling 2012, 52, 1027–1038. [Google Scholar] [CrossRef]
- Sledz, P.; Caflisch, A. Protein structure-based drug design: From docking to molecular dynamics. Curr. Opin. Struc. Biol. 2018, 48, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, J.J.; Zhao, X.; Mays, J.C.; Davoli, T. Not all cancers are created equal: Tissue specificity in cancer genes and pathways. Curr. Opin. Cell. Biol. 2020, 63, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive characterization of cancer driver genes and mutations. Cell 2018, 174, 1034–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, G.L.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Stevens, C.; et al. Patterns of somatic mutation in human cancer genomes. Nature 2007, 446, 153–158. [Google Scholar] [CrossRef] [Green Version]
- Boumahdi, S.; de Sauvage, F.J. The great escape: Tumour cell plasticity in resistance to targeted therapy. Nat. Rev. Drug Discov. 2020, 19, 39–56. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [Green Version]
- Jay, J.J.; Brouwer, C. Lollipops in the clinic: Information dense mutation plots for precision medicine. PLoS ONE 2016, 11, e0160519. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R. Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol. Res. 2016, 103, 26–48. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors: A 2020 update. Pharmacol. Res. 2020, 152, 104609. [Google Scholar] [CrossRef]
- Roskoski, R. A historical overview of protein kinases and their targeted small molecule inhibitors. Pharmacol. Res. 2015, 100, 1–23. [Google Scholar] [CrossRef]
- Helbig, G. Imatinib for the treatment of hypereosinophilic syndromes. Expert Rev. Clin. Immunol. 2018, 14, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Duma, N.; Santana-Davila, R.; Molina, J.R. Non-small cell lung cancer: Epidemiology, screening, diagnosis, and treatment. Mayo Clin. Proc. 2019, 94, 1623–1640. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, N.; Jogai, S.; Wallis, A. The revised lung adenocarcinoma classification-an imaging guide. J. Thorac. Dis 2014, 6, 537–546. [Google Scholar] [CrossRef]
- Li, C.; Lu, H. Adenosquamous carcinoma of the lung. Onco Targets Ther. 2018, 11, 4829–4835. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Zhuang, W.; Chen, L.; Yang, W.; Ou, W.B. Frontiers of ctDNA, targeted therapies, and immunotherapy in non-small-cell lung cancer. Transl. Lung Cancer Res. 2020, 9, 111–138. [Google Scholar] [CrossRef]
- Ruiz-Cordero, R.; Devine, W.P. Targeted therapy and checkpoint immunotherapy in lung cancer. Surg. Pathol. Clin. 2020, 13, 17–33. [Google Scholar] [CrossRef]
- Lim, Z.F.; Ma, P.C. Emerging insights of tumor heterogeneity and drug resistance mechanisms in lung cancer targeted therapy. J. Hematol. Oncol. 2019, 12, 134. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, F.R.; Scagliotti, G.V.; Mulshine, J.L.; Kwon, R.; Curran, W.J.; Wu, Y.L.; Paz-Ares, L. Lung cancer: Current therapies and new targeted treatments. Lancet 2017, 389, 299–311. [Google Scholar] [CrossRef]
- Xu, Z.Y.; Li, J.L. Comparative review of drug-drug interactions with epidermal growth factor receptor tyrosine kinase inhibitors for the treatment of non-small-cell lung cancer. Oncotargets Ther. 2019, 12, 5467–5484. [Google Scholar] [CrossRef] [Green Version]
- Herbst, R.S. Review of epidermal growth factor receptor biology. Int. J. Radiat. Oncol. 2004, 59, 21–26. [Google Scholar] [CrossRef]
- Burgering, B.M.T.; Coffer, P.J. Protein-Kinase-B (C-Akt) in Phosphatidylinositol-3-Oh Inase signal-transduction. Nature 1995, 376, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Mayer, I.A.; Arteaga, C.L. The PI3K/AKT pathway as a target for cancer treatment. Annu. Rev. Med. 2016, 67, 11–28. [Google Scholar] [CrossRef] [PubMed]
- Alroy, I.; Yarden, Y. The ErbB signaling network in embryogenesis and oncogenesis: Signal diversification through combinatorial ligand-receptor interactions. FEBS Lett. 1997, 410, 83–86. [Google Scholar] [CrossRef] [Green Version]
- Wee, P.; Wang, Z. Epidermal growth factor receptor cell proliferation signaling pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef] [Green Version]
- Bishop, A.L.; Hall, A. Rho GTPases and their effector proteins. Biochem. J. 2000, 348, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, B.J.; Zhou, H.; Lu, Q. Cdc42 signaling pathway inhibition as a therapeutic target in ras- related cancers. Curr. Med. Chem. 2017, 24, 3485–3507. [Google Scholar] [CrossRef]
- Nicholson, R.I.; Gee, J.M.; Harper, M.E. EGFR and cancer prognosis. Eur. J. Cancer 2001, 37, 9–15. [Google Scholar] [CrossRef]
- Hirsch, F.R.; Varella-Garcia, M.; Bunn, P.A., Jr.; Di Maria, M.V.; Veve, R.; Bremmes, R.M.; Baron, A.E.; Zeng, C.; Franklin, W.A. Epidermal growth factor receptor in non-small-cell lung carcinomas: Correlation between gene copy number and protein expression and impact on prognosis. J. Clin. Oncol. 2003, 21, 3798–3807. [Google Scholar] [CrossRef]
- Hsu, P.C.; Jablons, D.M.; Yang, C.T.; You, L. Epidermal Growth Factor Receptor (EGFR) pathway, Yes-Associated Protein (YAP) and the Regulation of Programmed Death-Ligand 1 (PD-L1) in Non-Small Cell Lung Cancer (NSCLC). Int. J. Mol. Sci. 2019, 20, 3821. [Google Scholar] [CrossRef] [Green Version]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef]
- Paez, J.G.; Janne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pao, W.; Miller, V.; Zakowski, M.; Doherty, J.; Politi, K.; Sarkaria, I.; Singh, B.; Heelan, R.; Rusch, V.; Fulton, L.; et al. EGF receptor gene mutations are common in lung cancers from "never smokers" and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. USA 2004, 101, 13306–13311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huse, M.; Kuriyan, J. The conformational plasticity of protein kinases. Cell 2002, 109, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Vyse, S.; Huang, P.H. Targeting EGFR exon 20 insertion mutations in non-small cell lung cancer. Signal Transduct. Target. Ther. 2019, 4, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, R.A.; Anderton, M.J.; Ashton, S.; Bethel, P.A.; Box, M.; Butterworth, S.; Colclough, N.; Chorley, C.G.; Chuaqui, C.; Cross, D.A.; et al. Structure- and reactivity-based development of covalent inhibitors of the activating and gatekeeper mutant forms of the epidermal growth factor receptor (EGFR). J. Med. Chem. 2013, 56, 7025–7048. [Google Scholar] [CrossRef]
- Pollack, V.A.; Savage, D.M.; Baker, D.A.; Tsaparikos, K.E.; Sloan, D.E.; Moyer, J.D.; Barbacci, E.G.; Pustilnik, L.R.; Smolarek, T.A.; Davis, J.A.; et al. Inhibition of epidermal growth factor receptor-associated tyrosine phosphorylation in human carcinomas with CP-358,774: Dynamics of receptor inhibition in situ and antitumor effects in athymic mice. J. Pharmacol. Exp. Ther. 1999, 291, 739–748. [Google Scholar]
- Wakeling, A.E.; Barker, A.J.; Davies, D.H.; Brown, D.S.; Green, L.R.; Cartlidge, S.A.; Woodburn, J.R. Specific inhibition of epidermal growth factor receptor tyrosine kinase by 4-anilinoquinazolines. Breast Cancer Res. Treat. 1996, 38, 67–73. [Google Scholar] [CrossRef]
- Ciardiello, F.; Caputo, R.; Bianco, R.; Damiano, V.; Pomatico, G.; De Placido, S.; Bianco, A.R.; Tortora, G. Antitumor effect and potentiation of cytotoxic drugs activity in human cancer cells by ZD-1839 (Iressa), an epidermal growth factor receptor-selective tyrosine kinase inhibitor. Clin. Cancer Res. 2000, 6, 2053–2063. [Google Scholar]
- Mulloy, R.; Ferrand, A.; Kim, Y.; Sordella, R.; Bell, D.W.; Haber, D.A.; Anderson, K.S.; Settleman, J. Epidermal growth factor receptor mutants from human lung cancers exhibit enhanced catalytic activity and increased sensitivity to gefitinib. Cancer Res. 2007, 67, 2325–2330. [Google Scholar] [CrossRef] [Green Version]
- Carey, K.D.; Garton, A.J.; Romero, M.S.; Kahler, J.; Thomson, S.; Ross, S.; Park, F.; Haley, J.D.; Gibson, N.; Sliwkowski, M.X. Kinetic analysis of epidermal growth factor receptor somatic mutant proteins shows increased sensitivity to the epidermal growth factor receptor tyrosine kinase inhibitor, erlotinib. Cancer Res. 2006, 66, 8163–8171. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Janne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005, 2, e73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eck, M.J.; Yun, C.H. Structural and mechanistic underpinnings of the differential drug sensitivity of EGFR mutations in non-small cell lung cancer. Biochim. Biophys. Acta 2010, 1804, 559–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, C.H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.K.; Meyerson, M.; Eck, M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar] [CrossRef] [Green Version]
- Yun, C.H.; Boggon, T.J.; Li, Y.; Woo, M.S.; Greulich, H.; Meyerson, M.; Eck, M.J. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: Mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell 2007, 11, 217–227. [Google Scholar] [CrossRef] [Green Version]
- Pazarentzos, E.; Giannikopoulos, P.; Hrustanovic, G.; St John, J.; Olivas, V.R.; Gubens, M.A.; Balassanian, R.; Weissman, J.; Polkinghorn, W.; Bivona, T.G. Oncogenic activation of the PI3-kinase p110beta isoform via the tumor-derived PIK3Cbeta(D1067V) kinase domain mutation. Oncogene 2016, 35, 1198–1205. [Google Scholar] [CrossRef]
- Dbouk, H.A.; Khalil, B.D.; Wu, H.; Shymanets, A.; Nurnberg, B.; Backer, J.M. Characterization of a tumor-associated activating mutation of the p110beta PI 3-kinase. PLoS ONE 2013, 8, e63833. [Google Scholar] [CrossRef] [Green Version]
- Hirsh, V. New developments in the treatment of advanced squamous cell lung cancer: Focus on afatinib. Onco Targets Ther. 2017, 10, 2513–2526. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Ambrogio, L.; Shimamura, T.; Kubo, S.; Takahashi, M.; Chirieac, L.R.; Padera, R.F.; Shapiro, G.I.; Baum, A.; Himmelsbach, F.; et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 2008, 27, 4702–4711. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Ko, J.; Cui, Z.; Abolhoda, A.; Ahn, J.S.; Ou, S.H.; Ahn, M.J.; Park, K. The EGFR T790M mutation in acquired resistance to an irreversible second-generation EGFR inhibitor. Mol. Cancer Ther. 2012, 11, 784–791. [Google Scholar] [CrossRef] [Green Version]
- Solca, F.; Dahl, G.; Zoephel, A.; Bader, G.; Sanderson, M.; Klein, C.; Kraemer, O.; Himmelsbach, F.; Haaksma, E.; Adolf, G.R. Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J. Pharmacol. Exp. Ther. 2012, 343, 342–350. [Google Scholar] [CrossRef]
- Wu, S.G.; Liu, Y.N.; Tsai, M.F.; Chang, Y.L.; Yu, C.J.; Yang, P.C.; Yang, J.C.; Wen, Y.F.; Shih, J.Y. The mechanism of acquired resistance to irreversible EGFR tyrosine kinase inhibitor-afatinib in lung adenocarcinoma patients. Oncotarget 2016, 7, 12404–12413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, Y.; Azuma, K.; Nagai, H.; Kim, Y.H.; Togashi, Y.; Sesumi, Y.; Chiba, M.; Shimoji, M.; Sato, K.; Tomizawa, K.; et al. Characterization of EGFR T790M, L792F, and C797S mutations as mechanisms of acquired resistance to afatinib in lung cancer. Mol. Cancer Ther. 2017, 16, 357–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, V.A.; Hirsh, V.; Cadranel, J.; Chen, Y.M.; Park, K.; Kim, S.W.; Zhou, C.; Su, W.C.; Wang, M.; Sun, Y.; et al. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): A phase 2b/3 randomised trial. Lancet Oncol. 2012, 13, 528–538. [Google Scholar] [CrossRef]
- Pirazzoli, V.; Nebhan, C.; Song, X.; Wurtz, A.; Walther, Z.; Cai, G.; Zhao, Z.; Jia, P.; de Stanchina, E.; Shapiro, E.M.; et al. Acquired resistance of EGFR-mutant lung adenocarcinomas to afatinib plus cetuximab is associated with activation of mTORC1. Cell Rep. 2014, 7, 999–1008. [Google Scholar] [CrossRef] [Green Version]
- Curto, M.; Cole, B.K.; Lallemand, D.; Liu, C.H.; McClatchey, A.I. Contact-dependent inhibition of EGFR signaling by Nf2/Merlin. J. Cell Biol. 2007, 177, 893–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, T.; Lopez-Lago, M.; Giancotti, F.G. Merlin/NF-2 mediates contact inhibition of growth by suppressing recruitment of Rac to the plasma membrane. J. Cell Biol. 2005, 171, 361–371. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Morrison, H.; Sherman, L.S.; Legg, J.; Banine, F.; Isacke, C.; Haipek, C.A.; Gutmann, D.H.; Ponta, H.; Herrlich, P. The NF2 tumor suppressor gene product, merlin, mediates contact inhibition of growth through interactions with CD44. Genes Dev. 2001, 15, 968–980. [Google Scholar] [CrossRef] [Green Version]
- Sainio, M.; Zhao, F.; Heiska, L.; Turunen, O.; den Bakker, M.; Zwarthoff, E.; Lutchman, M.; Rouleau, G.A.; Jaaskelainen, J.; Vaheri, A.; et al. Neurofibromatosis 2 tumor suppressor protein colocalizes with ezrin and CD44 and associates with actin-containing cytoskeleton. J. Cell Sci. 1997, 110, 2249–2260. [Google Scholar] [PubMed]
- Gladden, A.B.; Hebert, A.M.; Schneeberger, E.E.; McClatchey, A.I. The NF2 tumor suppressor, Merlin, regulates epidermal development through the establishment of a junctional polarity complex. Dev. Cell 2010, 19, 727–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lallemand, D.; Curto, M.; Saotome, I.; Giovannini, M.; McClatchey, A.I. NF2 deficiency promotes tumorigenesis and metastasis by destabilizing adherens junctions. Genes Dev. 2003, 17, 1090–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Pan, D. The hippo signaling pathway in development and disease. Dev. Cell 2019, 50, 264–282. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Sekido, Y. NF2/Merlin inactivation and potential therapeutic targets in mesothelioma. Int. J. Mol. Sci. 2018, 19, 988. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Cooper, J.; Zhou, L.; Yang, C.; Erdjument-Bromage, H.; Zagzag, D.; Snuderl, M.; Ladanyi, M.; Hanemann, C.O.; Zhou, P.; et al. Merlin/NF2 loss-driven tumorigenesis linked to CRL4(DCAF1)-mediated inhibition of the hippo pathway kinases Lats1 and 2 in the nucleus. Cancer Cell 2014, 26, 48–60. [Google Scholar] [CrossRef] [Green Version]
- Hikasa, H.; Sekido, Y.; Suzuki, A. Merlin/NF2-Lin28B-let-7 is a tumor-suppressive pathway that is cell-density dependent and hippo independent. Cell Rep. 2016, 14, 2950–2961. [Google Scholar] [CrossRef] [Green Version]
- Sekido, Y.; Bader, S.A.; Carbone, D.P.; Johnson, B.E.; Minna, J.D. Molecular analysis of the HuD gene encoding a paraneoplastic encephalomyelitis antigen in human lung cancer cell lines. Cancer Res. 1994, 54, 4988–4992. [Google Scholar]
- Bianchi, A.B.; Mitsunaga, S.I.; Cheng, J.Q.; Klein, W.M.; Jhanwar, S.C.; Seizinger, B.; Kley, N.; Klein-Szanto, A.J.; Testa, J.R. High frequency of inactivating mutations in the neurofibromatosis type 2 gene (NF2) in primary malignant mesotheliomas. Proc. Natl. Acad. Sci. USA 1995, 92, 10854–10858. [Google Scholar] [CrossRef] [Green Version]
- Gendreau, S.B.; Ventura, R.; Keast, P.; Laird, A.D.; Yakes, F.M.; Zhang, W.; Bentzien, F.; Cancilla, B.; Lutman, J.; Chu, F.; et al. Inhibition of the T790M gatekeeper mutant of the epidermal growth factor receptor by EXEL-7647. Clin. Cancer Res. 2007, 13, 3713–3723. [Google Scholar] [CrossRef] [Green Version]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef]
- Wu, B.; Rockel, J.S.; Lagares, D.; Kapoor, M. Ephrins and eph receptor signaling in tissue repair and fibrosis. Curr. Rheumatol. Rep. 2019, 21, 23. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef] [PubMed]
- Pietanza, M.C.; Lynch, T.J., Jr.; Lara, P.N., Jr.; Cho, J.; Yanagihara, R.H.; Vrindavanam, N.; Chowhan, N.M.; Gadgeel, S.M.; Pennell, N.A.; Funke, R.; et al. XL647--a multitargeted tyrosine kinase inhibitor: Results of a phase II study in subjects with non-small cell lung cancer who have progressed after responding to treatment with either gefitinib or erlotinib. J. Thorac. Oncol. 2012, 7, 219–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietanza, M.C.; Gadgeel, S.M.; Dowlati, A.; Lynch, T.J.; Salgia, R.; Rowland, K.M., Jr.; Wertheim, M.S.; Price, K.A.; Riely, G.J.; Azzoli, C.G.; et al. Phase II study of the multitargeted tyrosine kinase inhibitor XL647 in patients with non-small-cell lung cancer. J. Thorac. Oncol. 2012, 7, 856–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cross, D.; Ashton, S.; Nebhan, C.; Eberlein, C.; Finlay, M.R.V.; Hughes, G.; Jacobs, V.; Mellor, M.; Brewer, M.R.; Meador, C.; et al. AZD9291: An irreversible, potent and selective third generation tyrosine kinase inhibitor (TKI) targeting EGFR activating (EGFRm plus ) and resistance (T790M) mutations in advanced lung adenocarcinoma. Mol. Cancer Ther. 2013, 12. [Google Scholar] [CrossRef]
- Cross, D.A.E.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.V.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-Mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, L.K.; Chang, Y.L.; Shih, J.Y. Primary resistance to osimertinib despite acquired T790M. Respirol. Case Rep. 2020, 8, e00532. [Google Scholar] [CrossRef]
- Thress, K.S.; Paweletz, C.P.; Felip, E.; Cho, B.C.; Stetsonl, D.; Dougherty, B.; Lai, Z.W.; Markovets, A.; Vivancos, A.; Kuang, Y.; et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat. Med. 2015, 21, 560–562. [Google Scholar] [CrossRef] [Green Version]
- Zheng, D.; Hu, M.; Bai, Y.; Zhu, X.; Lu, X.; Wu, C.; Wang, J.; Liu, L.; Wang, Z.; Ni, J.; et al. EGFR G796D mutation mediates resistance to osimertinib. Oncotarget 2017, 8, 49671–49679. [Google Scholar] [CrossRef] [Green Version]
- Ou, S.H.I.; Cui, J.; Schrock, A.B.; Goldberg, M.E.; Zhu, V.W.; Albacker, L.; Stephens, P.J.; Miller, V.A.; Ali, S.M. Emergence of novel and dominant acquired EGFR solvent-front mutations at Gly796 (G796S/R) together with C797S/G and L792F/H mutations in one EGFR (L858R/T790M) NSCLC patient who progressed on osimertinib. Lung Cancer 2019, 138, 141-141. [Google Scholar] [CrossRef]
- Bersanelli, M.; Minari, R.; Bordi, P.; Gnetti, L.; Bozzetti, C.; Squadrilli, A.; Lagrasta, C.A.M.; Bottarelli, L.; Osipova, G.; Capelletto, E.; et al. L718Q Mutation as New Mechanism of Acquired Resistance to AZD9291 in EGFR-Mutated NSCLC. J. Thorac. Oncol. 2016, 11, E121–E123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ercan, D.; Choi, H.G.; Yun, C.H.; Capelletti, M.; Xie, T.; Eck, M.J.; Gray, N.S.; Janne, P.A. EGFR mutations and resistance to irreversible pyrimidine-based EGFR inhibitors. Clin. Cancer Res. 2015, 21, 3913–3923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callegari, D.; Ranaghan, K.E.; Woods, C.J.; Minari, R.; Tiseo, M.; Mor, M.; Mulholland, A.J.; Lodola, A. L718Q mutant EGFR escapes covalent inhibition by stabilizing a non-reactive conformation of the lung cancer drug osimertinib. Chem. Sci. 2018, 9, 2740–2749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Wang, Y.Y.; Zhai, Y.C.; Wang, J. Non-small cell lung cancer harboring a rare EGFR L747P mutation showing intrinsic resistance to both gefitinib and osimertinib (AZD9291): A case report. Thorac. Cancer 2018, 9, 745–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, T.; Zhou, X.Y.; Li, P.; Qi, C.; Ling, Y. EGFR L747P mutation in one lung adenocarcinoma patient responded to afatinib treatment: A case report. J. Thorac. Dis. 2018, 10, E802–E805. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.K.; Ko, J.C.; Yang, J.C.H.; Shih, J.Y. Afatinib is effective in the treatment of lung adenocarcinoma with uncommon EGFR p.L747P and p.L747S mutations. Lung Cancer 2019, 133, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.T.; Ning, W.W.; Li, J.; Huang, J.A. Exon 19 L747P mutation presented as a primary resistance to EGFR-TKI: A case report. J. Thorac. Dis. 2016, 8, E542–E546. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.H.; Xie, X.X.; Sun, D.J.; Geng, J.Z.; Fu, F.H.; Zhang, L.M.; Wang, H.B. EGFR mutation L747P led to gefitinib resistance and accelerated liver metastases in a Chinese patient with lung adenocarcinoma. Int. J. Clin. Exp. Patho. 2015, 8, 8603–8606. [Google Scholar]
- Yamaguchi, F.; Fukuchi, K.; Yamazaki, Y.; Takayasu, H.; Tazawa, S.; Tateno, H.; Kato, E.; Wakabayashi, A.; Fujimori, M.; Iwasaki, T.; et al. Acquired resistance L747S mutation in an epidermal growth factor receptor-tyrosine kinase inhibitor-naive patient: A report of three cases. Oncol. Lett. 2014, 7, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Leonetti, A.; Sharma, S.; Minari, R.; Perego, P.; Giovannetti, E.; Tiseo, M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br. J. Cancer 2019, 121, 725–737. [Google Scholar] [CrossRef]
- Mehlman, C.; Cadranel, J.; Rousseau-Bussac, G.; Lacave, R.; Pujals, A.; Girard, N.; Callens, C.; Gounant, V.; Theou-Anton, N.; Friard, S.; et al. Resistance mechanisms to osimertinib in EGFR-mutated advanced non-small-cell lung cancer: A multicentric retrospective French study. Lung Cancer 2019, 137, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.O.; Cha, M.Y.; Kim, M.; Song, J.Y.; Lee, J.H.; Kim, Y.H.; Lee, Y.M.; Suh, K.H.; Son, J. Discovery of HM61713 as an orally available and mutant EGFR selective inhibitor. Cancer Res. 2014, 74. [Google Scholar] [CrossRef]
- Kim, E.S. Olmutinib: First global approval. Drugs 2016, 76, 1153–1157. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Lee, D.H.; Han, J.Y.; Lee, J.; Cho, B.C.; Kang, J.H.; Lee, K.H.; Cho, E.K.; Kim, J.S.; Min, Y.J.; et al. Safety, tolerability, and anti-tumor activity of olmutinib in non-small cell lung cancer with T790M mutation: A single arm, open label, phase 1/2 trial. Lung Cancer 2019, 135, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Guo, X.; To, K.K.W.; Chen, Z.; Fang, X.; Luo, M.; Ma, C.; Xu, J.; Yan, S.; Fu, L. Olmutinib (HM61713) reversed multidrug resistance by inhibiting the activity of ATP-binding cassette subfamily G member 2 in vitro and in vivo. Acta Pharm. Sin. B 2018, 8, 563–574. [Google Scholar] [CrossRef]
- Quintieri, L.; Fantin, M.; Vizler, C. Identification of molecular determinants of tumor sensitivity and resistance to anticancer drugs. Adv. Exp. Med. Biol. 2007, 593, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Song, H.N.; Jung, K.S.; Yoo, K.H.; Cho, J.; Lee, J.Y.; Lim, S.H.; Kim, H.S.; Sun, J.M.; Lee, S.H.; Ahn, J.S.; et al. Acquired C797S mutation upon treatment with a T790M-Specific third-generation EGFR inhibitor (HM61713) in non-small cell lung cancer. J. Thorac. Oncol. 2016, 11, E45–E47. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R., Jr. Small molecule inhibitors targeting the EGFR/ErbB family of protein-tyrosine kinases in human cancers. Pharmacol. Res. 2019, 139, 395–411. [Google Scholar] [CrossRef]
- Russo, A.; Franchina, T.; Ricciardi, G.R.R.; Smiroldo, V.; Picciotto, M.; Zanghi, M.; Rolfo, C.; Adamo, V. Third generation EGFR TKIs in EGFR-mutated NSCLC: Where are we now and where are we going. Crit Rev. Oncol. Hematol. 2017, 117, 38–47. [Google Scholar] [CrossRef]
- Meador, C.B.; Hata, A.N. Acquired resistance to targeted therapies in NSCLC: Updates and evolving insights. Pharmacol. Ther. 2020. [Google Scholar] [CrossRef]
- Baraibar, I.; Mezquita, L.; Gil-Bazo, I.; Planchard, D. Novel drugs targeting EGFR and HER2 exon 20 mutations in metastatic NSCLC. Crit. Rev. Oncol. Hematol. 2020, 148, 102906. [Google Scholar] [CrossRef] [PubMed]
- Bottaro, D.P.; Rubin, J.S.; Faletto, D.L.; Chan, A.M.L.; Kmiecik, T.E.; Vandewoude, G.F.; Aaronson, S.A. Identification of the hepatocyte growth-factor receptor as the C-Met protooncogene product. Science 1991, 251, 802–804. [Google Scholar] [CrossRef] [PubMed]
- Naldini, L.; Vigna, E.; Narsimhan, R.P.; Gaudino, G.; Zarnegar, R.; Michalopoulos, G.K.; Comoglio, P.M. Hepatocyte growth-factor (Hgf) stimulates the tyrosine kinase-activity of the receptor encoded by the protooncogene C-Met. Oncogene 1991, 6, 501–504. [Google Scholar] [PubMed]
- Graziani, A.; Gramaglia, D.; Cantley, L.C.; Comoglio, P.M. The tyrosine-phosphorylated hepatocyte growth-factor scatter factor receptor associates with phosphatidylinositol 3-Kinase. J. Biol. Chem. 1991, 266, 22087–22090. [Google Scholar]
- Fan, S.J.; Ma, Y.X.; Wang, J.A.; Yuan, R.Q.; Meng, Q.H.; Cao, Y.J.; Laterra, J.J.; Goldberg, I.D.; Rosen, E.M. The cytokine hepatocyte growth factor/scatter factor inhibits apoptosis and enhances DNA repair by a common mechanism involving signaling through phosphatidyl inositol 3 ’ kinase. Oncogene 2000, 19, 2212–2223. [Google Scholar] [CrossRef] [Green Version]
- Salgia, R. MET in lung cancer: Biomarker selection based on scientific rationale. Mol. Cancer Ther. 2017, 16, 555–565. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.Z.; Xia, M.F.; Jin, K.; Wang, S.F.; Wei, H.; Fan, C.M.; Wu, Y.F.; Li, X.L.; Li, X.Y.; Li, G.Y.; et al. Function of the c-Met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities. Mol. Cancer 2018, 17. [Google Scholar] [CrossRef]
- Zhang, Y.W.; Wang, L.M.; Jove, R.; Vande Woude, G.F. Requirement of Stat3 signaling for HGF/SF-Met mediated tumorigenesis. Oncogene 2002, 21, 217–226. [Google Scholar] [CrossRef]
- Beau-Faller, M.; Ruppert, A.M.; Voegeli, A.C.; Neuville, A.; Meyer, N.; Guerin, E.; Legrain, M.; Mennecier, B.; Wihlm, J.M.; Massard, G.; et al. MET gene copy number in non-small cell lung cancer: Molecular analysis in a targeted tyrosine kinase inhibitor naive cohort. J. Thorac. Oncol. 2008, 3, 331–339. [Google Scholar] [CrossRef]
- Tong, J.H.; Yeung, S.F.; Chan, A.W.; Chung, L.Y.; Chau, S.L.; Lung, R.W.; Tong, C.Y.; Chow, C.; Tin, E.K.; Yu, Y.H.; et al. MET amplification and exon 14 splice site mutation define unique molecular subgroups of non-small cell lung carcinoma with poor prognosis. Clin. Cancer Res. 2016, 22, 3048–3056. [Google Scholar] [CrossRef] [Green Version]
- Cappuzzo, F.; Janne, P.A.; Skokan, M.; Finocchiaro, G.; Rossi, E.; Ligorio, C.; Zucali, P.A.; Terracciano, L.; Toschi, L.; Roncalli, M.; et al. MET increased gene copy number and primary resistance to gefitinib therapy in non-small-cell lung cancer patients. Ann. Oncol. 2009, 20, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Cappuzzo, F.; Marchetti, A.; Skokan, M.; Rossi, E.; Gajapathy, S.; Felicioni, L.; Del Grammastro, M.; Sciarrotta, M.G.; Buttitta, F.; Incarbone, M.; et al. Increased MET gene copy number negatively affects survival of surgically resected non-small-cell lung cancer patients. J. Clin. Oncol. 2009, 27, 1667–1674. [Google Scholar] [CrossRef] [Green Version]
- Tachibana, K.; Minami, Y.; Shiba-Ishii, A.; Kano, J.; Nakazato, Y.; Sato, Y.; Goya, T.; Noguchi, M. Abnormality of the hepatocyte growth factor/MET pathway in pulmonary adenocarcinogenesis. Lung Cancer 2012, 75, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Kubo, T.; Yamamoto, H.; Lockwood, W.W.; Valencia, I.; Soh, J.; Peyton, M.; Jida, M.; Otani, H.; Fujii, T.; Ouchida, M.; et al. MET gene amplification or EGFR mutation activate MET in lung cancers untreated with EGFR tyrosine kinase inhibitors. Int. J. Cancer 2009, 124, 1778–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, W.; Song, L.; Ai, T.; Zhang, Y.; Gao, Y.; Cui, J. Prognostic value of MET, cyclin D1 and MET gene copy number in non-small cell lung cancer. J. Biomed. Res. 2013, 27, 220–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; An, S.J.; Chen, Z.H.; Su, J.; Yan, H.H.; Wu, Y.L. MET expression plays differing roles in non-small-cell lung cancer patients with or without EGFR mutation. J. Thorac. Oncol. 2014, 9, 725–728. [Google Scholar] [CrossRef] [Green Version]
- Paik, P.K.; Drilon, A.; Fan, P.D.; Yu, H.; Rekhtman, N.; Ginsberg, M.S.; Borsu, L.; Schultz, N.; Berger, M.F.; Rudin, C.M.L.; et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov. 2015, 5, 842–849. [Google Scholar] [CrossRef] [Green Version]
- Ma, P.C. MET receptor juxtamembrane exon 14 alternative spliced variant: Novel cancer genomic predictive biomarker. Cancer Discov. 2015, 5, 802–805. [Google Scholar] [CrossRef] [Green Version]
- Ma, P.C.; Kijima, T.; Maulik, G.; Fox, E.A.; Sattler, M.; Griffin, J.D.; Johnson, B.E.; Salgia, R. c-MET mutational analysis in small cell lung cancer: Novel juxtamembrane domain mutations regulating cytoskeletal functions. Cancer Res. 2003, 63, 6272–6281. [Google Scholar]
- Parikh, P.K.; Ghate, M.D. Recent advances in the discovery of small molecule c-Met Kinase inhibitors. Eur. J. Med. Chem. 2018, 143, 1103–1138. [Google Scholar] [CrossRef]
- Pasquini, G.; Giaccone, G. C-MET inhibitors for advanced non-small cell lung cancer. Expert Opin. Investig. Drugs 2018, 27, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Cappuzzo, F.; Ou, S.I.; Camidge, D.R. Targeting MET in lung cancer: Will expectations finally be MET? J. Thorac. Oncol. 2017, 12, 15–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mo, H.N.; Liu, P. Targeting MET in cancer therapy. Chronic. Dis. Transl. Med. 2017, 3, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Gozdzik-Spychalska, J.; Szyszka-Barth, K.; Spychalski, L.; Ramlau, K.; Wojtowicz, J.; Batura-Gabryel, H.; Ramlau, R. C-MET inhibitors in the treatment of lung cancer. Curr. Treat. Options Oncol. 2014, 15, 670–682. [Google Scholar] [CrossRef]
- Eathiraj, S.; Palma, R.; Volckova, E.; Hirschi, M.; France, D.S.; Ashwell, M.A.; Chan, T.C. Discovery of a novel mode of protein kinase inhibition characterized by the mechanism of inhibition of human mesenchymal-epithelial transition factor (c-Met) protein autophosphorylation by ARQ 197. J. Biol. Chem. 2011, 286, 20666–20676. [Google Scholar] [CrossRef] [Green Version]
- Gherardi, E.; Birchmeier, W.; Birchmeier, C.; Woude, G.V. Targeting MET in cancer: Rationale and progress. Nat. Rev. Cancer 2012, 12, 89–103. [Google Scholar] [CrossRef]
- Dussault, I.; Bellon, S.F. From Concept to Reality: The Long Road to c-Met and RON Receptor Tyrosine Kinase Inhibitors for the Treatment of Cancer. Anti Cancer Agent Med. Chem. 2009, 9, 221–229. [Google Scholar] [CrossRef]
- Zou, H.Y.; Li, Q.H.; Lee, J.H.; Arango, M.E.; McDonnell, S.R.; Yamazaki, S.; Koudriakova, T.B.; Alton, G.; Cui, J.J.; Kung, P.P.; et al. An orally available small-molecule inhibitor of c-met, PF-2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanisms. Cancer Res. 2007, 67, 4408–4417. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.J.; Tran-Dube, M.; Shen, H.; Nambu, M.; Kung, P.P.; Pairish, M.; Jia, L.; Meng, J.; Funk, L.; Botrous, I.; et al. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK). J. Med. Chem. 2011, 54, 6342–6363. [Google Scholar] [CrossRef]
- Kang, J.; Chen, H.J.; Wang, Z.; Liu, J.; Li, B.; Zhang, T.; Yang, Z.; Wu, Y.L.; Yang, J.J. Osimertinib and Cabozantinib Combinatorial Therapy in an EGFR-Mutant Lung Adenocarcinoma Patient with Multiple MET Secondary-Site Mutations after Resistance to Crizotinib. J. Thorac. Oncol. 2018, 13, e49–e53. [Google Scholar] [CrossRef] [Green Version]
- Heist, R.S.; Sequist, L.V.; Borger, D.; Gainor, J.F.; Arellano, R.S.; Le, L.P.; Dias-Santagata, D.; Clark, J.W.; Engelman, J.A.; Shaw, A.T.; et al. Acquired resistance to crizotinib in NSCLC with MET exon 14 skipping. J. Thorac. Oncol. 2016, 11, 1242–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, H.J.; Li, P.; Wu, C.L.; Zhou, X.Y.; Lu, H.J.; Zhou, T. Response and acquired resistance to crizotinib in Chinese patients with lung adenocarcinomas harboring MET Exon 14 splicing alternations. Lung Cancer 2016, 102, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Yang, J.J.; Zhang, X.C.; Zhang, Z.; Su, J.; Gou, L.Y.; Bai, Y.; Zhou, Q.; Yang, Z.; Han-Zhang, H.; et al. Acquired MET Y1248H and D1246N mutations mediate resistance to MET inhibitors in non-small cell lung cancer. Clin. Cancer Res. 2017, 23, 4929–4937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Peled, N.; Greer, J.; Wu, W.; Choi, P.; Berger, A.H.; Wong, S.; Jen, K.Y.; Seo, Y.; Hann, B.; et al. MET exon 14 mutation encodes an actionable therapeutic target in lung adenocarcinoma. Cancer Res. 2017, 77, 4498–4505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, S.H.I.; Young, L.; Schrock, A.B.; Johnson, A.; Klempner, S.J.; Zhu, V.W.; Miller, V.A.; Ali, S.M. Emergence of preexisting MET Y1230C mutation as a resistance mechanism to crizotinib in NSCLC with MET exon 14 skipping. J. Thorac. Oncol. 2017, 12, 137–140. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.; Ginalski, K.; Lesyng, B.; Nakaigawa, N.; Schmidt, L.; Zbar, B. Structural basis of oncogenic activation caused by point mutations in the kinase domain of the MET proto-oncogene: Modeling studies. Proteins 2001, 44, 32–43. [Google Scholar] [CrossRef]
- Liu, X.; Wang, Q.; Yang, G.; Marando, C.; Koblish, H.K.; Hall, L.M.; Fridman, J.S.; Behshad, E.; Wynn, R.; Li, Y.; et al. A novel kinase inhibitor, INCB28060, blocks c-MET-dependent signaling, neoplastic activities, and cross-talk with EGFR and HER-3. Clin. Cancer Res. 2011, 17, 7127–7138. [Google Scholar] [CrossRef] [Green Version]
- Baltschukat, S.; Engstler, B.S.; Huang, A.; Hao, H.X.; Tam, A.; Wang, H.Q.; Liang, J.; DiMare, M.T.; Bhang, H.C.; Wang, Y.; et al. Capmatinib (INC280) is active against models of non-small cell lung cancer and other cancer types with defined mechanisms of MET activation. Clin. Cancer Res. 2019, 25, 3164–3175. [Google Scholar] [CrossRef] [Green Version]
- Jia, H.; Dai, G.; Weng, J.; Zhang, Z.; Wang, Q.; Zhou, F.; Jiao, L.; Cui, Y.; Ren, Y.; Fan, S.; et al. Discovery of (S)-1-(1-(Imidazo[1,2-a]pyridin-6-yl)ethyl)-6-(1-methyl-1H-pyrazol-4-yl)-1H-[1,2, 3]triazolo[4,5-b]pyrazine (volitinib) as a highly potent and selective mesenchymal-epithelial transition factor (c-Met) inhibitor in clinical development for treatment of cancer. J. Med. Chem. 2014, 57, 7577–7589. [Google Scholar] [CrossRef]
- Gavine, P.R.; Ren, Y.; Han, L.; Lv, J.; Fan, S.; Zhang, W.; Xu, W.; Liu, Y.J.; Zhang, T.; Fu, H.; et al. Volitinib, a potent and highly selective c-Met inhibitor, effectively blocks c-Met signaling and growth in c-MET amplified gastric cancer patient-derived tumor xenograft models. Mol. Oncol. 2015, 9, 323–333. [Google Scholar] [CrossRef]
- Schuller, A.G.; Barry, E.R.; Jones, R.D.; Henry, R.E.; Frigault, M.M.; Beran, G.; Linsenmayer, D.; Hattersley, M.; Smith, A.; Wilson, J.; et al. The MET inhibitor AZD6094 (Savolitinib, HMPL-504) induces regression in papillary renal cell carcinoma patient-derived xenograft models. Clin. Cancer Res. 2015, 21, 2811–2819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, R.E.; Barry, E.R.; Castriotta, L.; Ladd, B.; Markovets, A.; Beran, G.; Ren, Y.; Zhou, F.; Adam, A.; Zinda, M.; et al. Acquired savolitinib resistance in non-small cell lung cancer arises via multiple mechanisms that converge on MET-independent mTOR and MYC activation. Oncotarget 2016, 7, 57651–57670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Hu, P.; Fang, D.; Ni, L.; Xu, J. Electrostatic explanation of D1228V/H/N-induced c-Met resistance and sensitivity to type I and type II kinase inhibitors in targeted gastric cancer therapy. J. Mol. Modeling 2019, 25, 13. [Google Scholar] [CrossRef] [PubMed]
- Maritano, D.; Accornero, P.; Bonifaci, N.; Ponzetto, C. Two mutations affecting conserved residues in the Met receptor operate via different mechanisms. Oncogene 2000, 19, 1354–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H. Anaplastic Lymphoma Kinase (ALK) receptor tyrosine kinase: A catalytic receptor with many faces. Int. J. Mol. Sci. 2018, 19, 3448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roskoski, R., Jr. Anaplastic lymphoma kinase (ALK): Structure, oncogenic activation, and pharmacological inhibition. Pharmacol. Res. 2013, 68, 68–94. [Google Scholar] [CrossRef]
- Guan, J.; Umapathy, G.; Yamazaki, Y.; Wolfstetter, G.; Mendoza, P.; Pfeifer, K.; Mohammed, A.; Hugosson, F.; Zhang, H.; Hsu, A.W.; et al. FAM150A and FAM150B are activating ligands for anaplastic lymphoma kinase. eLife 2015, 4, e09811. [Google Scholar] [CrossRef]
- Ducray, S.P.; Natarajan, K.; Garland, G.D.; Turner, S.D.; Egger, G. The transcriptional roles of ALK fusion proteins in tumorigenesis. Cancers 2019, 11, 1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, S.W.; Kirstein, M.N.; Valentine, M.B.; Dittmer, K.G.; Shapiro, D.N.; Saltman, D.L.; Look, A.T. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 1994, 263, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Box, J.K.; Paquet, N.; Adams, M.N.; Boucher, D.; Bolderson, E.; O’Byrne, K.J.; Richard, D.J. Nucleophosmin: From structure and function to disease development. BMC Mol. Biol. 2016, 17, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamant, L.; Dastugue, N.; Pulford, K.; Delsol, G.; Mariame, B. A new fusion gene TPM3-ALK in anaplastic large cell lymphoma created by a (1;2)(q25;p23) translocation. Blood 1999, 93, 3088–3095. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Choi, Y.L.; Togashi, Y.; Soda, M.; Hatano, S.; Inamura, K.; Takada, S.; Ueno, T.; Yamashita, Y.; Satoh, Y.; et al. KIF5B-ALK, a novel fusion oncokinase identified by an immunohistochemistry-based diagnostic system for ALK-positive lung cancer. Clin. Cancer Res. 2009, 15, 3143–3149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Togashi, Y.; Soda, M.; Sakata, S.; Sugawara, E.; Hatano, S.; Asaka, R.; Nakajima, T.; Mano, H.; Takeuchi, K. KLC1-ALK: A novel fusion in lung cancer identified using a formalin-fixed paraffin-embedded tissue only. PLoS ONE 2012, 7, e31323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, L.M.; Barila, G.; Liu, P.; Evdokimova, V.N.; Trivedi, S.; Panebianco, F.; Gandhi, M.; Carty, S.E.; Hodak, S.P.; Luo, J.; et al. Identification of the transforming STRN-ALK fusion as a potential therapeutic target in the aggressive forms of thyroid cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 4233–4238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, Y.; Kim, P.; Jung, Y.; Keum, J.; Kim, S.N.; Choi, Y.S.; Do, I.G.; Lee, J.; Choi, S.J.; Kim, S.; et al. Discovery of ALK-PTPN3 gene fusion from human non-small cell lung carcinoma cell line using next generation RNA sequencing. Genes Chromosomes Cancer 2012, 51, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Iyevleva, A.G.; Raskin, G.A.; Tiurin, V.I.; Sokolenko, A.P.; Mitiushkina, N.V.; Aleksakhina, S.N.; Garifullina, A.R.; Strelkova, T.N.; Merkulov, V.O.; Ivantsov, A.O.; et al. Novel ALK fusion partners in lung cancer. Cancer Lett. 2015, 362, 116–121. [Google Scholar] [CrossRef]
- Jiang, J.; Wu, X.; Tong, X.; Wei, W.; Chen, A.; Wang, X.; Shao, Y.W.; Huang, J. GCC2-ALK as a targetable fusion in lung adenocarcinoma and its enduring clinical responses to ALK inhibitors. Lung Cancer 2018, 115, 5–11. [Google Scholar] [CrossRef]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.I.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef]
- Horn, L.; Pao, W. EML4-ALK: Honing in on a new target in non-small-cell lung cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 4232–4235. [Google Scholar] [CrossRef]
- Lin, Y.T.; Yu, C.J.; Yang, J.C.H.; Shih, J.Y. Anaplastic Lymphoma Kinase (ALK) kinase domain mutation following ALK Inhibitor(s) failure in advanced ALK positive Non-Small-Cell Lung Cancer: Analysis and literature review. Clin. Lung Cancer 2016, 17, e77–e94. [Google Scholar] [CrossRef]
- Shaw, A.T.; Yeap, B.Y.; Solomon, B.J.; Riely, G.J.; Gainor, J.; Engelman, J.A.; Shapiro, G.I.; Costa, D.B.; Ou, S.H.I.; Butaney, M.; et al. Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: A retrospective analysis. Lancet Oncol. 2011, 12, 1004–1012. [Google Scholar] [CrossRef] [Green Version]
- Narayanan, D.; Gani, O.A.B.S.M.; Gruber, F.X.E.; Engh, R.A. Data driven polypharmacological drug design for lung cancer: Analyses for targeting ALK, MET, and EGFR. J. Cheminform 2017, 9, 43. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Johnson, T.W.; Bailey, S.; Brooun, A.; Bunker, K.D.; Burke, B.J.; Collins, M.R.; Cook, A.S.; Cui, J.J.; Dack, K.N.; et al. Design of potent and selective inhibitors to overcome clinical anaplastic lymphoma kinase mutations resistant to crizotinib. J. Med. Chem. 2014, 57, 1170–1187. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.L.; Soda, M.; Yamashita, Y.; Ueno, T.; Takashima, J.; Nakajima, T.; Yatabe, Y.; Takeuchi, K.; Hamada, T.; Haruta, H.; et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N. Engl. J. Med. 2010, 363, 1734–1739. [Google Scholar] [CrossRef] [PubMed]
- Gainor, J.F.; Dardaei, L.; Yoda, S.; Friboulet, L.; Leshchiner, I.; Katayama, R.; Dagogo-Jack, I.; Gadgeel, S.; Schultz, K.; Singh, M.; et al. Molecular mechanisms of resistance to first- and second-generation ALK inhibitors in ALK-rearranged lung cancer. Cancer Discov. 2016, 6, 1118–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katayama, R.; Shaw, A.T.; Khan, T.M.; Mino-Kenudson, M.; Solomon, B.J.; Halmos, B.; Jessop, N.A.; Wain, J.C.; Yeo, A.T.; Benes, C.; et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Sci. Transl. Med. 2012, 4, 120ra117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Kim, T.M.; Kim, D.W.; Go, H.; Keam, B.; Lee, S.H.; Ku, J.L.; Chung, D.H.; Heo, D.S. Heterogeneity of genetic changes associated with acquired crizotinib resistance in ALK-rearranged lung cancer. J. Thorac. Oncol. 2013, 8, 415–422. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.; Kim, D.W.; Kotsakis, A.; Deng, S.; Lira, P.; Ho, S.N.; Lee, N.V.; Vizcarra, P.; Cao, J.Q.; Christensen, J.G.; et al. Multiplexed deep sequencing analysis of ALK kinase domain identifies resistance mutations in relapsed patients following crizotinib treatment. Genomics 2013, 102, 157–162. [Google Scholar] [CrossRef] [Green Version]
- Ignatius Ou, S.H.; Azada, M.; Hsiang, D.J.; Herman, J.M.; Kain, T.S.; Siwak-Tapp, C.; Casey, C.; He, J.; Ali, S.M.; Klempner, S.J.; et al. Next-generation sequencing reveals a Novel NSCLC ALK F1174V mutation and confirms ALK G1202R mutation confers high-level resistance to alectinib (CH5424802/RO5424802) in ALK-rearranged NSCLC patients who progressed on crizotinib. J. Thorac. Oncol. 2014, 9, 549–553. [Google Scholar]
- Friboulet, L.; Li, N.; Katayama, R.; Lee, C.C.; Gainor, J.F.; Crystal, A.S.; Michellys, P.Y.; Awad, M.M.; Yanagitani, N.; Kim, S.; et al. The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discov. 2014, 4, 662–673. [Google Scholar] [CrossRef] [Green Version]
- Saber, A.; Van der Wekken, A.J.; Kok, K.; Terpstra, M.M.; Bosman, L.J.; Mastik, M.F.; Timens, W.; Schuuring, E.; Hiltermann, T.J.N.; Groen, H.J.M.; et al. Genomic aberrations in crizotinib resistant lung adenocarcinoma samples identified by transcriptome sequencing. PLoS ONE 2016, 11, e0153065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ai, X.; Niu, X.; Chang, L.; Chen, R.; Ou, S.-H.I.; Lu, S. Next generation sequencing reveals a novel ALK G1128A mutation resistant to crizotinib in an ALK-Rearranged NSCLC patient. Lung Cancer 2018, 123, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Bresler, S.C.; Weiser, D.A.; Huwe, P.J.; Park, J.H.; Krytska, K.; Ryles, H.; Laudenslager, M.; Rappaport, E.F.; Wood, A.C.; McGrady, P.W.; et al. ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer Cell 2014, 26, 682–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyokawa, G.; Hirai, F.; Inamasu, E.; Yoshida, T.; Nosaki, K.; Takenaka, T.; Yamaguchi, M.; Seto, T.; Takenoyama, M.; Ichinose, Y. Secondary mutations at I1171 in the ALK gene confer resistance to both Crizotinib and Alectinib. J. Thorac. Oncol. 2014, 9, 86–87. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, H.; Tsukaguchi, T.; Hiroshima, S.; Kodama, T.; Kobayashi, T.; Fukami, T.A.; Oikawa, N.; Tsukuda, T.; Ishii, N.; Aoki, Y. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell 2011, 19, 679–690. [Google Scholar] [CrossRef] [Green Version]
- Kodama, T.; Tsukaguchi, T.; Yoshida, M.; Kondoh, O.; Sakamoto, H. Selective ALK inhibitor alectinib with potent antitumor activity in models of crizotinib resistance. Cancer Lett. 2014, 351, 215–221. [Google Scholar] [CrossRef]
- Paik, J.; Dhillon, S. Alectinib: A Review in Advanced, ALK-Positive NSCLC. Drugs 2018, 78, 1247–1257. [Google Scholar] [CrossRef]
- Ou, S.-H.I.; Greenbowe, J.; Khan, Z.U.; Azada, M.C.; Ross, J.S.; Stevens, P.J.; Ali, S.M.; Miller, V.A.; Gitlitz, B. I1171 missense mutation (particularly I1171N) is a common resistance mutation in ALK-positive NSCLC patients who have progressive disease while on alectinib and is sensitive to ceritinib. Lung Cancer 2015, 88, 231–234. [Google Scholar] [CrossRef]
- Ou, S.H.I.; Klempner, S.J.; Greenbowe, J.R.; Azada, M.; Schrock, A.B.; Ali, S.M.; Ross, J.S.; Stephens, P.J.; Miller, V.A. Identification of a novel HIP1-ALK fusion variant in Non-Small-Cell Lung Cancer (NSCLC) and discovery of ALK I1171 (I1171N/S) mutations in two ALK-rearranged NSCLC patients with resistance to Alectinib. J. Thorac. Oncol. 2014, 9, 1821–1825. [Google Scholar] [CrossRef] [Green Version]
- Ou, S.-H.; Milliken, J.C.; Azada, M.C.; Miller, V.A.; Ali, S.M.; Klempner, S.J. ALK F1174V mutation confers sensitivity while ALK I1171 mutation confers resistance to alectinib. The importance of serial biopsy post progression. Lung Cancer 2016, 91, 70–72. [Google Scholar] [CrossRef]
- He, M.; Li, W.; Zheng, Q.; Zhang, H. A molecular dynamics investigation into the mechanisms of alectinib resistance of three ALK mutants. J. Cell. Biochem. 2018, 119, 5332–5342. [Google Scholar] [CrossRef] [PubMed]
- Katayama, R.; Friboulet, L.; Koike, S.; Lockerman, E.L.; Khan, T.M.; Gainor, J.F.; Iafrate, A.J.; Takeuchi, K.; Taiji, M.; Okuno, Y.; et al. Two novel ALK mutations mediate acquired resistance to the next-generation ALK inhibitor alectinib. Clin. Cancer Res. 2014, 20, 5686–5696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soria, J.C.; Tan, D.S.W.; Chiari, R.; Wu, Y.L.; Paz-Ares, L.; Wolf, J.; Geater, S.L.; Orlov, S.; Cortinovis, D.; Yu, C.J.; et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): A randomised, open-label, phase 3 study. Lancet 2017, 389, 917–929. [Google Scholar] [CrossRef]
- Toyokawa, G.; Inamasu, E.; Shimamatsu, S.; Yoshida, T.; Nosaki, K.; Hirai, F.; Yamaguchi, M.; Seto, T.; Takenoyama, M.; Ichinose, Y. Identification of a Novel ALK G1123S Mutation in a Patient with ALK-rearranged Non-small-cell Lung Cancer Exhibiting Resistance to Ceritinib. J. Thorac. Oncol. 2015, 10, 55–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, M.Y.; Li, W.K.; Meiler, J.; Zheng, Q.C.; Zhang, H.X. Insight on mutation-induced resistance to anaplastic lymphoma kinase inhibitor ceritinib from molecular dynamics simulations. Biopolymers 2019, 110, e23257. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Ho, C.C.; Shih, J.Y. Multiple acquired resistance mutations of the ALK tyrosine kinase domainafter sequential use of ALK inhibitors. J. Thorac. Oncol. 2017, 12, e49–e51. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [Green Version]
- Cazzola, M. Introduction to a review series on myeloproliferative neoplasms. Blood 2017, 129, 659. [Google Scholar] [CrossRef]
- Houshmand, M.; Simonetti, G.; Circosta, P.; Gaidano, V.; Cignetti, A.; Martinelli, G.; Saglio, G.; Gale, R.P. Chronic myeloid leukemia stem cells. Leukemia 2019, 33, 1543–1556. [Google Scholar] [CrossRef] [Green Version]
- Hallek, M. Chronic lymphocytic leukemia: 2015 Update on diagnosis, risk stratification, and treatment. Am. J. Hematol. 2015, 90, 446–460. [Google Scholar] [CrossRef]
- Austen, B.; Skowronska, A.; Baker, C.; Powell, J.E.; Gardiner, A.; Oscier, D.; Majid, A.; Dyer, M.; Siebert, R.; Taylor, A.M.; et al. Mutation status of the residual ATM allele is an important determinant of the cellular response to chemotherapy and survival in patients with chronic lymphocytic leukemia containing an 11q deletion. J. Clin. Oncol. 2007, 25, 5448–5457. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.L.; Lawrence, M.S.; Wan, Y.Z.; Stojanov, P.; Sougnez, C.; Stevenson, K.; Werner, L.; Sivachenko, A.; DeLuca, D.S.; Zhang, L.; et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N. Engl. J. Med. 2011, 365, 2497–2506. [Google Scholar] [CrossRef] [PubMed]
- Rose-Zerilli, M.J.J.; Forster, J.; Parker, H.; Parker, A.; Rodriguez, A.E.; Chaplin, T.; Gardiner, A.; Steele, A.J.; Collins, A.; Young, B.D.; et al. ATM mutation rather than BIRC3 deletion and/or mutation predicts reduced survival in 11q-deleted chronic lymphocytic leukemia: Data from the UK LRF CLL4 trial. Haematologica 2014, 99, 736–742. [Google Scholar] [CrossRef] [PubMed]
- Puente, X.S.; Lopez-Otin, C. The evolutionary biography of chronic lymphocytic leukemia. Nat. Genet. 2013, 45, 229–231. [Google Scholar] [CrossRef] [PubMed]
- Juarez-Salcedo, L.M.; Castillo, J.J. Lymphoplasmacytic lymphoma and marginal zone lymphoma. Hematol. Oncol. Clin. N. Am. 2019, 33, 639–656. [Google Scholar] [CrossRef]
- Diamond, B.; Kumar, A. Mantle cell lymphoma: Current and emerging treatment strategies and unanswered questions. Hematol. Oncol. Clin. N. Am. 2019, 33, 613–626. [Google Scholar] [CrossRef]
- Konopka, J.B.; Watanabe, S.M.; Witte, O.N. An alteration of the human c-abl protein in K562 leukemia cells unmasks associated tyrosine kinase activity. Cell 1984, 37, 1035–1042. [Google Scholar] [CrossRef]
- Pui, C.-H.; Roberts, K.G.; Yang, J.J.; Mullighan, C.G. Philadelphia chromosome-like acute lymphoblastic leukemia. Clin. Lymphoma Myeloma Leuk. 2017, 17, 464–470. [Google Scholar] [CrossRef]
- Wang, J.Y.J. The capable ABL: What is its biological function? Mol. Cell. Biol. 2014, 34, 1188–1197. [Google Scholar] [CrossRef] [Green Version]
- Kharbanda, S.; Ren, R.; Pandey, P.; Shafman, T.D.; Feller, S.M.; Weichselbaum, R.R.; Kufe, D.W. Activation of the c-Abl tyrosine kinase in the stress response to DNA-damaging agents. Nature 1995, 376, 785–788. [Google Scholar] [CrossRef]
- Lewis, J.M.; Baskaran, R.; Taagepera, S.; Schwartz, M.A.; Wang, J.Y. Integrin regulation of c-Abl tyrosine kinase activity and cytoplasmic-nuclear transport. Proc. Natl. Acad. Sci. USA 1996, 93, 15174–15179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barila, D.; Superti-Furga, G. An intramolecular SH3-domain interaction regulates c-Abl activity. Nat. Genet. 1998, 18, 280–282. [Google Scholar] [CrossRef] [PubMed]
- Nowell, P.C.; Hungerford, D.A. Chromosome studies on normal and leukemic human leukocytes. J. Natl. Cancer Inst. 1960, 25, 85–109. [Google Scholar] [PubMed]
- Nowell, P.C.; Hungerford, D.A. Minute chromosome in human chronic granulocytic leukemia. Science 1960, 132, 1497-1497. [Google Scholar]
- Rowley, J.D. New consistent chromosomal abnormality in chronic myelogenous leukemia identified by quinacrine fluorescence and giemsa staining. Nature 1973, 243, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Kurzrock, R.; Kantarjian, H.M.; Druker, B.J.; Talpaz, M. Philadelphia chromosome-positive leukemias: From basic mechanisms to molecular therapeutics. Ann. Intern. Med. 2003, 138, 819–830. [Google Scholar] [CrossRef]
- Laurent, E.; Talpaz, M.; Kantarjian, H.; Kurzrock, R. The BCR gene and Philadelphia chromosome-positive leukemogenesis. Cancer Res. 2001, 61, 2343–2355. [Google Scholar]
- Arlinghaus, R.B. The involvement of Bcr in leukemias with the Philadelphia chromosome. Crit. Rev. Oncog. 1998, 9, 1–18. [Google Scholar] [CrossRef]
- Lugo, T.G.; Pendergast, A.M.; Muller, A.J.; Witte, O.N. Tyrosine kinase-activity and transformation potency of Bcr-Abl oncogene products. Science 1990, 247, 1079–1082. [Google Scholar] [CrossRef]
- Bedi, A.; Zehnbauer, B.A.; Barber, J.P.; Sharkis, S.J.; Jones, R.J. Inhibition of apoptosis by Bcr-Abl in chronic myeloid-leukemia. Blood 1994, 83, 2038–2044. [Google Scholar] [CrossRef] [Green Version]
- Sawyers, C.L.; Mclaughlin, J.; Witte, O.N. Genetic requirement for ras in the transformation of fibroblasts and hematopoietic-cells by the Bcr-Abl oncogene. J. Exp. Med. 1995, 181, 307–313. [Google Scholar] [CrossRef] [Green Version]
- Tauchi, T.; Okabe, S.; Miyazawa, K.; Ohyashiki, K. The tetramerization domain-independent Ras activation by BCR-ABL oncoprotein in hematopoietic cells. Int. J. Oncol. 1998, 12, 1269–1276. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.D.; Kurzrock, R.; Qiu, X.G.; Estrov, Z.; Ku, S.; Dulski, K.M.; Wang, J.Y.J.; Talpaz, M. Reduced focal adhesion kinase and paxillin phosphorylation in BCR-ABL-transfected cells. Cancer 2002, 95, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, A.; Miyazawa, K.; Ohyashiki, K.; Tauchi, T.; Boswell, H.S.; Broxmeyer, H.E.; Toyama, K. Tyrosine phosphorylation and activation of Focal Adhesion Kinase (P125(Fak)) by Bcr-Abl oncoprotein. Exp. Hematol. 1995, 23, 1153–1159. [Google Scholar] [PubMed]
- Ilaria, R.L.; VanEtten, R.A. P210 and P190(BCR/ABL) induce the tyrosine phosphorylation and DNA binding activity of multiple specific STAT family members. J. Biol. Chem. 1996, 271, 31704–31710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WilsonRawls, J.; Xie, S.H.; Liu, J.X.; Laneuville, P.; Arlinghaus, R.B. P210 Bcr-Abl interacts with the interleukin 3 receptor beta(c) subunit and constitutively induces its tyrosine phosphorylation. Cancer Res. 1996, 56, 4549. [Google Scholar]
- Takeda, N.; Shibuya, M.; Maru, Y. The BCR-ABL oncoprotein potentially interacts with the xeroderma pigmentosum group B protein. Proc. Natl. Acad. Sci. USA 1999, 96, 203–207. [Google Scholar] [CrossRef] [Green Version]
- Slupianek, A.; Schmutte, C.; Tombline, G.; Nieborowska-Skorska, M.; Hoser, G.; Nowicki, M.O.; Pierce, A.J.; Fishel, R.; Skorski, T. BCR/ABL regulates mammalian RecA homologs, resulting in drug resistance. Mol. Cell 2001, 8, 795–806. [Google Scholar] [CrossRef]
- Kalaycio, M.E. Chronic myelogenous leukemia: The news you have and haven’t heard. Cleve. Clin. J. Med. 2001, 68, 913–927. [Google Scholar] [CrossRef]
- Trela, E.; Glowacki, S.; Blasiak, J. Therapy of chronic myeloid leukemia: Twilight of the imatinib era? ISRN Oncol. 2014, 2014, 596483. [Google Scholar] [CrossRef] [Green Version]
- Mauro, M.J.; Druker, B.J. STI571: Targeting BCR-ABL as therapy for CML. Oncologist 2001, 6, 233–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagar, B.; Bornmann, W.G.; Pellicena, P.; Schindler, T.; Veach, D.R.; Miller, W.T.; Clarkson, B.; Kuriyan, J. Crystal structures of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and imatinib (STI-571). Cancer Res. 2002, 62, 4236–4243. [Google Scholar] [PubMed]
- Schindler, T.; Bornmann, W.; Pellicena, P.; Miller, W.T.; Clarkson, B.; Kuriyan, J. Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science 2000, 289, 1938–1942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorre, M.E.; Mohammed, M.; Ellwood, K.; Hsu, N.; Paquette, R.; Rao, P.N.; Sawyers, C.L. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 2001, 293, 876–880. [Google Scholar] [CrossRef] [Green Version]
- Milojkovic, D.; Apperley, J. Mechanisms of resistance to imatinib and second-generation tyrosine inhibitors in chronic myeloid leukemia. Clin. Cancer Res. 2009, 15, 7519–7527. [Google Scholar] [CrossRef] [Green Version]
- Schnittger, S.; Bacher, U.; Dicker, F.; Kern, W.; Alpermann, T.; Haferlach, T.; Haferlach, C. Associations between imatinib resistance conferring mutations and Philadelphia positive clonal cytogenetic evolution in CML. Genes Chromosomes Cancer 2010, 49, 910–918. [Google Scholar] [CrossRef]
- Tanaka, R.; Kimura, S.; Ashihara, E.; Yoshimura, M.; Takahashi, N.; Wakita, H.; Itoh, K.; Nishiwaki, K.; Suzuki, K.; Nagao, R.; et al. Rapid automated detection of ABL kinase domain mutations in imatinib-resistant patients. Cancer Lett. 2011, 312, 228–234. [Google Scholar] [CrossRef]
- Gibbons, D.L.; Pricl, S.; Kantarjian, H.; Cortes, J.; Quintas-Cardama, A. The rise and fall of gatekeeper mutations? The BCR-ABL1 T315I paradigm. Cancer 2012, 118, 293–299. [Google Scholar] [CrossRef]
- Xu, H.L.; Wang, Z.J.; Liang, X.M.; Li, X.; Shi, Z.; Zhou, N.; Bao, J.K. In silico identification of novel kinase inhibitors targeting wild-type and T315I mutant ABL1 from FDA-approved drugs. Mol. Biosyst. 2014, 10, 1524–1537. [Google Scholar] [CrossRef]
- Young, M.A.; Shah, N.P.; Chao, L.H.; Seeliger, M.; Milanov, Z.V.; Biggs, W.H., 3rd; Treiber, D.K.; Patel, H.K.; Zarrinkar, P.P.; Lockhart, D.J.; et al. Structure of the kinase domain of an imatinib-resistant Abl mutant in complex with the Aurora kinase inhibitor VX-680. Cancer Res. 2006, 66, 1007–1014. [Google Scholar] [CrossRef] [Green Version]
- Branford, S.; Rudzki, Z.; Walsh, S.; Parkinson, I.; Grigg, A.; Szer, J.; Taylor, K.; Herrmann, R.; Seymour, J.F.; Arthur, C.; et al. Detection of BCR-ABL mutations in patients with CML treated with imatinib is virtually always accompanied by clinical resistance, and mutations in the ATP phosphate-binding loop (P-loop) are associated with a poor prognosis. Blood 2003, 102, 276–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, D.; Saunders, V.; Lyons, A.B.; Branford, S.; Grigg, A.; To, L.B.; Hughes, T. In vitro sensitivity to imatinib-induced inhibition of ABL kinase activity is predictive of molecular response in patients with de novo CML. Blood 2005, 106, 2520–2526. [Google Scholar] [CrossRef] [Green Version]
- Jabbour, E.; Kantarjian, H.; Jones, D.; Talpaz, M.; Bekele, N.; O’Brien, S.; Zhou, X.; Luthra, R.; Garcia-Manero, G.; Giles, F.; et al. Frequency and clinical significance of BCR-ABL mutations in patients with chronic myeloid leukemia treated with imatinib mesylate. Leukemia 2006, 20, 1767–1773. [Google Scholar] [CrossRef] [PubMed]
- Cang, S.; Liu, D. P-loop mutations and novel therapeutic approaches for imatinib failures in chronic myeloid leukemia. J. Hematol. Oncol. 2008, 1, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roumiantsev, S.; Shah, N.P.; Gorre, M.E.; Nicoll, J.; Brasher, B.B.; Sawyers, C.L.; Van Etten, R.A. Clinical resistance to the kinase inhibitor STI-571 in chronic myeloid leukemia by mutation of Tyr-253 in the Abl kinase domain P-loop. Proc. Natl. Acad. Sci. USA 2002, 99, 10700–10705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagar, B.; Hantschel, O.; Seeliger, M.; Davies, J.M.; Weiss, W.I.; Superti-Furga, G.; Kuriyan, J. Organization of the SH3-SH2 unit in active and inactive forms of the c-Abl tyrosine kinase. Mol. Cell 2006, 21, 787–798. [Google Scholar] [CrossRef]
- Smith, K.M.; Yacobi, R.; Van Etten, R.A. Autoinhibition of Bcr-Abl through its SH3 domain. Mol. Cell 2003, 12, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Colicelli, J. ABL tyrosine kinases: Evolution of function, regulation, and specificity. Sci. Signal. 2010, 3. [Google Scholar] [CrossRef] [Green Version]
- Von Bubnoff, N.; Schneller, F.; Peschel, C.; Duyster, J. BCR-ABL gene mutations in relation to clinical resistance of Philadelphia-chromosome-positive leukaemia to STI571: A prospective study. Lancet 2002, 359, 487–491. [Google Scholar] [CrossRef]
- Radich, J.P.; Mauro, M.J. Tyrosine kinase inhibitor treatment for newly diagnosed chronic myeloid leukemia. Hematol. Oncol. Clin. N. Am. 2017, 31, 577–587. [Google Scholar] [CrossRef]
- Kennedy, J.A.; Hobbs, G. Tyrosine kinase inhibitors in the treatment of chronic-phase CML: Strategies for frontline decision-making. Curr. Hematol. Malig. Rep. 2018, 13, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Chopade, P.; Akard, L.P. Improving outcomes in chronic myeloid leukemia over time in the era of tyrosine kinase inhibitors. Clin. Lymph. Myelom. Leuk. 2018, 18, 710–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padmanabhan, S.; Ravella, S.; Curiel, T.; Giles, F. Current status of therapy for chronic myeloid leukemia: A review of drug development. Future Oncol. 2008, 4, 359–377. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.P.; Tran, C.; Lee, F.Y.; Chen, P.; Norris, D.; Sawyers, C.L. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science 2004, 305, 399–401. [Google Scholar] [CrossRef] [Green Version]
- Lombardo, L.J.; Lee, F.Y.; Chen, P.; Norris, D.; Barrish, J.C.; Behnia, K.; Castaneda, S.; Cornelius, L.A.; Das, J.; Doweyko, A.M.; et al. Discovery of N-(2-chloro-6-methyl- phenyl)-2-(6-(4-(2-hydroxyethyl)- piperazin-1-yl)-2-methylpyrimidin-4- ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J. Med. Chem. 2004, 47, 6658–6661. [Google Scholar] [CrossRef] [PubMed]
- Brattas, M.K.; Reikvam, H.; Tvedt, T.H.A.; Bruserud, O. Dasatinib as an investigational drug for the treatment of Philadelphia chromosome-positive acute lymphoblastic leukemia in adults. Expert Opin. Investig. Drugs 2019, 28, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Schittenhelm, M.M.; Shiraga, S.; Schroeder, A.; Corbin, A.S.; Griffith, D.; Lee, F.Y.; Bokemeyer, C.; Deininger, M.W.; Druker, B.J.; Heinrich, M.C. Dasatinib (BMS-354825), a dual SRC/ABL kinase inhibitor, inhibits the kinase activity of wild-type, juxtamembrane, and activation loop mutant KIT isoforms associated with human malignancies. Cancer Res. 2006, 66, 473–481. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, M.; Garg, R.J.; Cortes, J.; Quintas-Cardama, A. Tyrosine kinase inhibitors: The first decade. Curr. Hematol. Malig. Rep. 2010, 5, 70–80. [Google Scholar] [CrossRef]
- Kantarjian, H.; Ottmann, O.; Cortes, J.; Wassmann, B.; Jones, D.; Hochhaus, A.; Alland, L.; Dugan, M.; Albitar, M.; Giles, F. AMN107, a novel aminopyrimidine inhibitor of Bcr-Abl, has significant activity in imatinib-resistant Bcr-Abl positive chronic myeloid leukemia (CML). J. Clin. Oncol. 2005, 23, 195s-195s. [Google Scholar] [CrossRef]
- Golemovic, M.; Giles, F.J.; Beran, M.; Cortes, J.; Manshouri, T.; Kantarjian, H.; Verstovsek, S. AMN107, a novel aminopyrimidine inhibitor of BCR-ABL, has pre-clinical activity against imatinib mesylate-resistant chronic myeloid leukemia (CML). Blood 2004, 104, 547a-547a. [Google Scholar] [CrossRef]
- Giles, F.G.; Ottmann, O.T.; Cortes, J.; Wassmann, B.; Bhalla, K.; Jones, D.; Hochhaus, A.; Rae, P.E.; Tanaka, C.; Alland, L.; et al. AMN107, a novel aminopyrimidine inhibitor of Bcr-Abl, has significant activity in imatinib-resistant Bcr-Abl positive chronic myeloid leukemia (CML). Brit. J. Haematol. 2005, 129, 3. [Google Scholar]
- Weisberg, E.; Manley, P.W.; Breitenstein, W.; Bruggen, J.; Cowan-Jacob, S.W.; Ray, A.; Huntly, B.; Fabbro, D.; Fendrich, G.; Hall-Meyers, E.; et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell 2005, 7, 129–141. [Google Scholar] [CrossRef] [Green Version]
- Weisberg, E.; Manley, P.; Mestan, J.; Cowan-Jacob, S.; Ray, A.; Griffin, J.D. AMN107 (nilotinib): A novel and selective inhibitor of BCR-ABL (vol 94, pg 1765, 2016). Brit. J. Cancer 2019, 121, 282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Bubnoff, N.; Gorantla, S.H.P.; Kancha, R.K.; Lordick, F.; Peschel, C.; Duyster, J. The systemic mastocytosis-specific activating cKit mutation D816V can be inhibited by the tyrosine kinase inhibitor AMN107. Leukemia 2005, 19, 1670–1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gleixner, K.V.; Mayerhofer, M.; Aichberger, K.J.; Derdak, S.; Sonneck, K.; Bohm, A.; Gruze, A.; Samorapoompichit, P.; Manley, P.W.; Fabbro, D.; et al. PKC412 inhibits in vitro growth of neoplastic human mast cells expressing the D816V-mutated variant of KIT: Comparison with AMN107, imatinib, and cladribine (2CdA) and evaluation of cooperative drug effects. Blood 2006, 107, 752–759. [Google Scholar] [CrossRef] [Green Version]
- Montemurro, M.; Schoffski, P.; Reichardt, P.; Gelderblom, H.; Schutte, J.; Hartmann, J.T.; von Moos, R.; Seddon, B.; Joensuu, H.; Wendtner, C.M.; et al. Nilotinib in the treatment of advanced gastrointestinal stromal tumours resistant to both imatinib and sunitinib. Eur. J. Cancer 2009, 45, 2293–2297. [Google Scholar] [CrossRef] [PubMed]
- Sugase, T.; Takahashi, T.; Ishikawa, T.; Ichikawa, H.; Kanda, T.; Hirota, S.; Nakajima, K.; Tanaka, K.; Miyazaki, Y.; Makino, T.; et al. Surgical resection of recurrent gastrointestinal stromal tumor after interruption of long-term nilotinib therapy. Surg. Case Rep. 2016, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, K.; Chen, Z.; Zhang, X.M. Two new KIT exon 13 mutations in one gastric gastrointestinal stromal tumor (GIST). Int. J. Clin. Exp. Patho. 2017, 10, 8863–8867. [Google Scholar]
- Boschelli, D.H.; Wang, Y.D.; Ye, F.; Wu, B.; Zhang, N.; Dutia, M.; Powell, D.W.; Wissner, A.; Arndt, K.; Weber, J.M.; et al. Synthesis and Src kinase inhibitory activity of a series of 4-phenylamino-3-quinolinecarbonitriles. J. Med. Chem. 2001, 44, 822–833. [Google Scholar] [CrossRef]
- Golas, J.M.; Arndt, K.; Etienne, C.; Lucas, J.; Nardin, D.; Gibbons, J.; Frost, P.; Ye, F.; Boschelli, D.H.; Boschelli, F. SKI-606, a 4-anilino-3-quinolinecarbonitrile dual inhibitor of Src and Abl kinases, is a potent antiproliferative agent against chronic myelogenous leukemia cells in culture and causes regression of K562 xenografts in nude mice. Cancer Res. 2003, 63, 375–381. [Google Scholar]
- Cortes, J.E.; Kantarjian, H.M.; Brummendorf, T.H.; Kim, D.W.; Turkina, A.G.; Shen, Z.X.; Pasquini, R.; Khoury, H.J.; Arkin, S.; Volkert, A.; et al. Safety and efficacy of bosutinib (SKI-606) in chronic phase Philadelphia chromosome-positive chronic myeloid leukemia patients with resistance or intolerance to imatinib. Blood 2011, 118, 4567–4576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kantarjian, H.M.; Cortes, J.E.; Kim, D.-W.; Khoury, H.J.; Brummendorf, T.H.; Porkka, K.; Martinelli, G.; Durrant, S.; Leip, E.; Kelly, V.; et al. Bosutinib safety and management of toxicity in leukemia patients with resistance or intolerance to imatinib and other tyrosine kinase inhibitors. Blood 2014, 123, 1309–1318. [Google Scholar] [CrossRef] [PubMed]
- Gambacorti-Passerini, C.; Kantarjian, H.M.; Kim, D.W.; Khoury, H.J.; Turkina, A.G.; Brummendorf, T.H.; Matczak, E.; Bardy-Bouxin, N.; Shapiro, M.; Turnbull, K.; et al. Long-term efficacy and safety of bosutinib in patients with advanced leukemia following resistance/intolerance to imatinib and other tyrosine kinase inhibitors. Am. J. Hematol. 2015, 90, 755–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Gutierrez, V.; Martinez-Trillos, A.; Lopez Lorenzo, J.L.; Bautista, G.; Martin Mateos, M.L.; Alvarez-Larran, A.; Iglesias Perez, A.; Romo Collado, A.; Fernandez, A.; Portero, A.; et al. Bosutinib shows low cross intolerance, in chronic myeloid leukemia patients treated in fourth line. Results of the Spanish compassionate use program. Am. J. Hematol. 2015, 90, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Isfort, S.; Brummendorf, T.H. Bosutinib in chronic myeloid leukemia: Patient selection and perspectives. J. Blood Med. 2018, 9, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Puttini, M.; Coluccia, A.M.L.; Boschelli, F.; Cleris, L.; Marchesi, E.; Donella-Deana, A.; Ahmed, S.; Redaelli, S.; Piazza, R.; Magistroni, V.; et al. In vitro and in vivo activity of SKI-606, a novel Src-Abl inhibitor, against imatinib-resistant Bcr-Abl+ neoplastic cells. Cancer Res. 2006, 66, 11314–11322. [Google Scholar] [CrossRef] [Green Version]
- Levinson, N.M.; Boxer, S.G. Structural and spectroscopic analysis of the kinase inhibitor bosutinib and an isomer of bosutinib binding to the Abl tyrosine kinase domain. PLoS ONE 2012, 7, e29828. [Google Scholar] [CrossRef]
- Abbas, R.; Hsyu, P.H. Clinical Pharmacokinetics and Pharmacodynamics of Bosutinib. Clin. Pharm. 2016, 55, 1191–1204. [Google Scholar] [CrossRef]
- Redaelli, S.; Piazza, R.; Rostagno, R.; Magistroni, V.; Perini, P.; Marega, M.; Gambacorti-Passerini, C.; Boschelli, F. Activity of bosutinib, dasatinib, and nilotinib against 18 imatinib-resistant BCR/ABL mutants. J. Clin. Oncol. 2009, 27, 469–471. [Google Scholar] [CrossRef]
- Bradeen, H.A.; Eide, C.A.; O’Hare, T.; Johnson, K.J.; Willis, S.G.; Lee, F.Y.; Druker, B.J.; Deininger, M.W. Comparison of imatinib mesylate, dasatinib (BMS-354825), and nilotinib (AMN107) in an N-ethyl-N-nitrosourea (ENU)-based mutagenesis screen: High efficacy of drug combinations. Blood 2006, 108, 2332–2338. [Google Scholar] [CrossRef] [Green Version]
- Redaelli, S.; Mologni, L.; Rostagno, R.; Piazza, R.; Magistroni, V.; Ceccon, M.; Viltadi, M.; Flynn, D.; Gambacorti-Passerini, C. Three novel patient-derived BCR/ABL mutants show different sensitivity to second and third generation tyrosine kinase inhibitors. Am. J. Hematol. 2012, 87, 125–128. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A.; Wiestner, A. Targeting B cell receptor signalling in cancer: Preclinical and clinical advances. Nat. Rev. Cancer 2018, 18, 148–167. [Google Scholar] [CrossRef]
- Pal Singh, S.; Dammeijer, F.; Hendriks, R.W. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol. Cancer 2018, 17, 57. [Google Scholar] [CrossRef] [PubMed]
- Lam, K.P.; Kuhn, R.; Rajewsky, K. In vivo ablation of surface immunoglobulin on mature B cells by inducible gene targeting results in rapid cell death. Cell 1997, 90, 1073–1083. [Google Scholar] [CrossRef] [Green Version]
- Rolli, V.; Gallwitz, M.; Wossning, T.; Flemming, A.; Schamel, W.W.A.; Zurn, C.; Reth, M. Amplification of B cell antigen receptor signaling by a Syk/ITAM positive feedback loop. Mol. Cell 2002, 10, 1057–1069. [Google Scholar] [CrossRef]
- Inabe, K.; Ishiai, M.; Scharenberg, A.M.; Freshney, N.; Downward, J.; Kurosaki, T. Vav3 modulates B cell receptor responses by regulating phosphoinositide 3-kinase activation. J. Exp. Med. 2002, 195, 189–200. [Google Scholar] [CrossRef]
- Saito, K.; Scharenberg, A.M.; Kinet, J.P. Interaction between the Btk PH domain and phosphatidylinositol-3,4,5-trisphosphate directly regulates Btk. J. Biol. Chem. 2001, 276, 16201–16206. [Google Scholar] [CrossRef] [Green Version]
- Rawlings, D.J.; Scharenberg, A.M.; Park, H.; Wahl, M.I.; Lin, S.; Kato, R.M.; Fluckiger, A.C.; Witte, O.N.; Kinet, J.P. Activation of BTK by a phosphorylation mechanism initiated by SRC family kinases. Science 1996, 271, 822–825. [Google Scholar] [CrossRef] [PubMed]
- Saouaf, S.J.; Mahajan, S.; Rowley, R.B.; Kut, S.A.; Fargnoli, J.; Burkhardt, A.L.; Tsukada, S.; Witte, O.N.; Bolen, J.B. Temporal differences in the activation of three classes of non-transmembrane protein tyrosine kinases following B-cell antigen receptor surface engagement. Proc. Natl. Acad. Sci. USA 1994, 91, 9524–9528. [Google Scholar] [CrossRef] [Green Version]
- Yamanashi, Y.; Kakiuchi, T.; Mizuguchi, J.; Yamamoto, T.; Toyoshima, K. Association of B cell antigen receptor with protein tyrosine kinase Lyn. Science 1991, 251, 192–194. [Google Scholar] [CrossRef]
- Mahajan, S.; Fargnoli, J.; Burkhardt, A.L.; Kut, S.A.; Saouaf, S.J.; Bolen, J.B. Src family protein tyrosine kinases induce autoactivation of Bruton’s tyrosine kinase. Mol. Cell. Biol. 1995, 15, 5304–5311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.; Wahl, M.I.; Afar, D.E.; Turck, C.W.; Rawlings, D.J.; Tam, C.; Scharenberg, A.M.; Kinet, J.P.; Witte, O.N. Regulation of Btk function by a major autophosphorylation site within the SH3 domain. Immunity 1996, 4, 515–525. [Google Scholar] [CrossRef] [Green Version]
- Feng, S.; Chen, J.K.; Yu, H.; Simon, J.A.; Schreiber, S.L. Two binding orientations for peptides to the Src SH3 domain: Development of a general model for SH3-ligand interactions. Science 1994, 266, 1241–1247. [Google Scholar] [CrossRef]
- Goudreau, N.; Cornille, F.; Duchesne, M.; Parker, F.; Tocque, B.; Garbay, C.; Roques, B.P. NMR structure of the N-terminal SH3 domain of GRB2 and its complex with a proline-rich peptide from Sos. Nat. Struct. Biol. 1994, 1, 898–907. [Google Scholar] [CrossRef] [PubMed]
- Koyama, S.; Yu, H.; Dalgarno, D.C.; Shin, T.B.; Zydowsky, L.D.; Schreiber, S.L. Structure of the PI3K SH3 domain and analysis of the SH3 family. Cell 1993, 72, 945–952. [Google Scholar] [CrossRef]
- Wahl, M.I.; Fluckiger, A.C.; Kato, R.M.; Park, H.; Witte, O.N.; Rawlings, D.J. Phosphorylation of two regulatory tyrosine residues in the activation of Bruton’s tyrosine kinase via alternative receptors. Proc. Natl. Acad. Sci. USA 1997, 94, 11526–11533. [Google Scholar] [CrossRef] [Green Version]
- Tsukada, S.; Saffran, D.C.; Rawlings, D.J.; Parolini, O.; Allen, R.C.; Klisak, I.; Sparkes, R.S.; Kubagawa, H.; Mohandas, T.; Quan, S. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell 1993, 72, 279–290. [Google Scholar] [CrossRef]
- Valiaho, J.; Smith, C.I.E.; Vihinen, M. BTKbase: The mutation database for X-linked agammaglobulinemia. Hum. Mutat. 2006, 27, 1209–1217. [Google Scholar] [CrossRef]
- Singh, S.P.; Pillai, S.Y.; de Bruijn, M.J.W.; Stadhouders, R.; Corneth, O.B.J.; van den Ham, H.J.; Muggen, A.; van Ijcken, W.; Slinger, E.; Kuil, A.; et al. Cell lines generated from a chronic lymphocytic leukemia mouse model exhibit constitutive Btk and Akt signaling. Oncotarget 2017, 8, 71981–71995. [Google Scholar] [CrossRef]
- Kil, L.P.; de Bruijn, M.J.; van Hulst, J.A.; Langerak, A.W.; Yuvaraj, S.; Hendriks, R.W. Bruton’s tyrosine kinase mediated signaling enhances leukemogenesis in a mouse model for chronic lymphocytic leukemia. Am. J. Blood Res. 2013, 3, 71–83. [Google Scholar]
- Cinar, M.; Hamedani, F.; Mo, Z.; Cinar, B.; Amin, H.M.; Alkan, S. Bruton tyrosine kinase is commonly overexpressed in mantle cell lymphoma and its attenuation by Ibrutinib induces apoptosis. Leuk. Res. 2013, 37, 1271–1277. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.Y.; Francesco, M.; De Rooij, M.F.M.; Magadala, P.; Steggerda, S.M.; Huang, M.M.; Kuil, A.; Herman, S.E.M.; Chang, S.; Pals, S.T.; et al. Egress of CD19(+)CD5(+) cells into peripheral blood following treatment with the Bruton tyrosine kinase inhibitor ibrutinib in mantle cell lymphoma patients. Blood 2013, 122, 2412–2424. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Zhou, Y.; Liu, X.; Xu, L.; Cao, Y.; Manning, R.J.; Patterson, C.J.; Buhrlage, S.J.; Gray, N.; Tai, Y.T.; et al. A mutation in MYD88 (L265P) supports the survival of lymphoplasmacytic cells by activation of Bruton tyrosine kinase in Waldenstrom macroglobulinemia. Blood 2013, 122, 1222–1232. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.E.; Ngo, V.N.; Lenz, G.; Tolar, P.; Young, R.M.; Romesser, P.B.; Kohlhammer, H.; Lamy, L.; Zhao, H.; Yang, Y.; et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 2010, 463, 88–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.O. Development of BTK inhibitors for the treatment of B-cell malignancies. Arch. Pharm. Res. 2019, 42, 171–181. [Google Scholar] [CrossRef]
- Pan, Z.Y.; Scheerens, H.; Li, S.J.; Schultz, B.E.; Sprengeler, P.A.; Burrill, L.C.; Mendonca, R.V.; Sweeney, M.D.; Scott, K.C.K.; Grothaus, P.G.; et al. Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase. Chemmedchem 2007, 2, 58–61. [Google Scholar] [CrossRef]
- Honigberg, L.A.; Smith, A.M.; Sirisawad, M.; Verner, E.; Loury, D.; Chang, B.; Li, S.; Pan, Z.; Thamm, D.H.; Miller, R.A.; et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc. Natl. Acad. Sci. USA 2010, 107, 13075–13080. [Google Scholar] [CrossRef] [Green Version]
- Akinleye, A.; Avvaru, P.; Furqan, M.; Song, Y.; Liu, D. Phosphatidylinositol 3-kinase (PI3K) inhibitors as cancer therapeutics. J. Hematol. Oncol. 2013, 6, 88. [Google Scholar] [CrossRef] [Green Version]
- Parmar, S.; Patel, K.; Pinilla-Ibarz, J. Ibrutinib (imbruvica): A novel targeted therapy for chronic lymphocytic leukemia. Pharm. Ther. 2014, 39, 483–519. [Google Scholar]
- Cheng, S.; Guo, A.; Lu, P.; Ma, J.; Coleman, M.; Wang, Y.L. Functional characterization of BTK(C481S) mutation that confers ibrutinib resistance: Exploration of alternative kinase inhibitors. Leukemia 2015, 29, 895–900. [Google Scholar] [CrossRef] [PubMed]
- Woyach, J.A.; Furman, R.R.; Liu, T.M.; Ozer, H.G.; Zapatka, M.; Ruppert, A.S.; Xue, L.; Li, D.H.; Steggerda, S.M.; Versele, M.; et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N. Engl. J. Med. 2014, 370, 2286–2294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furman, R.R.; Cheng, S.; Lu, P.; Setty, M.; Perez, A.R.; Guo, A.; Racchumi, J.; Xu, G.; Wu, H.; Ma, J.; et al. Ibrutinib resistance in chronic lymphocytic leukemia. N. Engl. J. Med. 2014, 370, 2352–2354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Galanina, N.; Guo, A.; Lee, J.; Kadri, S.; Van Slambrouck, C.; Long, B.; Wang, W.; Ming, M.; Furtado, L.V.; et al. Identification of a structurally novel BTK mutation that drives ibrutinib resistance in CLL. Oncotarget 2016, 7, 68833–68841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazi, J.U.; Ronnstrand, L. The role of SRC family kinases in FLT3 signaling. Int. J. Biochem. Cell Biol. 2019, 107, 32–37. [Google Scholar] [CrossRef]
- Gilliland, D.G.; Griffin, J.D. The roles of FLT3 in hematopoiesis and leukemia. Blood 2002, 100, 1532–1542. [Google Scholar] [CrossRef] [Green Version]
- Moriki, T.; Maruyama, H.; Maruyama, I.N. Activation of preformed EGF receptor dimers by ligand-induced rotation of the transmembrane domain. J. Mol. Biol. 2001, 311, 1011–1026. [Google Scholar] [CrossRef]
- Griffith, J.; Black, J.; Faerman, C.; Swenson, L.; Wynn, M.; Lu, F.; Lippke, J.; Saxena, K. The structural basis for autoinhibition of FLT3 by the juxtamembrane domain. Mol. Cell 2004, 13, 169–178. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [Green Version]
- Kiyoi, H.; Ohno, R.; Ueda, R.; Saito, H.; Naoe, T. Mechanism of constitutive activation of FLT3 with internal tandem duplication in the juxtamembrane domain. Oncogene 2002, 21, 2555–2563. [Google Scholar] [CrossRef] [Green Version]
- Horiike, S.; Yokota, S.; Nakao, M.; Iwai, T.; Sasai, Y.; Kaneko, H.; Taniwaki, M.; Kashima, K.; Fujii, H.; Abe, T.; et al. Tandem duplications of the FLT3 receptor gene are associated with leukemic transformation of myelodysplasia. Leukemia 1997, 11, 1442–1446. [Google Scholar] [CrossRef] [Green Version]
- Iwai, T.; Yokota, S.; Nakao, M.; Okamoto, T.; Taniwaki, M.; Onodera, N.; Watanabe, A.; Kikuta, A.; Tanaka, A.; Asami, K.; et al. Internal tandem duplication of the FLT3 gene and clinical evaluation in childhood acute myeloid leukemia. The Children’s Cancer and Leukemia Study Group, Japan. Leukemia 1999, 13, 38–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiyoi, H.; Naoe, T.; Yokota, S.; Nakao, M.; Minami, S.; Kuriyama, K.; Takeshita, A.; Saito, K.; Hasegawa, S.; Shimodaira, S.; et al. Internal tandem duplication of FLT3 associated with leukocytosis in acute promyelocytic leukemia. Leukemia Study Group of the Ministry of Health and Welfare (Kohseisho). Leukemia 1997, 11, 1447–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokota, S.; Kiyoi, H.; Nakao, M.; Iwai, T.; Misawa, S.; Okuda, T.; Sonoda, Y.; Abe, T.; Kahsima, K.; Matsuo, Y.; et al. Internal tandem duplication of the FLT3 gene is preferentially seen in acute myeloid leukemia and myelodysplastic syndrome among various hematological malignancies. A study on a large series of patients and cell lines. Leukemia 1997, 11, 1605–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meshinchi, S.; Woods, W.G.; Stirewalt, D.L.; Sweetser, D.A.; Buckley, J.D.; Tjoa, T.K.; Bernstein, I.D.; Radich, J.P. Prevalence and prognostic significance of Flt3 internal tandem duplication in pediatric acute myeloid leukemia. Blood 2001, 97, 89–94. [Google Scholar] [CrossRef]
- Abu-Duhier, F.M.; Goodeve, A.C.; Wilson, G.A.; Care, R.S.; Peake, I.R.; Reilly, J.T. Identification of novel FLT-3 Asp835 mutations in adult acute myeloid leukaemia. Br. J. Haematol. 2001, 113, 983–988. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Wang, Q.; Chin, C.S.; Salerno, S.; Damon, L.E.; Levis, M.J.; Perl, A.E.; Travers, K.J.; Wang, S.; Hunt, J.P.; et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature 2012, 485, 260–263. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, H.; Hanawa, H.; Uchida, N.; Inamai, M.; Sawaguchi, K.; Mitamura, Y.; Shimada, T.; Dan, K.; Inokuchi, K. Multistep pathogenesis of leukemia via the MLL-AF4 chimeric gene/Flt3 gene tyrosine kinase domain (TKD) mutation-related enhancement of S100A6 expression. Exp. Hematol. 2009, 37, 701–714. [Google Scholar] [CrossRef]
- Chen, Y.; Pan, Y.; Guo, Y.; Zhao, W.; Ho, W.T.; Wang, J.; Xu, M.; Yang, F.C.; Zhao, Z.J. Tyrosine kinase inhibitors targeting FLT3 in the treatment of acute myeloid leukemia. Stem. Cell Investig. 2017, 4, 48. [Google Scholar] [CrossRef] [Green Version]
- Leick, M.B.; Levis, M.J. The Future of Targeting FLT3 Activation in AML. Curr. Hematol. Malig. Rep. 2017, 12, 153–167. [Google Scholar] [CrossRef]
- Lowinger, T.B.; Riedl, B.; Dumas, J.; Smith, R.A. Design and discovery of small molecules targeting raf-1 kinase. Curr. Pharm. Des. 2002, 8, 2269–2278. [Google Scholar] [CrossRef]
- Wilhelm, S.; Carter, C.; Lynch, M.; Lowinger, T.; Dumas, J.; Smith, R.A.; Schwartz, B.; Simantov, R.; Kelley, S. Discovery and development of sorafenib: A multikinase inhibitor for treating cancer. Nat. Rev. Drug Discov. 2006, 5, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Adnane, L.; Trail, P.A.; Taylor, I.; Wilhelm, S.M. Sorafenib (BAY 43-9006, Nexavar (R)), a dual-action inhibitor that targets RAF/MEK/ERK pathway in tumor cells and tyrosine kinases VEGFR/PDGFR in tumor vasculature. Methods Enzymol. 2006, 407, 597–612. [Google Scholar] [CrossRef] [PubMed]
- Auclair, D.; Miller, D.; Yatsula, V.; Pickett, W.; Carter, C.; Chang, Y.; Zhang, X.; Wilkie, D.; Burd, A.; Shi, H.; et al. Antitumor activity of sorafenib in FLT3-driven leukemic cells. Leukemia 2007, 21, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef] [Green Version]
- Carlomagno, F.; Anaganti, S.; Guida, T.; Salvatore, G.; Troncone, G.; Wilhelm, S.M.; Santoro, M. BAY 43-9006 inhibition of oncogenic RET mutants. J. Natl. Cancer Inst. 2006, 98, 326–334. [Google Scholar] [CrossRef] [Green Version]
- Hasskarl, J. Sorafenib: Targeting multiple tyrosine kinases in cancer. Recent Results Cancer Res. 2014, 201, 145–164. [Google Scholar] [CrossRef]
- Man, C.H.; Fung, T.K.; Ho, C.; Han, H.H.C.; Chow, H.C.H.; Ma, A.C.H.; Choi, W.W.L.; Lok, S.; Cheung, A.M.S.; Eaves, C.; et al. Sorafenib treatment of FLT3-ITD+ acute myeloid leukemia: Favorable initial outcome and mechanisms of subsequent nonresponsiveness associated with the emergence of a D835 mutation. Blood 2012, 119, 5133–5143. [Google Scholar] [CrossRef]
- Baker, S.D.; Zimmerman, E.I.; Wang, Y.D.; Orwick, S.; Zatechka, D.S.; Buaboonnam, J.; Neale, G.A.; Olsen, S.R.; Enemark, E.J.; Shurtleff, S.; et al. Emergence of polyclonal FLT3 tyrosine kinase domain mutations during sequential therapy with sorafenib and sunitinib in FLT3-ITD-Positive acute myeloid leukemia. Clin. Cancer Res. 2013, 19, 5758–5768. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.C.; Lin, K.; Stecula, A.; Sali, A.; Shah, N.P. FLT3 D835 mutations confer differential resistance to type II FLT3 inhibitors. Leukemia 2015, 29, 2390–2392. [Google Scholar] [CrossRef] [Green Version]
- Chao, Q.; Sprankle, K.G.; Grotzfeld, R.M.; Lai, A.G.; Carter, T.A.; Velasco, A.M.; Gunawardane, R.N.; Cramer, M.D.; Gardner, M.F.; James, J.; et al. Identification of N-(5-tert-Butyl-isoxazol-3-yl)-N ’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,1-b][1,3]benzothiazol-2-yl]phenyl}urea dihydrochloride (AC220), a uniquely potent, selective, and efficacious FMS-Like tyrosine Kinase-3 (FLT3) inhibitor. J. Med. Chem. 2009, 52, 7808–7816. [Google Scholar] [CrossRef]
- Zarrinkar, P.P.; Gunawardane, R.N.; Cramer, M.D.; Gardner, M.F.; Brigham, D.; Belli, B.; Karaman, M.W.; Pratz, K.W.; Pallares, G.; Chao, Q.; et al. AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML). Blood 2009, 114, 2984–2992. [Google Scholar] [CrossRef] [PubMed]
- Naqvi, K.; Ravandi, F. FLT3 inhibitor quizartinib (AC220). Leuk Lymphoma 2019, 60, 1866–1876. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Ge, Z.; Chen, B. Quizartinib (AC220): A promising option for acute myeloid leukemia. Drug Des. Devel Ther. 2019, 13, 1117–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zorn, J.A.; Wang, Q.; Fujimura, E.; Barros, T.; Kuriyan, J. Crystal structure of the FLT3 kinase domain bound to the inhibitor Quizartinib (AC220). PLoS ONE 2015, 10, e0121177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albers, C.; Leischner, H.; Verbeek, M.; Yu, C.; Illert, A.L.; Peschel, C.; von Bubnoff, N.; Duyster, J. The secondary FLT3-ITD F691L mutation induces resistance to AC220 in FLT3-ITD+ AML but retains in vitro sensitivity to PKC412 and Sunitinib. Leukemia 2013, 27, 1416–1418. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.S.; Faisal, A.; de Castro, D.G.; Bavetsias, V.; Sun, C.; Atrash, B.; Valenti, M.; Brandon, A.D.; Avery, S.; Mair, D.; et al. Selective FLT3 inhibition of FLT3-ITD+ acute myeloid leukaemia resulting in secondary D835Y mutation: A model for emerging clinical resistance patterns. Leukemia 2012, 26, 1462–1470. [Google Scholar] [CrossRef] [PubMed]
- Chow, L.Q.M.; Eckhardt, S.G. Sunitinib: From rational design to clinical efficacy. J. Clin. Oncol. 2007, 25, 884–896. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E.D.; Keating, G.M. Sunitinib. Drugs 2006, 66, 2255–2266. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Levesque, M.P.; Dummer, R.; Mangana, J. Targeting complex, adaptive responses in melanoma therapy. Cancer Treat. Rev. 2020, 86, 101997. [Google Scholar] [CrossRef]
- Klener, P.; Klanova, M. Drug Resistance in Non-Hodgkin Lymphomas. Int. J. Mol. Sci. 2020, 21, 2081. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Yue, C. Gastrointestinal stromal tumor. J. Gastrointest. Oncol. 2012, 3, 189–208. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, M.; Lasota, J. Gastrointestinal stromal tumors--definition, clinical, histological, immunohistochemical, and molecular genetic features and differential diagnosis. Virchows Arch. 2001, 438, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Khoshnood, A. Gastrointestinal stromal tumor—A review of clinical studies. J. Oncol. Pharm. Pract. 2019, 25, 1473–1485. [Google Scholar] [CrossRef] [PubMed]
- Sanders, K.M.; Koh, S.D.; Ward, S.M. Interstitial cells of Cajal as pacemakers in the gastrointestinal tract. Annu. Rev. Physiol. 2006, 68, 307–343. [Google Scholar] [CrossRef]
- Hirota, S.; Isozaki, K.; Moriyama, Y.; Hashimoto, K.; Nishida, T.; Ishiguro, S.; Kawano, K.; Hanada, M.; Kurata, A.; Takeda, M.; et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998, 279, 577–580. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Corless, C.L.; Duensing, A.; McGreevey, L.; Chen, C.J.; Joseph, N.; Singer, S.; Griffith, D.J.; Haley, A.; Town, A.; et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science 2003, 299, 708–710. [Google Scholar] [CrossRef]
- Ashman, L.K. The biology of stem cell factor and its receptor C-kit. Int. J. Biochem. Cell Biol. 1999, 31, 1037–1051. [Google Scholar] [CrossRef]
- Wang, H.; Boussouar, A.; Mazelin, L.; Tauszig-Delamasure, S.; Sun, Y.; Goldschneider, D.; Paradisi, A.; Mehlen, P. The Proto-oncogene c-Kit Inhibits Tumor Growth by Behaving as a Dependence Receptor. Mol. Cell 2018, 72, 413–425. [Google Scholar] [CrossRef] [Green Version]
- Abbaspour Babaei, M.; Kamalidehghan, B.; Saleem, M.; Huri, H.Z.; Ahmadipour, F. Receptor tyrosine kinase (c-Kit) inhibitors: A potential therapeutic target in cancer cells. Drug Des. Devel. Ther. 2016, 10, 2443–2459. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Wu, Y.L.; Chen, B.J.; Zhang, W.; Tanaka, Y.; Sugiyama, H. The C-kit receptor-mediated signal transduction and tumor-related diseases. Int. J. Biol. Sci. 2013, 9, 435–443. [Google Scholar] [CrossRef]
- Corless, C.L.; McGreevey, L.; Town, A.; Schroeder, A.; Bainbridge, T.; Harrell, P.; Fletcher, J.A.; Heinrich, M.C. KIT gene deletions at the intron 10-exon 11 boundary in GI stromal tumors. J. Mol. Diagn. 2004, 6, 366–370. [Google Scholar] [CrossRef]
- Corless, C.L. Assessing the prognosis of gastrointestinal stromal tumors: A growing role for molecular testing. Am. J. Clin. Pathol. 2004, 122, 11–13. [Google Scholar] [CrossRef] [PubMed]
- Isozaki, K.; Terris, B.; Belghiti, J.; Schiffmann, S.; Hirota, S.; Vanderwinden, J.M. Germline-activating mutation in the kinase domain of KIT gene in familial gastrointestinal stromal tumors. Am. J. Pathol. 2000, 157, 1581–1585. [Google Scholar] [CrossRef] [Green Version]
- Yarden, Y.; Kuang, W.J.; Yang-Feng, T.; Coussens, L.; Munemitsu, S.; Dull, T.J.; Chen, E.; Schlessinger, J.; Francke, U.; Ullrich, A. Human proto-oncogene c-kit: A new cell surface receptor tyrosine kinase for an unidentified ligand. EMBO J. 1987, 6, 3341–3351. [Google Scholar] [CrossRef]
- Schwaab, J.; Schnittger, S.; Sotlar, K.; Walz, C.; Fabarius, A.; Pfirrmann, M.; Kohlmann, A.; Grossmann, V.; Meggendorfer, M.; Horny, H.P.; et al. Comprehensive mutational profiling in advanced systemic mastocytosis. Blood 2013, 122, 2460–2466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinrich, M.C.; Griffith, D.J.; Druker, B.J.; Wait, C.L.; Ott, K.A.; Zigler, A.J. Inhibition of c-kit receptor tyrosine kinase activity by STI 571, a selective tyrosine kinase inhibitor. Blood 2000, 96, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Craig, J.W.; Hasserjian, R.P.; Kim, A.S.; Aster, J.C.; Pinkus, G.S.; Hornick, J.L.; Steensma, D.P.; Coleman Lindsley, R.; DeAngelo, D.J.; Morgan, E.A. Detection of the KIT(D816V) mutation in myelodysplastic and/or myeloproliferative neoplasms and acute myeloid leukemia with myelodysplasia-related changes predicts concurrent systemic mastocytosis. Mod. Pathol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, T.; Preiss, B.; Broesby-Olsen, S.; Vestergaard, H.; Friis, L.; Moller, M.B. Systemic mastocytosis is uncommon in KIT D816V mutation positive core-binding factor acute myeloid leukemia. Leukemia Lymphoma 2012, 53, 1338–1344. [Google Scholar] [CrossRef] [PubMed]
- Niinuma, T.; Suzuki, H.; Sugai, T. Molecular characterization and pathogenesis of gastrointestinal stromal tumor. Transl. Gastroenterol. Hepatol. 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Wozniak, A.; Rutkowski, P.; Schoffski, P.; Ray-Coquard, I.; Hostein, I.; Schildhaus, H.U.; Le Cesne, A.; Bylina, E.; Limon, J.; Blay, J.Y.; et al. Tumor genotype is an independent prognostic factor in primary gastrointestinal stromal tumors of gastric origin: A european multicenter analysis based on conticaGIST. Clin. Cancer Res. 2014, 20, 6105–6116. [Google Scholar] [CrossRef] [Green Version]
- Croom, K.F.; Perry, C.M. Imatinib mesylate: In the treatment of gastrointestinal stromal tumours. Drugs 2003, 63, 513–522. [Google Scholar] [CrossRef]
- Cools, J.; DeAngelo, D.J.; Gotlib, J.; Stover, E.H.; Legare, R.D.; Cortes, J.; Kutok, J.; Clark, J.; Galinsky, I.; Griffin, J.D.; et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N. Engl. J. Med. 2003, 348, 1201–1214. [Google Scholar] [CrossRef] [PubMed]
- Rajasekaran, R.; Sethumadhavan, R. Exploring the cause of drug resistance by the detrimental missense mutations in KIT receptor: Computational approach. Amino Acids 2010, 39, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Corless, C.L.; Schroeder, A.; Griffith, D.; Town, A.; McGreevey, L.; Harrell, P.; Shiraga, S.; Bainbridge, T.; Morich, J.; Heinrich, M.C. PDGFRA mutations in gastrointestinal stromal tumors: Frequency, spectrum and in vitro sensitivity to imatinib. J. Clin. Oncol. 2005, 23, 5357–5364. [Google Scholar] [CrossRef]
- Liang, L.; Yan, X.E.; Yin, Y.; Yun, C.H. Structural and biochemical studies of the PDGFRA kinase domain. Biochem. Bioph. Res. Commun. 2016, 477, 667–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, J.; Kim, S.Y.; Kim, Y.J.; Sim, M.H.; Kim, S.T.; Kim, N.K.D.; Kim, K.; Park, W.; Kim, J.H.; Jang, K.T.; et al. Emergence of CTNNB1 mutation at acquired resistance to KIT inhibitor in metastatic melanoma. Clin. Transl. Oncol. 2017, 19, 1247–1252. [Google Scholar] [CrossRef]
- Noujaim, J.; Gonzalez, D.; Thway, K.; Jones, R.L.; Judson, I. p. (L576P) -KIT mutation in GIST: Favorable prognosis and sensitive to imatinib? Cancer Biol. Ther. 2016, 17, 543–545. [Google Scholar] [CrossRef] [Green Version]
- Conca, E.; Negri, T.; Gronchi, A.; Fumagalli, E.; Tamborini, E.; Pavan, G.M.; Fermeglia, M.; Pierotti, M.A.; Pricl, S.; Pilotti, S. Activate and resist: L576P-KIT in GIST. Mol. Cancer Ther. 2009, 8, 2491–2495. [Google Scholar] [CrossRef] [Green Version]
- Pai, S.G.; Carneiro, B.A.; Mota, J.M.; Costa, R.; Leite, C.A.; Barroso-Sousa, R.; Kaplan, J.B.; Chae, Y.K.; Giles, F.J. Wnt/beta-catenin pathway: Modulating anticancer immune response. J. Hematol. Oncol. 2017, 10, 101. [Google Scholar] [CrossRef] [Green Version]
- Chiarini, F.; Paganelli, F.; Martelli, A.M.; Evangelisti, C. The role played by wnt/beta-catenin signaling pathway in acute lymphoblastic leukemia. Int. J. Mol. Sci. 2020, 21, 1098. [Google Scholar] [CrossRef] [Green Version]
- Provost, E.; Yamamoto, Y.; Lizardi, I.; Stern, J.; D’Aquila, T.G.; Gaynor, R.B.; Rimm, D.L. Functional correlates of mutations in beta-catenin exon 3 phosphorylation sites. J. Biol. Chem. 2003, 278, 31781–31789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, D.; Shibahara, J.; Sakuma, T.; Isobe, M.; Teshima, S.; Mori, M.; Oda, K.; Nakagawa, S.; Taketani, Y.; Ishikawa, S.; et al. Beta-catenin (CTNNB1) S33C mutation in ovarian microcystic stromal tumors. American J. Surg. Pathol. 2011, 35, 1429–1440. [Google Scholar] [CrossRef] [PubMed]
- Dziadziuszko, R.; Le, A.T.; Wrona, A.; Jassem, J.; Camidge, D.R.; Varella-Garcia, M.; Aisner, D.L.; Doebele, R.C. An activating KIT mutation induces crizotinib resistance in ROS1-Positive lung cancer. J. Thorac. Oncol. 2016, 11, 1273–1281. [Google Scholar] [CrossRef] [Green Version]
- Birchmeier, C.; Sharma, S.; Wigler, M. Expression and rearrangement of the Ros1 gene in human glioblastoma cells. Proc. Natl. Acad. Sci. USA 1987, 84, 9270–9274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbour, K.C.; Riely, G.J. Systemic therapy for locally advanced and metastatic non-small cell lung cancer a review. J. Am. Med. Assoc. 2019, 322, 764–774. [Google Scholar] [CrossRef]
- Asao, T.; Takahashi, F.; Takahashi, K. Resistance to molecularly targeted therapy in non-small-cell lung cancer. Respir. Investig. 2019, 57, 20–26. [Google Scholar] [CrossRef]
- Skoulidis, F.; Heymach, J.V. Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat. Rev. Cancer 2019, 19, 495–509. [Google Scholar] [CrossRef]
- Arai, Y.; Totoki, Y.; Takahashi, H.; Nakamura, H.; Hama, N.; Kohno, T.; Tsuta, K.; Yoshida, A.; Asamura, H.; Mutoh, M.; et al. Mouse model for ros1-rearranged lung cancer. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Bergethon, K.; Shaw, A.T.; Ou, S.H.; Katayama, R.; Lovly, C.M.; McDonald, N.T.; Massion, P.P.; Siwak-Tapp, C.; Gonzalez, A.; Fang, R.; et al. ROS1 rearrangements define a unique molecular class of lung cancers. J. Clin. Oncol. 2012, 30, 863–870. [Google Scholar] [CrossRef] [Green Version]
- Davies, K.D.; Le, A.T.; Theodoro, M.F.; Skokan, M.C.; Aisner, D.L.; Berge, E.M.; Terracciano, L.M.; Cappuzzo, F.; Incarbone, M.; Roncalli, M.; et al. Identifying and targeting ROS1 gene fusions in non-small cell lung cancer. Clin. Cancer Res. 2012, 18, 4570–4579. [Google Scholar] [CrossRef] [Green Version]
- Shaw, A.T.; Ou, S.H.I.; Bang, Y.J.; Camidge, D.R.; Solomon, B.J.; Salgia, R.; Riely, G.J.; Varella-Garcia, M.; Shapiro, G.I.; Costa, D.B.; et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N. Engl. J. Med. 2014, 371, 1963–1971. [Google Scholar] [CrossRef] [Green Version]
- Shaw, A.T.; Kim, D.W.; Mehra, R.; Tan, D.S.W.; Felip, E.; Chow, L.Q.M.; Camidge, D.R.; Vansteenkiste, J.; Sharma, S.; De Pas, T.; et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. N. Engl. J. Med. 2014, 370, 1189–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazieres, J.; Zalcman, G.; Crino, L.; Biondani, P.; Barlesi, F.; Filleron, T.; Dingemans, A.M.C.; Lena, H.; Monnet, I.; Rothschild, S.I.; et al. Crizotinib therapy for advanced lung adenocarcinoma and a ROS1 rearrangement: Results from the EUROS1 cohort. J. Clin. Oncol. 2015, 33, 992–999. [Google Scholar] [CrossRef]
- Mort, R.L.; Jackson, I.J.; Patton, E.E. The melanocyte lineage in development and disease. Development 2015, 142, 1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastian, B.C. The molecular pathology of melanoma: An integrated taxonomy of melanocytic neoplasia. Annu. Rev. Pathol. 2014, 9, 239–271. [Google Scholar] [CrossRef] [Green Version]
- Shain, A.H.; Bastian, B.C. From melanocytes to melanomas. Nat. Rev. Cancer 2016, 16, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Haugh, A.M.; Johnson, D.B. Management of V600E and V600K BRAF-Mutant Melanoma. Curr. Treat. Options Oncol. 2019, 20, 81. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, C.G.; Andelkovic, V.; Ladwa, R.; Pavlakis, N.; Zhou, C.; Hirsch, F.; Richard, D.; O’Byrne, K. Targeting BRAF mutations in non-small cell lung cancer. Transl. Lung Cancer Res. 2019, 8, 1119–1124. [Google Scholar] [CrossRef]
- Curtin, J.A.; Fridlyand, J.; Kageshita, T.; Patel, H.N.; Busam, K.J.; Kutzner, H.; Cho, K.H.; Aiba, S.; Brocker, E.B.; LeBoit, P.E.; et al. Distinct sets of genetic alterations in melanoma. N. Engl. J. Med. 2005, 353, 2135–2147. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Cantwell-Dorris, E.R.; O’Leary, J.J.; Sheils, O.M. BRAFV600E: Implications for carcinogenesis and molecular therapy. Mol. Cancer Ther. 2011, 10, 385–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lassam, N.; Bickford, S. Loss of c-kit expression in cultured melanoma cells. Oncogene 1992, 7, 51–56. [Google Scholar] [PubMed]
- Shain, A.H.; Yeh, I.; Kovalyshyn, I.; Sriharan, A.; Talevich, E.; Gagnon, A.; Dummer, R.; North, J.; Pincus, L.; Ruben, B.; et al. The genetic evolution of melanoma from precursor lesions. N. Engl. J. Med. 2015, 373, 1926–1936. [Google Scholar] [CrossRef] [PubMed]
- Philpott, C.; Tovell, H.; Frayling, I.M.; Cooper, D.N.; Upadhyaya, M. The NF1 somatic mutational landscape in sporadic human cancers. Hum. Genom. 2017, 11, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maertens, O.; Johnson, B.; Hollstein, P.; Frederick, D.T.; Cooper, Z.A.; Messiaen, L.; Bronson, R.T.; McMahon, M.; Granter, S.; Flaherty, K.; et al. Elucidating distinct roles for NF1 in melanomagenesis. Cancer Discov. 2013, 3, 338–349. [Google Scholar] [CrossRef] [Green Version]
- Franken, M.G.; Leeneman, B.; Gheorghe, M.; Uyl-de Groot, C.A.; Haanen, J.; van Baal, P.H.M. A systematic literature review and network meta-analysis of effectiveness and safety outcomes in advanced melanoma. Eur. J. Cancer 2019, 123, 58–71. [Google Scholar] [CrossRef]
- Tsai, J.; Lee, J.T.; Wang, W.; Zhang, J.; Cho, H.; Mamo, S.; Bremer, R.; Gillette, S.; Kong, J.; Haass, N.K.; et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc. Natl. Acad. Sci. USA 2008, 105, 3041–3046. [Google Scholar] [CrossRef] [Green Version]
- Bollag, G.; Hirth, P.; Tsai, J.; Zhang, J.; Ibrahim, P.N.; Cho, H.; Spevak, W.; Zhang, C.; Zhang, Y.; Habets, G.; et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 2010, 467, 596–599. [Google Scholar] [CrossRef]
- Li, H.F.; Chen, Y.; Rao, S.S.; Chen, X.M.; Liu, H.C.; Qin, J.H.; Tang, W.F.; Yue, W.; Zhou, X.; Lu, T. Recent advances in the research and development of B-Raf inhibitors. Curr. Med. Chem. 2010, 17, 1618–1634. [Google Scholar] [CrossRef]
- Smith, R.A.; Dumas, J.; Adnane, L.; Wilhelm, S.M. Recent advances in the research and development of RAF kinase inhibitors. Curr. Top. Med. Chem. 2006, 6, 1071–1089. [Google Scholar] [CrossRef]
- Yang, H.; Higgins, B.; Kolinsky, K.; Packman, K.; Go, Z.; Iyer, R.; Kolis, S.; Zhao, S.; Lee, R.; Grippo, J.F.; et al. RG7204 (PLX4032), a selective BRAFV600E inhibitor, displays potent antitumor activity in preclinical melanoma models. Cancer Res. 2010, 70, 5518–5527. [Google Scholar] [CrossRef] [Green Version]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef] [Green Version]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyman, D.M.; Puzanov, I.; Subbiah, V.; Faris, J.E.; Chau, I.; Blay, J.Y.; Wolf, J.; Raje, N.S.; Diamond, E.L.; Hollebecque, A.; et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N. Engl. J. Med. 2015, 373, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Mazieres, J.; Cropet, C.; Montane, L.; Barlesi, F.; Souquet, P.J.; Quantin, X.; Dubos-Arvis, C.; Otto, J.; Favier, L.; Avrillon, V.; et al. Vemurafenib in non-small-cell lung cancer patients with BRAF(V600) and BRAF(nonV600) mutations. Ann. Oncol. 2020, 31, 289–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subbiah, V.; Puzanov, I.; Blay, J.Y.; Chau, I.; Lockhart, A.C.; Raje, N.S.; Wolf, J.; Baselga, J.; Meric-Bernstam, F.; Roszik, J.; et al. Pan-cancer efficacy of vemurafenib in BRAFV600-mutant non-melanoma cancers. Cancer Discov. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roskoski, R., Jr. RAF protein-serine/threonine kinases: Structure and regulation. Biochem. Biophys. Res. Commun. 2010, 399, 313–317. [Google Scholar] [CrossRef]
- Luebker, S.A.; Koepsell, S.A. Diverse mechanisms of BRAF inhibitor resistance in melanoma identified in clinical and preclinical studies. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Wagle, N.; Emery, C.; Berger, M.F.; Davis, M.J.; Sawyer, A.; Pochanard, P.; Kehoe, S.M.; Johannessen, C.M.; Macconaill, L.E.; Hahn, W.C.; et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J. Clin. Oncol. 2011, 29, 3085–3096. [Google Scholar] [CrossRef] [Green Version]
- Van Allen, E.M.; Wagle, N.; Sucker, A.; Treacy, D.J.; Johannessen, C.M.; Goetz, E.M.; Place, C.S.; Taylor-Weiner, A.; Whittaker, S.; Kryukov, G.V.; et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014, 4, 94–109. [Google Scholar] [CrossRef] [Green Version]
- Rizos, H.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Fung, C.; Hyman, J.; Haydu, L.E.; Mijatov, B.; Becker, T.M.; Boyd, S.C.; et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: Spectrum and clinical impact. Clin. Cancer Res. 2014, 20, 1965–1977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwong, L.N.; Boland, G.M.; Frederick, D.T.; Helms, T.L.; Akid, A.T.; Miller, J.P.; Jiang, S.; Cooper, Z.A.; Song, X.; Seth, S.; et al. Co-clinical assessment identifies patterns of BRAF inhibitor resistance in melanoma. J. Clin. Investig. 2015, 125, 1459–1470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.Y.; Menzies, A.M.; Rizos, H. Mechanisms and strategies to overcome resistance to molecularly targeted therapy for melanoma. Cancer 2017, 123, 2118–2129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.D.; Lee, N.H. Aberrant RNA splicing in cancer and drug resistance. Cancers 2018, 10, 458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulikakos, P.I.; Persaud, Y.; Janakiraman, M.; Kong, X.; Ng, C.; Moriceau, G.; Shi, H.; Atefi, M.; Titz, B.; Gabay, M.T.; et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 2011, 480, 387–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.; Landrette, S.F.; Wang, T.; Evans, P.; Bacchiocchi, A.; Bjornson, R.; Cheng, E.; Stiegler, A.L.; Gathiaka, S.; Acevedo, O.; et al. Identification of PLX4032-resistance mechanisms and implications for novel RAF inhibitors. Pigment Cell Melanoma Res. 2014, 27, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Hoogstraat, M.; Gadellaa-van Hooijdonk, C.G.; Ubink, I.; Besselink, N.J.; Pieterse, M.; Veldhuis, W.; van Stralen, M.; Meijer, E.F.; Willems, S.M.; Hadders, M.A.; et al. Detailed imaging and genetic analysis reveal a secondary BRAF(L505H) resistance mutation and extensive intrapatient heterogeneity in metastatic BRAF mutant melanoma patients treated with vemurafenib. Pigment Cell Melanoma Res. 2015, 28, 318–323. [Google Scholar] [CrossRef]
- Romano, E.; Pradervand, S.; Paillusson, A.; Weber, J.; Harshman, K.; Muehlethaler, K.; Speiser, D.; Peters, S.; Rimoldi, D.; Michielin, O. Identification of multiple mechanisms of resistance to vemurafenib in a patient with BRAFV600E-mutated cutaneous melanoma successfully rechallenged after progression. Clin. Cancer Res. 2013, 19, 5749–5757. [Google Scholar] [CrossRef] [Green Version]
- Mao, M.; Tian, F.; Mariadason, J.M.; Tsao, C.C.; Lemos, R., Jr.; Dayyani, F.; Gopal, Y.N.; Jiang, Z.Q.; Wistuba, II.; Tang, X.M.; et al. Resistance to BRAF inhibition in BRAF-mutant colon cancer can be overcome with PI3K inhibition or demethylating agents. Clin. Cancer Res. 2013, 19, 657–667. [Google Scholar] [CrossRef] [Green Version]
- Menzies, A.M.; Long, G.V.; Murali, R. Dabrafenib and its potential for the treatment of metastatic melanoma. Drug Des. Devel. Ther. 2012, 6, 391–405. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Targeting oncogenic Raf protein-serine/threonine kinases in human cancers. Pharmacol. Res. 2018, 135, 239–258. [Google Scholar] [CrossRef] [PubMed]
- Falchook, G.S.; Long, G.V.; Kurzrock, R.; Kim, K.B.; Arkenau, T.H.; Brown, M.P.; Hamid, O.; Infante, J.R.; Millward, M.; Pavlick, A.C.; et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: A phase 1 dose-escalation trial. Lancet 2012, 379, 1893–1901. [Google Scholar] [CrossRef] [Green Version]
- Rheault, T.R.; Stellwagen, J.C.; Adjabeng, G.M.; Hornberger, K.R.; Petrov, K.G.; Waterson, A.G.; Dickerson, S.H.; Mook, R.A.; Laquerre, S.G.; King, A.J.; et al. Discovery of dabrafenib: a selective inhibitor of raf kinases with antitumor activity against B-Raf-driven tumors. Acs Med. Chem. Lett. 2013, 4, 358–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibney, G.T.; Zager, J.S. Clinical development of dabrafenib in BRAF mutant melanoma and other malignancies. Expert Opin. Drug Metab. Toxicol. 2013, 9, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Laquerre, S.; Amone, M.; Moss, K.; Yang, J.; Fisher, K.; Kane-Carson, L.S.; Smitheman, K.; Ward, J.; Heidrich, B.; Rheault, T.; et al. A selective Raf kinase inhibitor induces cell death and tumor regression of human cancer cell lines encoding B-Raf(V600E) mutation. Mol. Cancer Ther. 2009, 8. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Targeting ERK1/2 protein-serine/threonine kinases in human cancers. Pharmacol. Res. 2019, 142, 151–168. [Google Scholar] [CrossRef]
- Carlino, M.S.; Gowrishankar, K.; Saunders, C.A.B.; Pupo, G.M.; Snoyman, S.; Zhang, X.D.; Saw, R.; Becker, T.M.; Kefford, R.F.; Long, G.V.; et al. Antiproliferative effects of continued mitogen-activated protein kinase pathway inhibition following acquired resistance to BRAF and/or MEK inhibition in melanoma. Mol. Cancer Ther. 2013, 12, 1332–1342. [Google Scholar] [CrossRef] [Green Version]
- Kakadia, S.; Yarlagadda, N.; Awad, R.; Kundranda, M.; Niu, J.; Naraev, B.; Mina, L.; Dragovich, T.; Gimbel, M.; Mahmoud, F. Mechanisms of resistance to BRAF and MEK inhibitors and clinical update of US Food and Drug Administration-approved targeted therapy in advanced melanoma. Oncotargets Ther. 2018, 11, 7095–7107. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Fung, C.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Hyman, J.; Shahheydari, H.; Tembe, V.; Thompson, J.F.; Saw, R.P.; et al. Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therapy of BRAF-mutant metastatic melanoma. Nat. Commun. 2014, 5, 5694. [Google Scholar] [CrossRef] [Green Version]
- Lian, T.; Li, C.; Wang, H. Trametinib in the treatment of multiple malignancies harboring MEK1 mutations. Cancer Treat. Rev. 2019, 81, 101907. [Google Scholar] [CrossRef] [PubMed]
- Cocco, E.; Schram, A.M.; Kulick, A.; Misale, S.; Won, H.H.; Yaeger, R.; Razavi, P.; Ptashkin, R.; Hechtman, J.F.; Toska, E.; et al. Resistance to TRK inhibition mediated by convergent MAPK pathway activation. Nat. Med. 2019, 25, 1422–1427. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Rauch, J.; Kolch, W. Targeting MAPK signaling in cancer: Mechanisms of drug resistance and sensitivity. Int. J. Mol. Sci. 2020, 21, 1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, T.C.; Marsh, V.; Bernat, B.A.; Ballard, J.; Colwell, H.; Evans, R.J.; Parry, J.; Smith, D.; Brandhuber, B.J.; Gross, S.; et al. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin. Cancer Res. 2007, 13, 1576–1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, B.R.; Logie, A.; McKay, J.S.; Martin, P.; Steele, S.; Jenkins, R.; Cockerill, M.; Cartlidge, S.; Smith, P.D. AZD6244 (ARRY-142886), a potent inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1/2 kinases: Mechanism of action in vivo, pharmacokinetic/pharmacodynamic relationship, and potential for combination in preclinical models. Mol. Cancer Ther. 2007, 6, 2209–2219. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.W.; Patel, S.P. Profile of selumetinib and its potential in the treatment of melanoma. OncoTargets Ther. 2014, 7, 1631–1639. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Ding, W.; Zhang, L.; Tian, W.; Chen, S. Clinical response to sunitinib as a multitargeted tyrosine-kinase inhibitor (TKI) in solid cancers: A review of clinical trials. OncoTargets Ther. 2014, 7, 719–728. [Google Scholar] [CrossRef] [Green Version]
- Casaluce, F.; Sgambato, A.; Maione, P.; Sacco, P.C.; Santabarbara, G.; Gridelli, C. Selumetinib for the treatment of non-small cell lung cancer. Expert Opin. Investig. Drugs 2017, 26, 973–984. [Google Scholar] [CrossRef]
- Bernabe, R.; Patrao, A.; Carter, L.; Blackhall, F.; Dean, E. Selumetinib in the treatment of non-small-cell lung cancer. Future Oncol. 2016, 12, 2545–2560. [Google Scholar] [CrossRef]
- Engelman, J.A.; Chen, L.; Tan, X.H.; Crosby, K.; Guimaraes, A.R.; Upadhyay, R.; Maira, M.; McNamara, K.; Perera, S.A.; Song, Y.C.; et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat. Med. 2008, 14, 1351–1356. [Google Scholar] [CrossRef] [Green Version]
- Dreaden, E.C.; Kong, Y.W.; Morton, S.W.; Correa, S.; Choi, K.Y.; Shopsowitz, K.E.; Renggli, K.; Drapkin, R.; Yaffe, M.B.; Hammond, P.T. Tumor-targeted synergistic blockade of MAPK and PI3K from a layer-by-layer nanoparticle. Clin. Cancer Res. 2015, 21, 4410–4419. [Google Scholar] [CrossRef] [Green Version]
- Atefi, M.; von Euw, E.; Attar, N.; Ng, C.; Chu, C.; Guo, D.L.; Nazarian, R.; Chmielowski, B.; Glaspy, J.A.; Comin-Anduix, B.; et al. Reversing melanoma cross-resistance to BRAF and MEK inhibitors by Co-targeting the AKT/mTOR pathway. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Emery, C.M.; Vijayendran, K.G.; Zipser, M.C.; Sawyer, A.M.; Niu, L.; Kim, J.J.; Hatton, C.; Chopra, R.; Oberholzer, P.A.; Karpova, M.B.; et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc. Natl. Acad. Sci. USA 2009, 106, 20411–20416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poole, R.M. Pembrolizumab: First global approval. Drugs 2014, 74, 1973–1981. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.P.; Gerriets, V. Pembrolizumab. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamalis, A.; Garcha, M.; Jagdeo, J. Targeting the PD-1 pathway: A promising future for the treatment of melanoma. Arch. Dermatol. Res. 2014, 306, 511–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, P.A.; Heng, D.Y.C. Programmed Death 1 Pathway inhibition in Metastatic Renal Cell Cancer and Prostate Cancer. Curr. Oncol. Rep. 2013, 15, 98–104. [Google Scholar] [CrossRef]
- Sahni, S.; Valecha, G.; Sahni, A. Role of Anti-PD-1 Antibodies in Advanced Melanoma: The Era of Immunotherapy. Cureus 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol. 2018, 62, 29–39. [Google Scholar] [CrossRef]
- Simsek, M.; Tekin, S.B.; Bilici, M. Immunological Agents Used in Cancer Treatment. Eurasian J. Med. 2019, 51, 90–94. [Google Scholar] [CrossRef]
- Hodi, F.S. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 1290-1290. [Google Scholar] [CrossRef] [PubMed]
- Peggs, K.S.; Quezada, S.A.; Chambers, C.A.; Korman, A.J.; Allison, J.P. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J. Exp. Med. 2009, 206, 1717–1725. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.C.; Levine, J.H.; Cogdill, A.P.; Zhao, Y.; Anang, N.A.A.S.; Andrews, M.C.; Sharma, P.; Wang, J.; Wargo, J.A.; Pe’er, D.; et al. Distinct cellular mechanisms underlie Anti-CTLA-4 and Anti-PD-1 checkpoint blockade. Cell 2017, 170, 1120–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Bach, E.A.; Aguet, M.; Schreiber, R.D. The IFN gamma receptor: A paradigm for cytokine receptor signaling. Annu. Rev. Immunol. 1997, 15, 563–591. [Google Scholar] [CrossRef]
- Muller, M.; Briscoe, J.; Laxton, C.; Guschin, D.; Ziemiecki, A.; Silvennoinen, O.; Harpur, A.G.; Barbieri, G.; Witthuhn, B.A.; Schindler, C.; et al. The protein-tyrosine kinase Jak1 complements defects in interferon-alpha/beta and interferon-gamma signal-transduction. Nature 1993, 366, 129–135. [Google Scholar] [CrossRef]
- Zhuang, Y.; Liu, C.; Liu, J.; Li, G. Resistance mechanism of PD-1/PD-L1 blockade in the cancer-immunity cycle. Onco. Targets Ther. 2020, 13, 83–94. [Google Scholar] [CrossRef] [Green Version]
- Robarge, K.D.; Brunton, S.A.; Castanedo, G.M.; Cui, Y.; Dina, M.S.; Goldsmith, R.; Gould, S.E.; Guichert, O.; Gunzner, J.L.; Halladay, J.; et al. GDC-0449-A potent inhibitor of the hedgehog pathway. Bioorganic Med. Chem. Lett. 2009, 19, 5576–5581. [Google Scholar] [CrossRef]
- Huang, P.X.; Zheng, S.D.; Wierbowski, B.M.; Kim, Y.; Nedelcu, D.; Aravena, L.; Liu, J.; Kruse, A.C.; Salic, A. Structural basis of smoothened activation in hedgehog signaling. Cell 2018, 174, 312–324. [Google Scholar] [CrossRef] [Green Version]
- Alcedo, J.; Ayzenzon, M.; VonOhlen, T.; Noll, M.; Hooper, J.E. The Drosophila smoothened gene encodes a seven-pass membrane protein, a putative receptor for the hedgehog signal. Cell 1996, 86, 221–232. [Google Scholar] [CrossRef] [Green Version]
- Van den Heuvel, M.; Ingham, P.W. Smoothened encodes a receptor-like serpentine protein required for hedgehog signalling. Nature 1996, 382, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Carballo, G.B.; Honorato, J.R.; de Lopes, G.P.F.; Spohr, T. A highlight on Sonic hedgehog pathway. Cell Commun. Signal. 2018, 16, 11. [Google Scholar] [CrossRef] [PubMed]
- Skoda, A.M.; Simovic, D.; Karin, V.; Kardum, V.; Vranic, S.; Serman, L. The role of the Hedgehog signaling pathway in cancer: A comprehensive review. Bosn. J. Basic Med. Sci. 2018, 18, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Yauch, R.L.; Gould, S.E.; Scales, S.J.; Tang, T.; Tian, H.; Ahn, C.P.; Marshall, D.; Fu, L.; Januario, T.; Kallop, D.; et al. A paracrine requirement for hedgehog signalling in cancer. Nature 2008, 455, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Fattahi, S.; Langroudi, M.P.; Akhavan-Niaki, H. Hedgehog signaling pathway: Epigenetic regulation and role in disease and cancer development. J. Cell. Physiol. 2018, 233, 5726–5735. [Google Scholar] [CrossRef] [PubMed]
- Kinzler, K.W.; Ruppert, J.M.; Bigner, S.H.; Vogelstein, B. The GLI gene is a member of the Kruppel family of zinc finger proteins. Nature 1988, 332, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001, 15, 3059–3087. [Google Scholar] [CrossRef] [Green Version]
- Rubin, L.L.; de Sauvage, F.J. Targeting the Hedgehog pathway in cancer. Nat. Rev. Drug Discov. 2006, 5, 1026–1033. [Google Scholar] [CrossRef]
- Salaritabar, A.; Berindan-Neagoe, I.; Darvish, B.; Hadjiakhoondi, F.; Manayi, A.; Devi, K.P.; Barreca, D.; Orhan, I.E.; Suntar, I.; Farooqi, A.A.; et al. Targeting Hedgehog signaling pathway: Paving the road for cancer therapy. Pharmacol. Res. 2019, 141, 466–480. [Google Scholar] [CrossRef]
- Girardi, D.; Barrichello, A.; Fernandes, G.; Pereira, A. Targeting the hedgehog pathway in cancer: current evidence and future perspectives. Cells 2019, 8, 153. [Google Scholar] [CrossRef] [Green Version]
- Chahal, K.K.; Parle, M.; Abagyan, R. Hedgehog pathway and smoothened inhibitors in cancer therapies. Anti Cancer Drug 2018, 29, 387–401. [Google Scholar] [CrossRef]
- Lorusso, P.M.; Jimeno, A.; Dy, G.; Adjei, A.; Berlin, J.; Leichman, L.; Low, J.A.; Colburn, D.; Chang, I.; Cheeti, S.; et al. Pharmacokinetic dose-scheduling study of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with locally advanced or metastatic solid tumors. Clin. Cancer Res. 2011, 17, 5774–5782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keating, G.M. Vismodegib: In locally advanced or metastatic basal cell carcinoma. Drugs 2012, 72, 1535–1541. [Google Scholar] [CrossRef] [PubMed]
- Frampton, J.E.; Basset-Seguin, N. Vismodegib: A review in advanced basal cell carcinoma. Drugs 2018, 78, 1145–1156. [Google Scholar] [CrossRef] [PubMed]
- Fecher, L.A.; Sharfman, W.H. Advanced basal cell carcinoma, the hedgehog pathway, and treatment options—Role of smoothened inhibitors. Biologics 2015, 9, 129–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekulic, A.; Migden, M.R.; Oro, A.E.; Dirix, L.; Lewis, K.D.; Hainsworth, J.D.; Solomon, J.A.; Yoo, S.; Arron, S.T.; Friedlander, P.A.; et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N. Engl. J. Med. 2012, 366, 2171–2179. [Google Scholar] [CrossRef] [Green Version]
- Sharpe, H.J.; Pau, G.; Dijkgraaf, G.J.; Basset-Seguin, N.; Modrusan, Z.; Januario, T.; Tsui, V.; Durham, A.B.; Dlugosz, A.A.; Haverty, P.M.; et al. Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma. Cancer Cell 2015, 27, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Atwood, S.X.; Sarin, K.Y.; Whitson, R.J.; Li, J.R.; Kim, G.; Rezaee, M.; Ally, M.S.; Kim, J.; Yao, C.; Chang, A.L.; et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell 2015, 27, 342–353. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Wu, H.; Katritch, V.; Han, G.W.; Huang, X.P.; Liu, W.; Siu, F.Y.; Roth, B.L.; Cherezov, V.; Stevens, R.C. Structure of the human smoothened receptor bound to an antitumour agent. Nature 2013, 497, 338–343. [Google Scholar] [CrossRef] [Green Version]
- Pusztai, L.; Mazouni, C.; Anderson, K.; Wu, Y.; Symmans, W.F. Molecular classification of breast cancer: Limitations and potential. Oncologist 2006, 11, 868–877. [Google Scholar] [CrossRef]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, M.; Dahlman-Wright, K.; Gustafsson, J.A. Estrogen receptor alpha and beta in health and disease. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Gourdy, P.; Guillaume, M.; Fontaine, C.; Adlanmerini, M.; Montagner, A.; Laurell, H.; Lenfant, F.; Arnal, J.F. Estrogen receptor subcellular localization and cardiometabolism. Mol. Metab. 2018, 15, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Livezey, M.; Kim, J.E.; Shapiro, D.J. A new role for estrogen receptor alpha in cell proliferation and cancer: Activating the anticipatory unfolded protein response. Front. Endocrinol. 2018, 9, 325. [Google Scholar] [CrossRef] [Green Version]
- Klinge, C.M. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 2001, 29, 2905–2919. [Google Scholar] [CrossRef] [Green Version]
- Ayaz, G.; Yasar, P.; Olgun, C.E.; Karakaya, B.; Kars, G.; Razizadeh, N.; Yavuz, K.; Turan, G.; Muyan, M. Dynamic transcriptional events mediated by estrogen receptor alpha. Front. Biosci. 2019, 24, 245–276. [Google Scholar]
- Kladde, M.P.; Xu, M.; Simpson, R.T. Direct study of DNA-protein interactions in repressed and active chromatin in living cells. EMBO J. 1996, 15, 6290–6300. [Google Scholar] [CrossRef]
- Gronemeyer, H. Transcription activation by estrogen and progesterone receptors. Annu. Rev. Genet. 1991, 25, 89–123. [Google Scholar] [CrossRef]
- Weigel, N.L.; Zhang, Y. Ligand-independent activation of steroid hormone receptors. J. Mol. Med. 1998, 76, 469–479. [Google Scholar] [CrossRef]
- Berger, C.E.; Qian, Y.; Liu, G.; Chen, H.; Chen, X. p53, a target of estrogen receptor (ER) alpha, modulates DNA damage-induced growth suppression in ER-positive breast cancer cells. J. Biol. Chem. 2012, 287, 30117–30127. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.F.; Chambers, J.A.; Solomon, E. Complex regulation of the BRCA1 gene. J. Biol. Chem. 1997, 272, 20994–20997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, H.; Huang, D.; Yang, F.; Guan, X. Potential biomarkers of CDK4/6 inhibitors in hormone receptor-positive advanced breast cancer. Breast Cancer Res. Treat. 2018, 168, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.K.; Schiff, R. Estrogen-receptor biology: Continuing progress and therapeutic implications. J. Clin. Oncol. 2005, 23, 1616–1622. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, G.; Ma, C. Revisiting the estrogen receptor pathway and its role in endocrine therapy for postmenopausal women with estrogen receptor-positive metastatic breast cancer. Breast Cancer Res. Treat. 2015, 150, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Han, H.; Bajic, V.B. ERGDB: Estrogen Responsive Genes Database. Nucleic Acids Res. 2004, 32, 533–536. [Google Scholar] [CrossRef]
- Ikeda, K.; Horie-Inoue, K.; Inoue, S. Identification of estrogen-responsive genes based on the DNA binding properties of estrogen receptors using high-throughput sequencing technology. Acta Pharmacol. Sin. 2015, 36, 24–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamalakaran, S.; Radhakrishnan, S.K.; Beck, W.T. Identification of estrogen-responsive genes using a genome-wide analysis of promoter elements for transcription factor binding sites. J. Biol. Chem. 2005, 280, 21491–21497. [Google Scholar] [CrossRef] [Green Version]
- Greenwalt, I.; Zaza, N.; Das, S.; Li, B.D. Precision medicine and targeted therapies in breast cancer. Surg. Oncol. Clin. N. Am. 2020, 29, 51–62. [Google Scholar] [CrossRef]
- Herrada, J.; Iyer, R.B.; Atkinson, E.N.; Sneige, N.; Buzdar, A.U.; Hortobagyi, G.N. Relative value of physical examination, mammography, and breast sonography in evaluating the size of the primary tumor and regional lymph node metastases in women receiving neoadjuvant chemotherapy for locally advanced breast carcinoma. Clin. Cancer Res. 1997, 3, 1565–1569. [Google Scholar]
- Goldhirsch, A.; Gelber, R.D. Endocrine therapies of breast cancer. Semin. Oncol. 1996, 23, 494–505. [Google Scholar]
- Metwally, I.H.; Hamdy, O.; Elbalka, S.S.; Elbadrawy, M.; Elsaid, D.M. Oophorectomy as a hormonal ablation therapy in metastatic and recurrent breast cancer: Current indications and results. India J. Surg. Oncol. 2019, 10, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Abderrahman, B.; Jordan, V.C. Successful targeted therapies for breast cancer: The worcester foundation and future opportunities in women’s health. Endocrinology 2018, 159, 2980–2990. [Google Scholar] [CrossRef] [PubMed]
- Dustin, D.; Gu, G.W.; Fuqua, S.A.W. ESR1 mutations in breast cancer. Cancer 2019, 125, 3714–3728. [Google Scholar] [CrossRef] [PubMed]
- Haque, M.M.; Desai, K.V. Pathways to endocrine therapy resistance in breast cancer. Front. Endocrinol. 2019, 10. [Google Scholar] [CrossRef]
- Robinson, D.R.; Wu, Y.M.; Vats, P.; Su, F.Y.; Lonigro, R.J.; Cao, X.H.; Kalyana-Sundaram, S.; Wang, R.; Ning, Y.; Hodges, L.; et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat. Genet. 2013, 45, 1446–1451. [Google Scholar] [CrossRef] [Green Version]
- Jordan, V.C. Tamoxifen (ICI46,474) as a targeted therapy to treat and prevent breast cancer. Brit. J. Pharmacol. 2006, 147, S269–S276. [Google Scholar] [CrossRef] [Green Version]
- Jordan, V.C.; Koerner, S. Tamoxifen (Ici 46,474) and Human Carcinoma 8s Estrogen-Receptor. Eur. J. Cancer 1975, 11, 205–206. [Google Scholar] [CrossRef]
- Grainger, D.J.; Metcalfe, J.C. Tamoxifen: Teaching an old drug new tricks? Nat. Med. 1996, 2, 381–385. [Google Scholar] [CrossRef]
- Shiau, A.K.; Barstad, D.; Loria, P.M.; Cheng, L.; Kushner, P.J.; Agard, D.A.; Greene, G.L. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 1998, 95, 927–937. [Google Scholar] [CrossRef] [Green Version]
- Ascenzi, P.; Bocedi, A.; Marino, M. Structure-function relationship of estrogen receptor alpha and beta: Impact on human health. Mol. Asp. Med. 2006, 27, 299–402. [Google Scholar] [CrossRef]
- Merenbakh-Lamin, K.; Ben-Baruch, N.; Yeheskel, A.; Dvir, A.; Soussan-Gutman, L.; Jeselsohn, R.; Yelensky, R.; Brown, M.; Miller, V.A.; Sarid, D.; et al. D538G mutation in estrogen receptor-alpha: A novel mechanism for acquired endocrine resistance in breast cancer. Cancer Res. 2013, 73, 6856–6864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guttery, D.S.; Page, K.; Hills, A.; Woodley, L.; Marchese, S.D.; Rghebi, B.; Hastings, R.K.; Luo, J.L.; Pringle, J.H.; Stebbing, J.; et al. Noninvasive detection of activating estrogen receptor 1 (ESR1) mutations in estrogen receptor-positive metastatic breast cancer. Clin. Chem. 2015, 61, 974–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.; Im, S.A.; Gelmon, K.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S.; et al. Palbociclib and letrozole in advanced breast cancer. N. Engl. J. Med. 2016, 375, 1925–1936. [Google Scholar] [CrossRef] [PubMed]
- Goetz, M.P.; Toi, M.; Campone, M.; Sohn, J.; Paluch-Shimon, S.; Huober, J.; Park, I.H.; Tredan, O.; Chen, S.C.; Manso, L.; et al. MONARCH 3: Abemaciclib as initial therapy for advanced breast cancer. J. Clin. Oncol. 2017, 35, 3638–3646. [Google Scholar] [CrossRef] [PubMed]
- Rodrik-Outmezguine, V.S.; Okaniwa, M.; Yao, Z.; Novotny, C.J.; McWhirter, C.; Banaji, A.; Won, H.; Wong, W.; Berger, M.; de Stanchina, E.; et al. Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor. Nature 2016, 534, 272–276. [Google Scholar] [CrossRef] [Green Version]
- Murphy, C.G. The Role of CDK4/6 Inhibitors in Breast Cancer. Curr. Treat. Options Oncol. 2019, 20, 52. [Google Scholar] [CrossRef]
- Hamadeh, I.S.; Patel, J.N.; Rusin, S.; Tan, A.R. Personalizing aromatase inhibitor therapy in patients with breast cancer. Cancer Treat. Rev. 2018, 70, 47–55. [Google Scholar] [CrossRef]
- Ariazi, E.A.; Lewis-Wambi, J.S.; Gill, S.D.; Pyle, J.R.; Ariazi, J.L.; Kim, H.R.; Sharma, C.G.; Cordera, F.; Shupp, H.A.; Li, T.; et al. Emerging principles for the development of resistance to antihormonal therapy: implications for the clinical utility of fulvestrant. J. Steroid. Biochem. Mol. Biol. 2006, 102, 128–138. [Google Scholar] [CrossRef] [Green Version]
- Rao, R.D.; Cobleigh, M.A. Adjuvant endocrine therapy for breast cancer. Oncology (Williston Park) 2012, 26, 541–547, 550, 552. [Google Scholar]
- Huerta-Reyes, M.; Maya-Nunez, G.; Perez-Solis, M.A.; Lopez-Munoz, E.; Guillen, N.; Olivo-Marin, J.C.; Aguilar-Rojas, A. Treatment of Breast Cancer With Gonadotropin-Releasing Hormone Analogs. Front. Oncol. 2019, 9, 943. [Google Scholar] [CrossRef] [Green Version]
- Fribbens, C.; O’Leary, B.; Kilburn, L.; Hrebien, S.; Garcia-Murillas, I.; Beaney, M.; Cristofanilli, M.; Andre, F.; Loi, S.; Loibl, S.; et al. Plasma ESR1 mutations and the treatment of estrogen receptor-positive advanced breast cancer. J. Clin. Oncol. 2016, 34, 2961–2968. [Google Scholar] [CrossRef] [PubMed]
- Chandarlapaty, S.; Chen, D.; He, W.; Sung, P.; Samoila, A.; You, D.Q.; Bhatt, T.; Patel, P.; Voi, M.; Gnant, M.; et al. Prevalence of ESR1 mutations in Cell-Free DNA and outcomes in metastatic breast cancer a secondary analysis of the BOLERO-2 clinical trial. JAMA Oncol. 2016, 2, 1310–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spoerke, J.M.; Gendreau, S.; Walter, K.; Qiu, J.H.; Wilson, T.R.; Savage, H.; Aimi, J.; Derynck, M.K.; Chen, M.; Chan, I.T.; et al. Heterogeneity and clinical significance of ESR1 mutations in ER-positive metastatic breast cancer patients receiving fulvestrant. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Toy, W.; Weir, H.; Razavi, P.; Lawson, M.; Goeppert, A.U.; Mazzola, A.M.; Smith, A.; Wilson, J.; Morrow, C.; Wong, W.L.; et al. Activating ESR1 mutations differentially affect the efficacy of ER antagonists. Cancer Discov. 2017, 7, 277–287. [Google Scholar] [CrossRef] [Green Version]
- Takeshita, T.; Yamamoto, Y.; Yamamoto-Ibusuki, M.; Sueta, A.; Tomiguchi, M.; Murakami, K.; Omoto, Y.; Iwase, H. Prevalence of ESR1 E380Q mutation in tumor tissue and plasma from Japanese breast cancer patients. BMC Cancer 2017, 17. [Google Scholar] [CrossRef] [Green Version]
- Ross, D.S.; Zehir, A.; Brogi, E.; Konno, F.; Krystel-Whittemore, M.; Edelweiss, M.; Berger, M.F.; Toy, W.; Chandarlapaty, S.; Razavi, P.; et al. Immunohistochemical analysis of estrogen receptor in breast cancer with ESR1 mutations detected by hybrid capture-based next-generation sequencing. Mod. Pathol. 2019, 32, 81–87. [Google Scholar] [CrossRef]
- Najim, O.; Huizing, M.; Papadimitriou, K.; Trinh, X.B.; Pauwels, P.; Goethals, S.; Zwaenepoel, K.; Peterson, K.; Weyler, J.; Altintas, S.; et al. The prevalence of estrogen receptor-1 mutation in advanced breast cancer: The estrogen receptor one study (EROS1). Cancer Treat. Res. Commun. 2019, 19, 100123. [Google Scholar] [CrossRef]
- Toy, W.; Weir, H.; Razavi, P.; Berger, M.; Wong, W.L.; De Stanchina, E.; Baselga, J.; Chandarlapaty, S. Differential activity and SERD sensitivity of clinical ESR1 mutations. Cancer Res. 2016, 76. [Google Scholar] [CrossRef]
- Jeannot, E.; Darrigues, L.; Michel, M.; Stern, M.H.; Pierga, J.Y.; Rampanou, A.; Melaabi, S.; Benoist, C.; Bieche, I.; Vincent-Salomon, A.; et al. A single droplet digital PCR for ESR1 activating mutations detection in plasma. Oncogene 2020. [Google Scholar] [CrossRef]
- Khan, A.; Ashfaq Ur, R.; Junaid, M.; Li, C.D.; Saleem, S.; Humayun, F.; Shamas, S.; Ali, S.S.; Babar, Z.; Wei, D.Q. Dynamics insights into the gain of flexibility by Helix-12 in ESR1 as a mechanism of resistance to drugs in breast cancer cell lines. Front. Mol. Biosci. 2019, 6, 159. [Google Scholar] [CrossRef] [Green Version]
- Gucalp, A.; Traina, T.A. The Androgen Receptor: Is It a Promising Target? Ann. Surg. Oncol. 2017, 24, 2876–2880. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, A.O.; Trapman, J. Prostate cancer schemes for androgen escape. Nat. Med. 2000, 6, 628–629. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.H.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E.L. Androgen receptor: Structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin. 2015, 36, 3–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cleutjens, K.B.; van Eekelen, C.C.; van der Korput, H.A.; Brinkmann, A.O.; Trapman, J. Two androgen response regions cooperate in steroid hormone regulated activity of the prostate-specific antigen promoter. J. Biol. Chem. 1996, 271, 6379–6388. [Google Scholar] [CrossRef] [Green Version]
- Pang, S.; Taneja, S.; Dardashti, K.; Cohan, P.; Kaboo, R.; Sokoloff, M.; Tso, C.L.; Dekernion, J.B.; Belldegrun, A.S. Prostate tissue specificity of the prostate-specific antigen promoter isolated from a patient with prostate cancer. Hum. Gene Ther. 1995, 6, 1417–1426. [Google Scholar] [CrossRef]
- Shang, Y.; Myers, M.; Brown, M. Formation of the androgen receptor transcription complex. Mol. Cell 2002, 9, 601–610. [Google Scholar] [CrossRef]
- Maximum androgen blockade in advanced prostate cancer: An overview of 22 randomised trials with 3283 deaths in 5710 patients. Prostate Cancer Trialists’ Collaborative Group. Lancet 1995, 346, 265–269.
- Litvinov, I.V.; De Marzo, A.M.; Isaacs, J.T. Is the Achilles’ heel for prostate cancer therapy a gain of function in androgen receptor signaling? J. Clin. Endocrinol. Metab. 2003, 88, 2972–2982. [Google Scholar] [CrossRef] [Green Version]
- Singla, N.; Ghandour, R.A.; Raj, G.V. Investigational luteinizing hormone releasing hormone (LHRH) agonists and other hormonal agents in early stage clinical trials for prostate cancer. Expert Opin. Investig. Drugs 2019, 28, 249–259. [Google Scholar] [CrossRef]
- Feng, Q.; He, B. Androgen receptor signaling in the development of castration-resistant prostate cancer. Front. Oncol. 2019, 9, 858. [Google Scholar] [CrossRef] [Green Version]
- Hobisch, A.; Culig, Z.; Radmayr, C.; Bartsch, G.; Klocker, H.; Hittmair, A. Androgen receptor status of lymph node metastases from prostate cancer. Prostate 1996, 28, 129–135. [Google Scholar] [CrossRef]
- Taplin, M.E.; Bubley, G.J.; Ko, Y.J.; Small, E.J.; Upton, M.; Rajeshkumar, B.; Balk, S.P. Selection for androgen receptor mutations in prostate cancers treated with androgen antagonist. Cancer Res. 1999, 59, 2511–2515. [Google Scholar] [PubMed]
- Linja, M.J.; Savinainen, K.J.; Saramaki, O.R.; Tammela, T.L.; Vessella, R.L.; Visakorpi, T. Amplification and overexpression of androgen receptor gene in hormone-refractory prostate cancer. Cancer Res. 2001, 61, 3550–3555. [Google Scholar] [PubMed]
- Brown, R.S.; Edwards, J.; Dogan, A.; Payne, H.; Harland, S.J.; Bartlett, J.M.; Masters, J.R. Amplification of the androgen receptor gene in bone metastases from hormone-refractory prostate cancer. J. Pathol. 2002, 198, 237–244. [Google Scholar] [CrossRef]
- Culig, Z.; Hobisch, A.; Cronauer, M.V.; Cato, A.C.; Hittmair, A.; Radmayr, C.; Eberle, J.; Bartsch, G.; Klocker, H. Mutant androgen receptor detected in an advanced-stage prostatic carcinoma is activated by adrenal androgens and progesterone. Mol. Endocrinol. 1993, 7, 1541–1550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinkamp, M.P.; O’Mahony, O.A.; Brogley, M.; Rehman, H.; Lapensee, E.W.; Dhanasekaran, S.; Hofer, M.D.; Kuefer, R.; Chinnaiyan, A.; Rubin, M.A.; et al. Treatment-dependent androgen receptor mutations in prostate cancer exploit multiple mechanisms to evade therapy. Cancer Res. 2009, 69, 4434–4442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rees, I.; Lee, S.; Kim, H.; Tsai, F.T. The E3 ubiquitin ligase CHIP binds the androgen receptor in a phosphorylation-dependent manner. Biochim. Biophys. Acta 2006, 1764, 1073–1079. [Google Scholar] [CrossRef] [PubMed]
- Veldscholte, J.; Ris-Stalpers, C.; Kuiper, G.G.; Jenster, G.; Berrevoets, C.; Claassen, E.; van Rooij, H.C.; Trapman, J.; Brinkmann, A.O.; Mulder, E. A mutation in the ligand binding domain of the androgen receptor of human LNCaP cells affects steroid binding characteristics and response to anti-androgens. Biochem. Biophys. Res. Commun. 1990, 173, 534–540. [Google Scholar] [CrossRef] [Green Version]
- Fenton, M.A.; Shuster, T.D.; Fertig, A.M.; Taplin, M.E.; Kolvenbag, G.; Bubley, G.J.; Balk, S.P. Functional characterization of mutant androgen receptors from androgen-independent prostate cancer. Clin. Cancer Res. 1997, 3, 1383–1388. [Google Scholar]
- Hobisch, A.; Colleselli, K.; Ennemoser, O.; Horninger, W.; Poisel, S.; Janetschek, G.; Bartsch, G. Modified retroperitoneal lymphadenectomy for testicular tumor: Anatomical approach, operative technique and results. Eur. Urol. 1993, 23, 39–43. [Google Scholar] [CrossRef]
- Brown, T.R.; Lubahn, D.B.; Wilson, E.M.; French, F.S.; Migeon, C.J.; Corden, J.L. Functional characterization of naturally occurring mutant androgen receptors from subjects with complete androgen insensitivity. Mol. Endocrinol. 1990, 4, 1759–1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcelli, M.; Tilley, W.D.; Zoppi, S.; Griffin, J.E.; Wilson, J.D.; Mcphaul, M.J. Androgen resistance associated with a mutation of the androgen receptor at amino-acid 772 (Arg-]Cys) results from a combination of decreased messenger-ribonucleic-acid levels and impairment of receptor function. J. Clin. Endocr. Metab. 1991, 73, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Reichert, Z.R.; Hussain, M. Androgen receptor and beyond, targeting androgen signaling in castration-resistant prostate cancer. Cancer J. 2016, 22, 326–329. [Google Scholar] [CrossRef] [PubMed]
- Cabeza, M.; Sanchez-Marquez, A.; Garrido, M.; Silva, A.; Bratoeff, E. Recent advances in drug design and drug discovery for androgen-dependent diseases. Curr. Med. Chem. 2016, 23, 792–815. [Google Scholar] [CrossRef] [Green Version]
- Yap, T.A.; Carden, C.P.; Attard, G.; de Bono, J.S. Targeting CYP17: Established and novel approaches in prostate cancer. Curr. Opin. Pharmacol. 2008, 8, 449–457. [Google Scholar] [CrossRef]
- Hall, P.F. Cytochrome P-450 C21scc: One enzyme with two actions: Hydroxylase and lyase. J. Steroid Biochem. Mol. Biol. 1991, 40, 527–532. [Google Scholar] [CrossRef]
- Chen, Y.; Clegg, N.J.; Scher, H.I. Anti-androgens and androgen-depleting therapies in prostate cancer: New agents for an established target. Lancet Oncol. 2009, 10, 981–991. [Google Scholar] [CrossRef] [Green Version]
- Gupta, E.; Guthrie, T.; Tan, W. Changing paradigms in management of metastatic Castration Resistant Prostate Cancer (mCRPC). BMC Urol. 2014, 14, 55. [Google Scholar] [CrossRef] [Green Version]
- Potter, G.A.; Barrie, S.E.; Jarman, M.; Rowlands, M.G. Novel steroidal inhibitors of human cytochrome P45017 alpha (17 alpha-hydroxylase-C17,20-lyase): Potential agents for the treatment of prostatic cancer. J. Med. Chem. 1995, 38, 2463–2471. [Google Scholar] [CrossRef]
- Rowlands, M.G.; Barrie, S.E.; Chan, F.; Houghton, J.; Jarman, M.; Mccague, R.; Potter, G.A. Esters of 3-pyridylacetic acid that combine potent inhibition of 17-Alpha-Hydroxylase C-17,C-20-Lyase (Cytochrome P45017-Alpha) with resistance to esterase hydrolysis. J. Med. Chem. 1995, 38, 4191–4197. [Google Scholar] [CrossRef]
- McCague, R.; Rowlands, M.G.; Barrie, S.E.; Houghton, J. Inhibition of enzymes of estrogen and androgen biosynthesis by esters of 4-pyridylacetic acid. J. Med. Chem. 1990, 33, 3050–3055. [Google Scholar] [CrossRef] [PubMed]
- Attard, G.; Belldegrun, A.S.; de Bono, J.S. Selective blockade of androgenic steroid synthesis by novel lyase inhibitors as a therapeutic strategy for treating metastatic prostate cancer. BJU Int. 2005, 96, 1241–1246. [Google Scholar] [CrossRef] [PubMed]
- Ryan, C.J.; Smith, M.R.; de Bono, J.S.; Molina, A.; Logothetis, C.J.; de Souza, P.; Fizazi, K.; Mainwaring, P.; Piulats, J.M.; Ng, S.; et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N. Engl. J. Med. 2013, 368, 138–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, L.J. Enzalutamide: A review in castration-resistant prostate cancer. Drugs 2018, 78, 1913–1924. [Google Scholar] [CrossRef] [PubMed]
- Tran, C.; Ouk, S.; Clegg, N.J.; Chen, Y.; Watson, P.A.; Arora, V.; Wongvipat, J.; Smith-Jones, P.M.; Yoo, D.; Kwon, A.; et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 2009, 324, 787–790. [Google Scholar] [CrossRef] [Green Version]
- Hejmej, A.; Bilinska, B. The effects of flutamide on cell-cell junctions in the testis, epididymis, and prostate. Reprod. Toxicol. 2018, 81, 1–16. [Google Scholar] [CrossRef]
- Neri, R.; Peets, E.; Watnick, A. Anti-Androgenicity of Flutamide and its Metabolite Sch 16423. Biochem. Soc. T 1979, 7, 565–569. [Google Scholar] [CrossRef]
- Anahara, R.; Toyama, Y.; Mori, C. Review of the histological effects of the anti-androgen, flutamide, on mouse testis. Reprod. Toxicol. 2008, 25, 139–143. [Google Scholar] [CrossRef]
- Romanel, A.; Tandefelt, D.G.; Conteduca, V.; Jayaram, A.; Casiraghi, N.; Wetterskog, D.; Salvi, S.; Amadori, D.; Zafeiriou, Z.; Rescigno, P.; et al. Plasma AR and abiraterone-resistant prostate cancer. Sci. Transl. Med. 2015, 7. [Google Scholar] [CrossRef] [Green Version]
- Chen, E.J.; Sowalsky, A.G.; Gao, S.; Cai, C.M.; Voznesensky, O.; Schaefer, R.; Loda, M.; True, L.D.; Ye, H.H.; Troncoso, P.; et al. Abiraterone treatment in castration-resistant prostate cancer selects for progesterone responsive mutant androgen receptors. Clin. Cancer Res. 2015, 21, 1273–1280. [Google Scholar] [CrossRef] [Green Version]
- Azad, A.A.; Volik, S.V.; Wyatt, A.W.; Haegert, A.; Le Bihan, S.; Bell, R.H.; Anderson, S.A.; McConeghy, B.; Shukin, R.; Bazov, J.; et al. Androgen receptor gene aberrations in circulating cell-free DNA: Biomarkers of therapeutic resistance in castration-resistant prostate cancer. Clin. Cancer Res. 2015, 21, 2315–2324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sperger, J.M.; Strotman, L.N.; Welsh, A.; Casavant, B.P.; Chalmers, Z.; Horn, S.; Heninger, E.; Thiede, S.M.; Tokar, J.; Gibbs, B.K.; et al. Integrated analysis of multiple biomarkers from circulating tumor cells enabled by exclusion-based analyte isolation. Clin. Cancer Res. 2017, 23, 746–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korpal, M.; Korn, J.M.; Gao, X.; Rakiec, D.P.; Ruddy, D.A.; Doshi, S.; Yuan, J.; Kovats, S.G.; Kim, S.; Cooke, V.G.; et al. An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide). Cancer Discov. 2013, 3, 1030–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prekovic, S.; van Royen, M.E.; Voet, A.R.; Geverts, B.; Houtman, R.; Melchers, D.; Zhang, K.Y.; Van den Broeck, T.; Smeets, E.; Spans, L.; et al. The effect of F877L and T878A mutations on androgen receptor response to enzalutamide. Mol. Cancer Ther. 2016, 15, 1702–1712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guagnano, V.; Furet, P.; Spanka, C.; Bordas, V.; Le Douget, M.; Stamm, C.; Brueggen, J.; Jensen, M.R.; Schnell, C.; Schmid, H.; et al. Discovery of 3-(2,6-dichloro-3,5-dimethoxy-phenyl)-1-{6-[4-(4-ethyl-piperazin-1-yl)-phenylamin o]-pyrimidin-4-yl}-1-methyl-urea (NVP-BGJ398), a potent and selective inhibitor of the fibroblast growth factor receptor family of receptor tyrosine kinase. J. Med. Chem. 2011, 54, 7066–7083. [Google Scholar] [CrossRef]
- Nogova, L.; Sequist, L.V.; Perez Garcia, J.M.; Andre, F.; Delord, J.P.; Hidalgo, M.; Schellens, J.H.; Cassier, P.A.; Camidge, D.R.; Schuler, M.; et al. Evaluation of BGJ398, a fibroblast growth factor receptor 1-3 kinase inhibitor, in patients with advanced solid tumors harboring genetic alterations in fibroblast growth factor receptors: Results of a global phase i, dose-escalation and dose-expansion study. J. Clin. Oncol. 2017, 35, 157–165. [Google Scholar] [CrossRef]
- Harimoto, N.; Taguchi, K.; Shirabe, K.; Adachi, E.; Sakaguchi, Y.; Toh, Y.; Okamura, T.; Kayashima, H.; Taketomi, A.; Maehara, Y. The significance of fibroblast growth factor receptor 2 expression in differentiation of hepatocellular carcinoma. Oncology 2010, 78, 361–368. [Google Scholar] [CrossRef]
- Touat, M.; Ileana, E.; Postel-Vinay, S.; Andre, F.; Soria, J.C. Targeting FGFR signaling in cancer. Clin. Cancer Res. 2015, 21, 2684–2694. [Google Scholar] [CrossRef] [Green Version]
- Casadei, C.; Dizman, N.; Schepisi, G.; Cursano, M.C.; Basso, U.; Santini, D.; Pal, S.K.; De Giorgi, U. Targeted therapies for advanced bladder cancer: New strategies with FGFR inhibitors. Ther. Adv. Med. Oncol. 2019, 11, 1758835919890285. [Google Scholar] [CrossRef]
- Huynh, H.; Lee, L.Y.; Goh, K.Y.; Ong, R.; Hao, H.X.; Huang, A.L.; Wang, Y.Z.; Porta, D.G.; Chow, P.; Chung, A. Infigratinib mediates vascular normalization, impairs metastasis, and improves chemotherapy in hepatocellular carcinoma. Hepatology 2019, 69, 943–958. [Google Scholar] [CrossRef] [Green Version]
- Javle, M.; Lowery, M.; Shroff, R.T.; Weiss, K.H.; Springfeld, C.; Borad, M.J.; Ramanathan, R.K.; Goyal, L.; Sadeghi, S.; Macarulla, T.; et al. Phase II study of BGJ398 in patients with FGFR-Altered advanced cholangiocarcinoma. J. Clin. Oncol. 2018, 36, 276. [Google Scholar] [CrossRef] [PubMed]
- Javle, M.; Borbath, I.; Clarke, S.; Hitre, E.; Louvet, C.; Macarulla, T.; Oh, D.; Spratlin, J.; Valle, J.; Weiss, K.; et al. Phase 3 multicenter, open-label, randomized study of infigratinib versus gemcitabine plus cisplatin in the first-line treatment of patients with advanced cholangiocarcinoma with FGFR2 gene fusions/translocations: The PROOF trial. Ann. Oncol. 2019, 30. [Google Scholar] [CrossRef] [PubMed]
- Goyal, L.; Saha, S.K.; Liu, L.Y.; Siravegna, G.; Leshchiner, I.; Ahronian, L.G.; Lennerz, J.K.; Vu, P.; Deshpande, V.; Kambadakone, A.; et al. Polyclonal secondary FGFR2 mutations drive acquired resistance to FGFR inhibition in patients with FGFR2 fusion-positive cholangiocarcinoma. Cancer Discov. 2017, 7, 252–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasskarl, J. Everolimus. Recent Results Cancer Res. 2014, 201, 373–392. [Google Scholar] [CrossRef] [PubMed]
- Wagle, N.; Grabiner, B.C.; Van Allen, E.M.; Amin-Mansour, A.; Taylor-Weiner, A.; Rosenberg, M.; Gray, N.; Barletta, J.A.; Guo, Y.; Swanson, S.J.; et al. Response and acquired resistance to everolimus in anaplastic thyroid cancer. N. Engl. J. Med. 2014, 371, 1426–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenz, M.C.; Heitman, J. Tor mutations confer rapamycin resistance by preventing interaction with Fkbp12-rapamycin. J. Biol. Chem. 1995, 270, 27531–27537. [Google Scholar] [CrossRef] [Green Version]
- Zou, H.Y.; Li, Q.; Lee, J.H.; Arango, M.E.; Burgess, K.; Qiu, M.; Engstrom, L.D.; Yamazaki, S.; Parker, M.; Timofeevski, S.; et al. Sensitivity of selected human tumor models to PF-04217903, a novel selective c-Met kinase inhibitor. Mol. Cancer Ther. 2012, 11, 1036–1047. [Google Scholar] [CrossRef] [Green Version]
- Diamond, J.R.; Salgia, R.; Varella-Garcia, M.; Kanteti, R.; LoRusso, P.M.; Clark, J.W.; Xu, L.G.; Wilner, K.; Eckhardt, S.G.; Ching, K.A.; et al. Initial clinical sensitivity and acquired resistance to MET inhibition in MET-mutated papillary renal cell carcinoma. J. Clin. Oncol. 2013, 31, e254–e258. [Google Scholar] [CrossRef] [Green Version]
- Sebolt-Leopold, J.S. Advances in the development of cancer therapeutics directed against the RAS-mitogen-activated protein kinase pathway. Clin. Cancer Res. 2008, 14, 3651–3656. [Google Scholar] [CrossRef] [Green Version]
- Brown, A.P.; Carlson, T.C.G.; Loi, C.M.; Graziano, M.J. Pharmacodynamic and toxicokinetic evaluation of the novel MEK inhibitor, PD0325901, in the rat following oral and intravenous administration. Cancer Chemoth. Pharmacol. 2007, 59, 671–679. [Google Scholar] [CrossRef]
- Kohno, M.; Pouyssegur, J. Targeting the ERK signaling pathway in cancer therapy. Ann. Med. 2006, 38, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Thompson, N.; Lyons, J. Recent progress in targeting the Raf/MEK/ERK pathway with inhibitors in cancer drug discovery. Curr. Opin. Pharmacol. 2005, 5, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Hatzivassiliou, G.; Liu, B.; O’Brien, C.; Spoerke, J.M.; Hoeflich, K.P.; Haverty, P.M.; Soriano, R.; Forrest, W.F.; Heldens, S.; Chen, H.; et al. ERK inhibition overcomes acquired resistance to MEK inhibitors. Mol. Cancer Ther. 2012, 11, 1143–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Daouti, S.; Li, W.H.; Wen, Y.; Rizzo, C.; Higgins, B.; Packman, K.; Rosen, N.; Boylan, J.F.; Heimbrook, D.; et al. Identification of the MEK1(F129L) activating mutation as a potential mechanism of acquired resistance to MEK inhibition in human cancers carrying the B-RafV600E mutation. Cancer Res. 2011, 71, 5535–5545. [Google Scholar] [CrossRef] [Green Version]
- Jubb, H.C.; Saini, H.K.; Verdonk, M.L.; Forbes, S.A. COSMIC-3D provides structural perspectives on cancer genetics for drug discovery. Nat. Genet. 2018, 50, 1200–1202. [Google Scholar] [CrossRef]
- Bedard, P.L.; Hyman, D.M.; Davids, M.S.; Siu, L.L. Small molecules, big impact: 20 years of targeted therapy in oncology. Lancet 2020, 395, 1078–1088. [Google Scholar] [CrossRef]
- Yi, M.; Xu, L.; Jiao, Y.; Luo, S.; Li, A.; Wu, K. The role of cancer-derived microRNAs in cancer immune escape. J. Hematol. Oncol. 2020, 13, 25. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.P.; Singh, R.; Gupta, O.P.; Gupta, S.; Brahma Bhatt, M.L. Immunotherapy: Newer therapeutic armamentarium against cancer stem cells. J. Oncol. 2020, 2020, 3963561. [Google Scholar] [CrossRef]
- Guzel, E.; Okyay, T.M.; Yalcinkaya, B.; Karacaoglu, S.; Gocmen, M.; Akcakuyu, M.H. Tumor suppressor and oncogenic role of long non-coding RNAs in cancer. North. Clin. Istanb. 2020, 7, 81–86. [Google Scholar] [CrossRef]
- Behl, T.; Kumar, C.; Makkar, R.; Gupta, A.; Sachdeva, M. Intercalating the role of MicroRNAs in cancer: As enemy or protector. Asian Pac. J. Cancer Prev. 2020, 21, 593–598. [Google Scholar] [CrossRef]
- Jiang, W.; Xia, J.; Xie, S.; Zou, R.; Pan, S.; Wang, Z.W.; Assaraf, Y.G.; Zhu, X. Long non-coding RNAs as a determinant of cancer drug resistance: Towards the overcoming of chemoresistance via modulation of lncRNAs. Drug Resist. Updates 2020, 50, 100683. [Google Scholar] [CrossRef] [PubMed]
- Allegra, A.; Musolino, C.; Tonacci, A.; Pioggia, G.; Gangemi, S. Interactions between the MicroRNAs and microbiota in cancer development: Roles and therapeutic opportunities. Cancers 2020, 12, 805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galon, J.; Bruni, D. Tumor immunology and tumor evolution: Intertwined histories. Immunity 2020, 52, 55–81. [Google Scholar] [CrossRef] [PubMed]
- Egen, J.G.; Ouyang, W.; Wu, L.C. Human anti-tumor immunity: Insights from immunotherapy clinical trials. Immunity 2020, 52, 36–54. [Google Scholar] [CrossRef]
- Luo, Y.; Yang, J.; Yu, J.; Liu, X.; Yu, C.; Hu, J.; Shi, H.; Ma, X. Long non-coding RNAs: Emerging roles in the immunosuppressive tumor microenvironment. Front. Oncol. 2020, 10, 48. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Yang, Y.; Hong, W.; Huang, M.; Wu, M.; Zhao, X. Applications of genome editing technology in the targeted therapy of human diseases: Mechanisms, advances and prospects. Signal. Transduct. Target. Ther. 2020, 5, 1. [Google Scholar] [CrossRef]
- Zatloukalova, P.; Krejcir, R.; Valik, D.; Vojtesek, B. CRISPR-Cas9 as a tool in cancer therapy. Klin. Onkol. 2019, 32, 13–18. [Google Scholar] [CrossRef]
- Datta, P.; Ray, S. Nanoparticulate formulations of radiopharmaceuticals: Strategy to improve targeting and biodistribution properties. J. Label. Comp. Radiopharm. 2020. [Google Scholar] [CrossRef]
- Haider, M.; Abdin, S.M.; Kamal, L.; Orive, G. Nanostructured lipid carriers for delivery of chemotherapeutics: A review. Pharmaceutics 2020, 12, 288. [Google Scholar] [CrossRef] [Green Version]
- Song, W.; Das, M.; Chen, X. Nanotherapeutics for immuno-oncology: A crossroad for new paradigms. Trends Cancer 2020, 6, 288–298. [Google Scholar] [CrossRef]
- Habibi, N.; Quevedo, D.F.; Gregory, J.V.; Lahann, J. Emerging methods in therapeutics using multifunctional nanoparticles. Wiley Interdiscip Rev. Nanomed Nanobiotechnol. 2020, e1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chivere, V.T.; Kondiah, P.P.D.; Choonara, Y.E.; Pillay, V. Nanotechnology-based biopolymeric oral delivery platforms for advanced cancer treatment. Cancers 2020, 12, 522. [Google Scholar] [CrossRef] [PubMed] [Green Version]

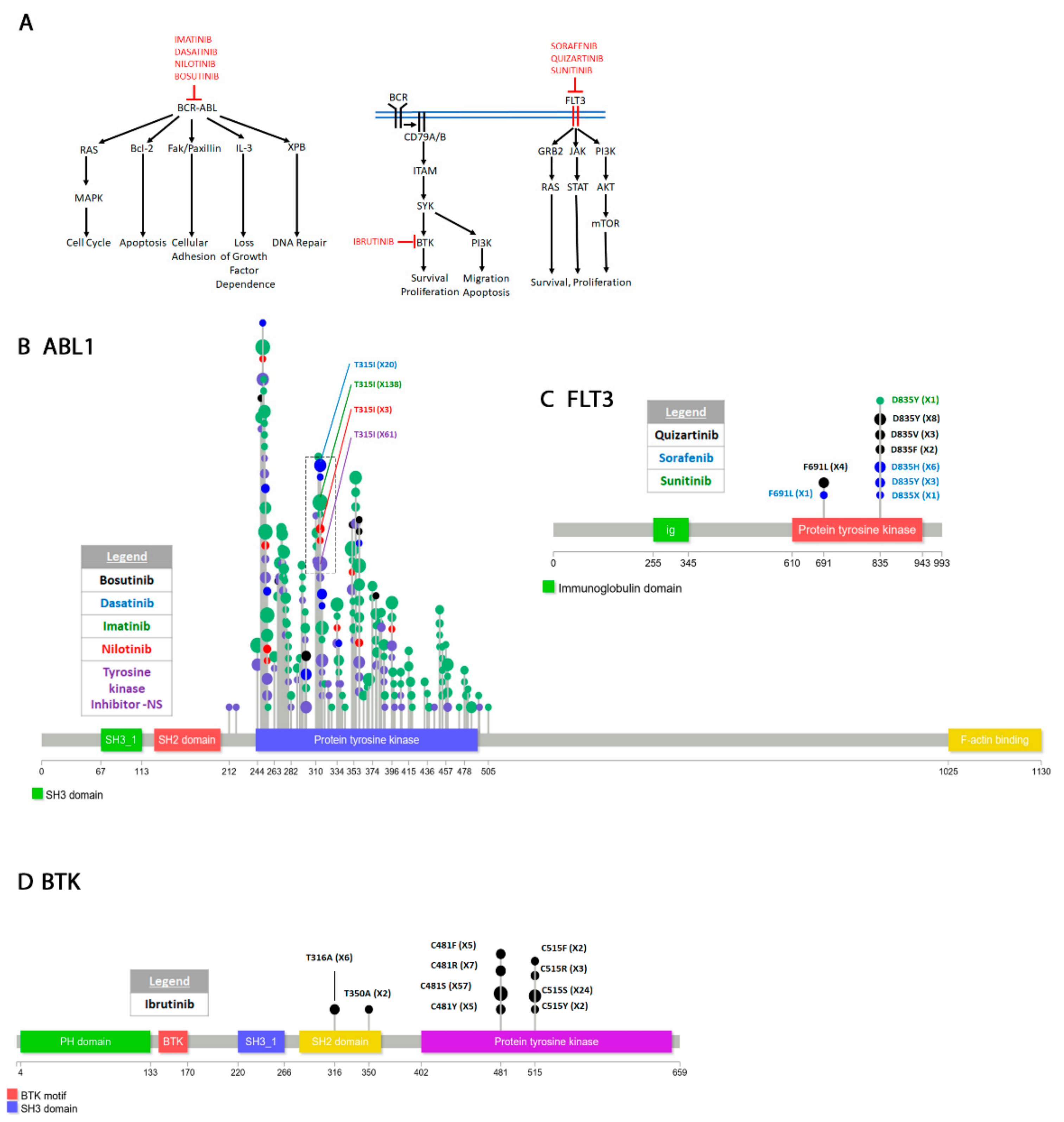
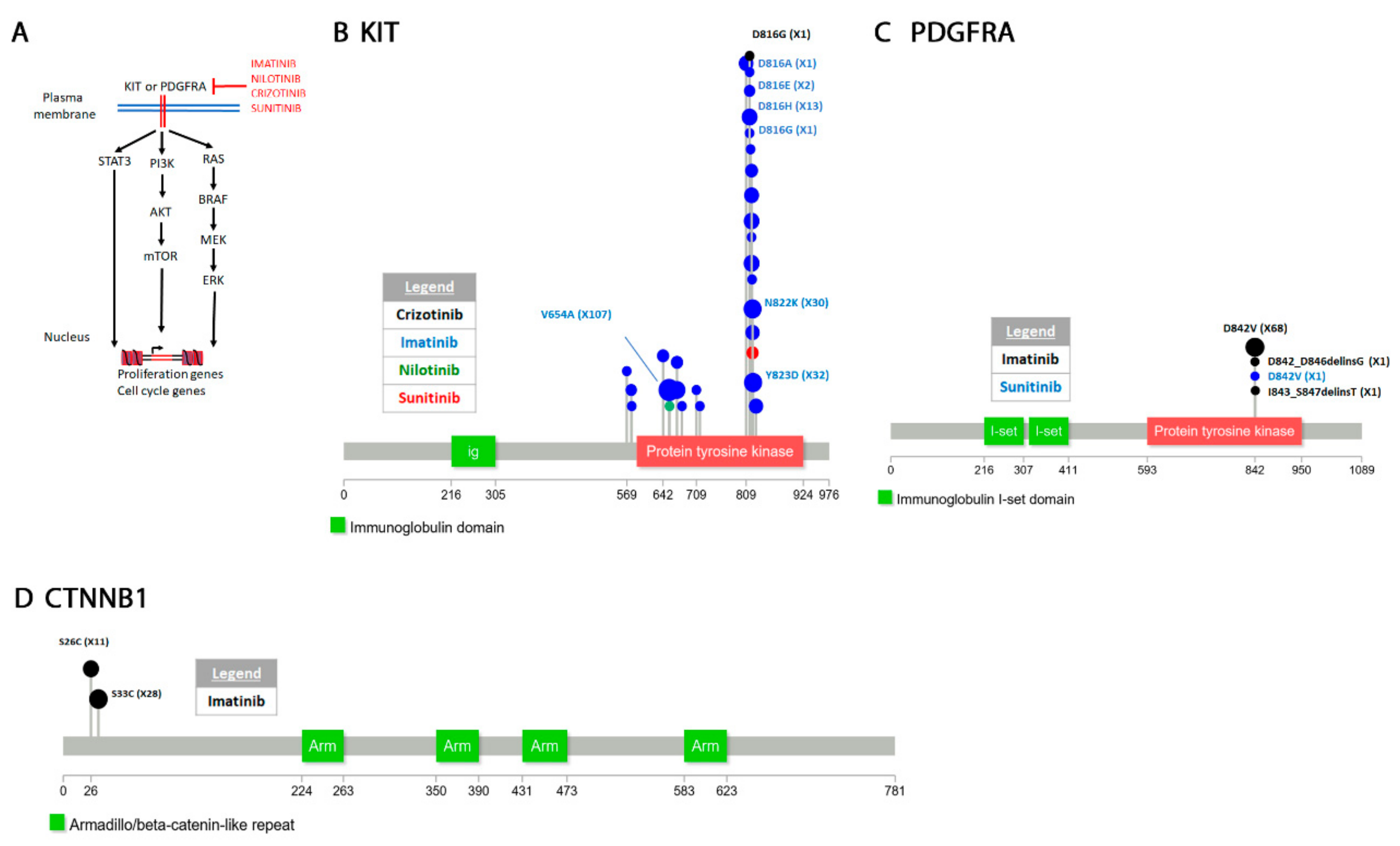
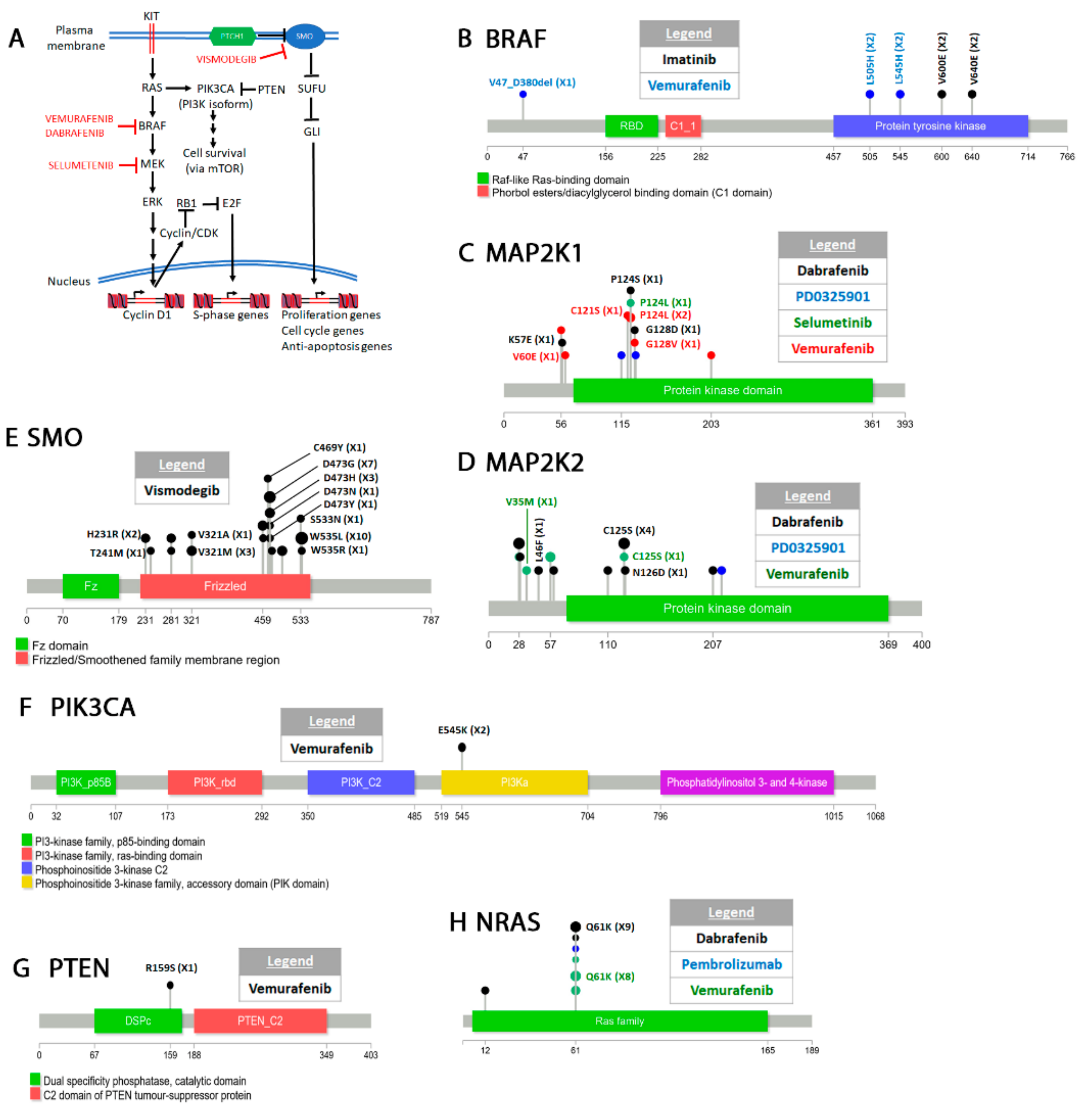



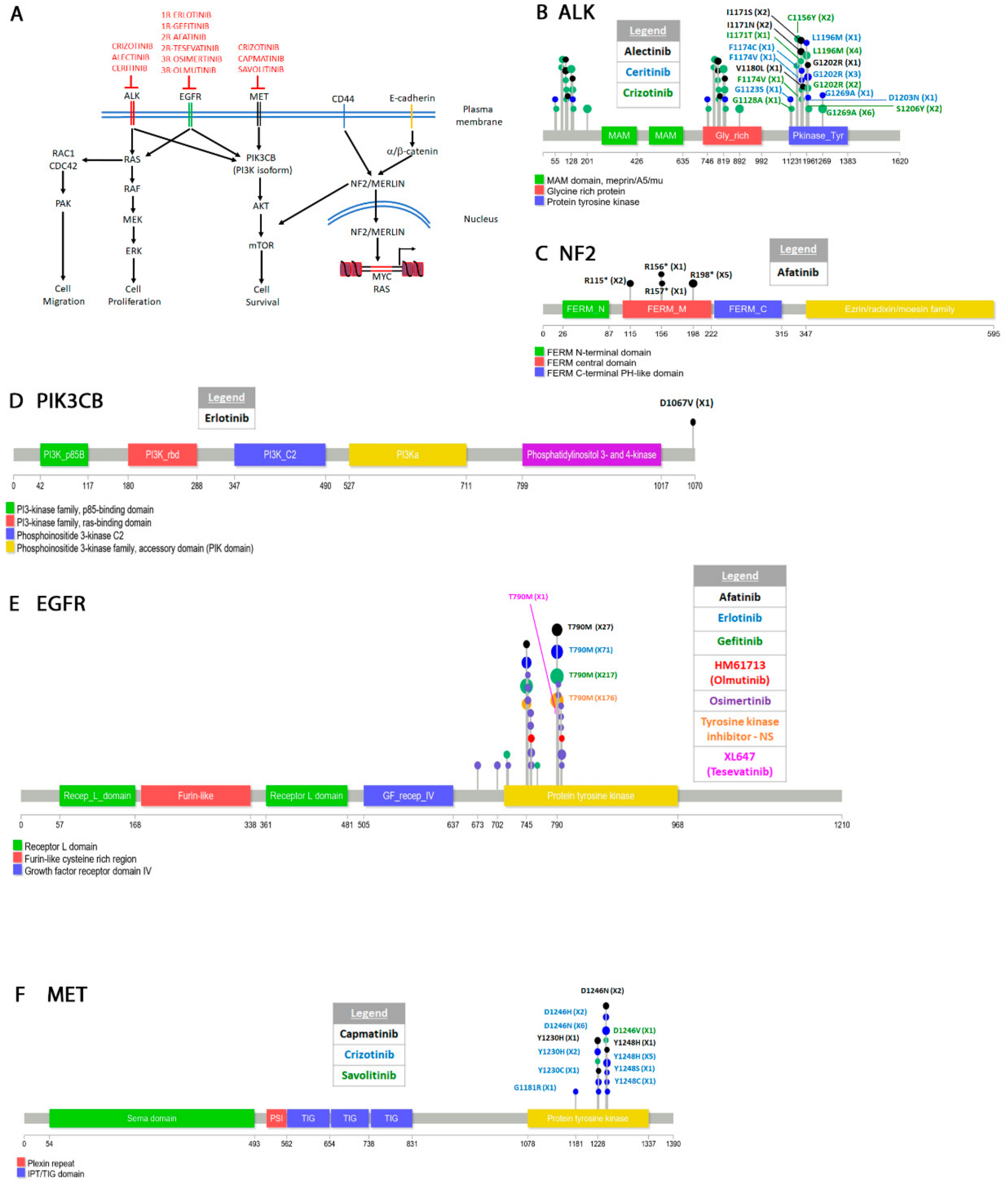{kind=link}
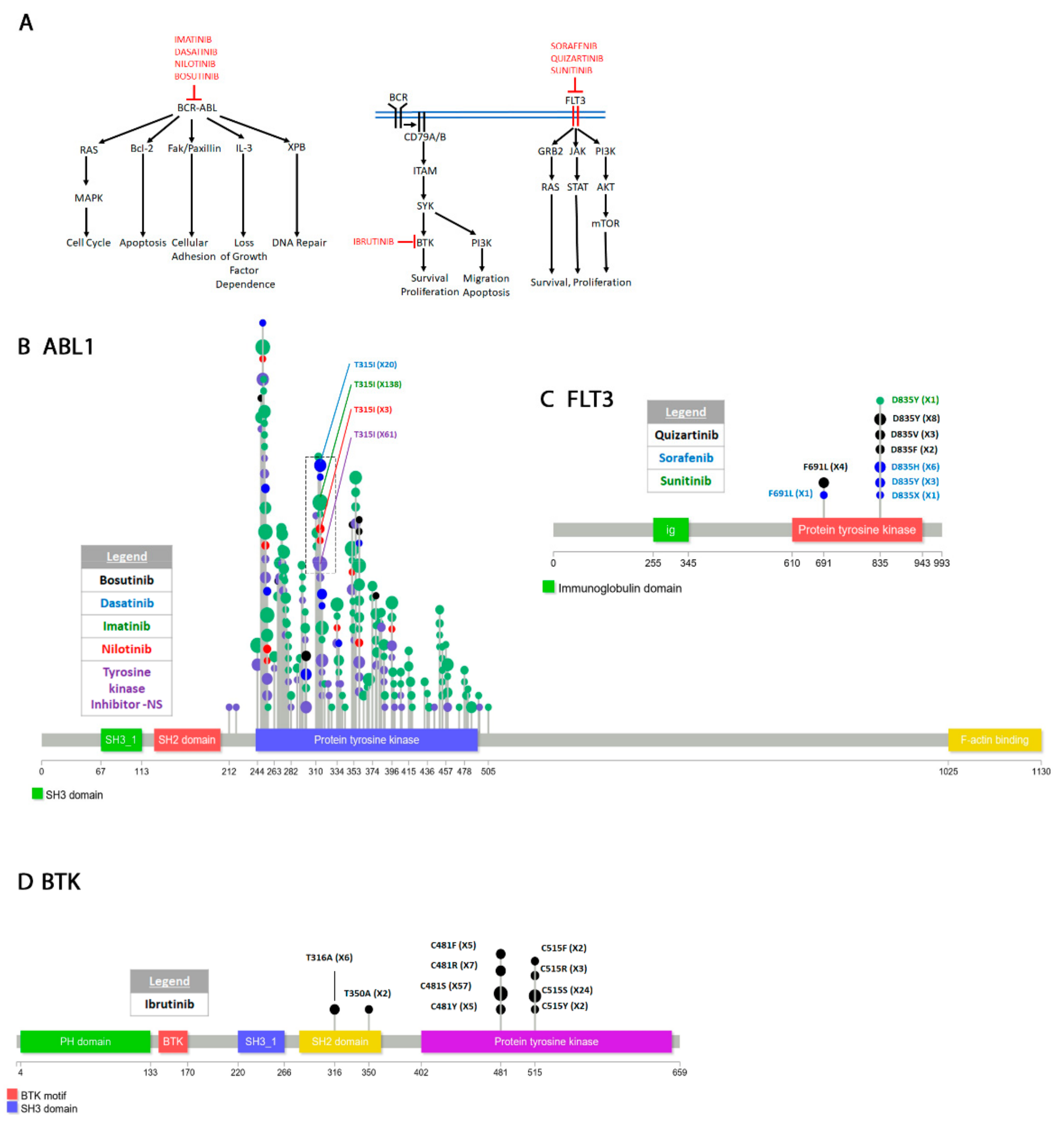{kind=link}
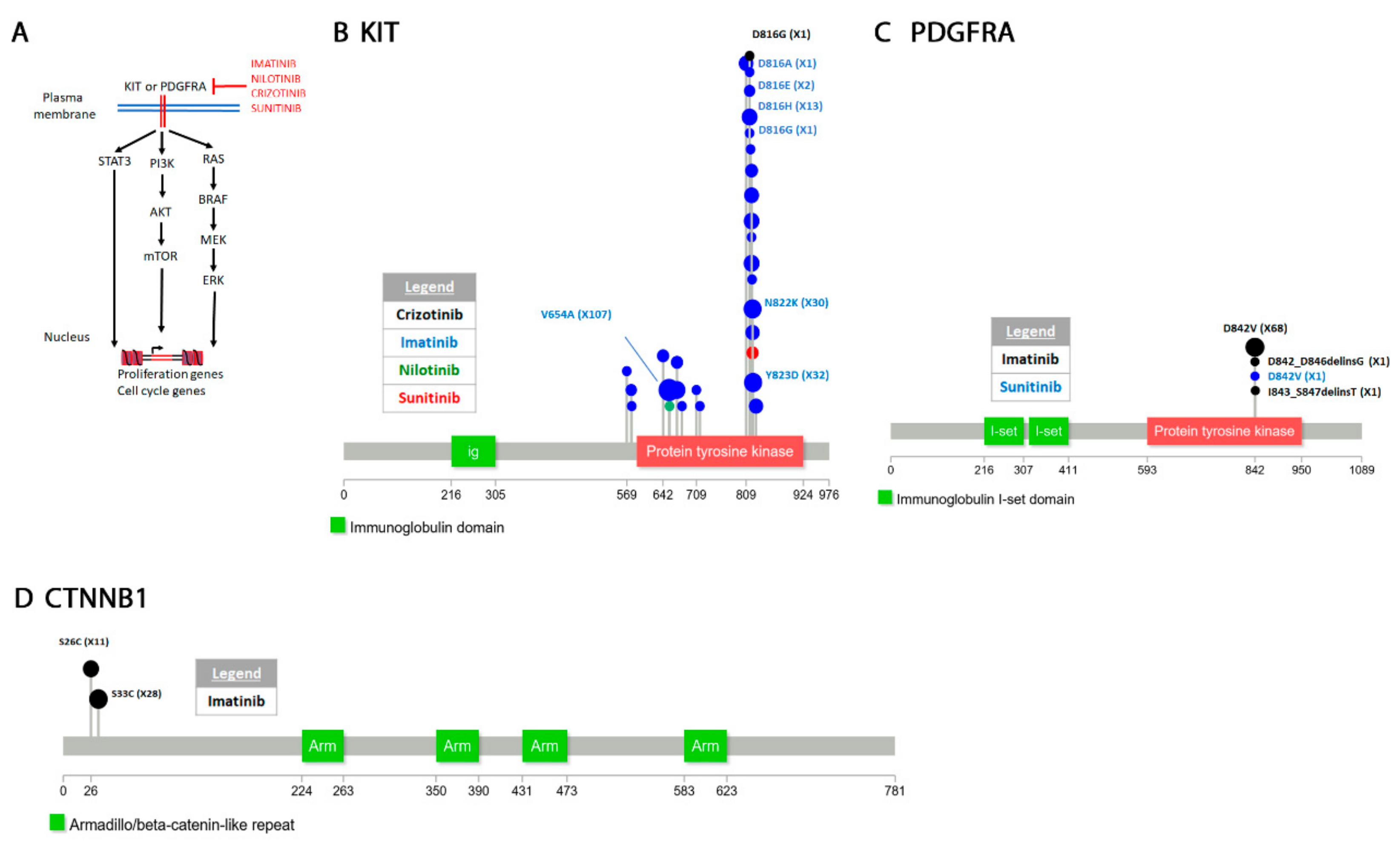{kind=link}
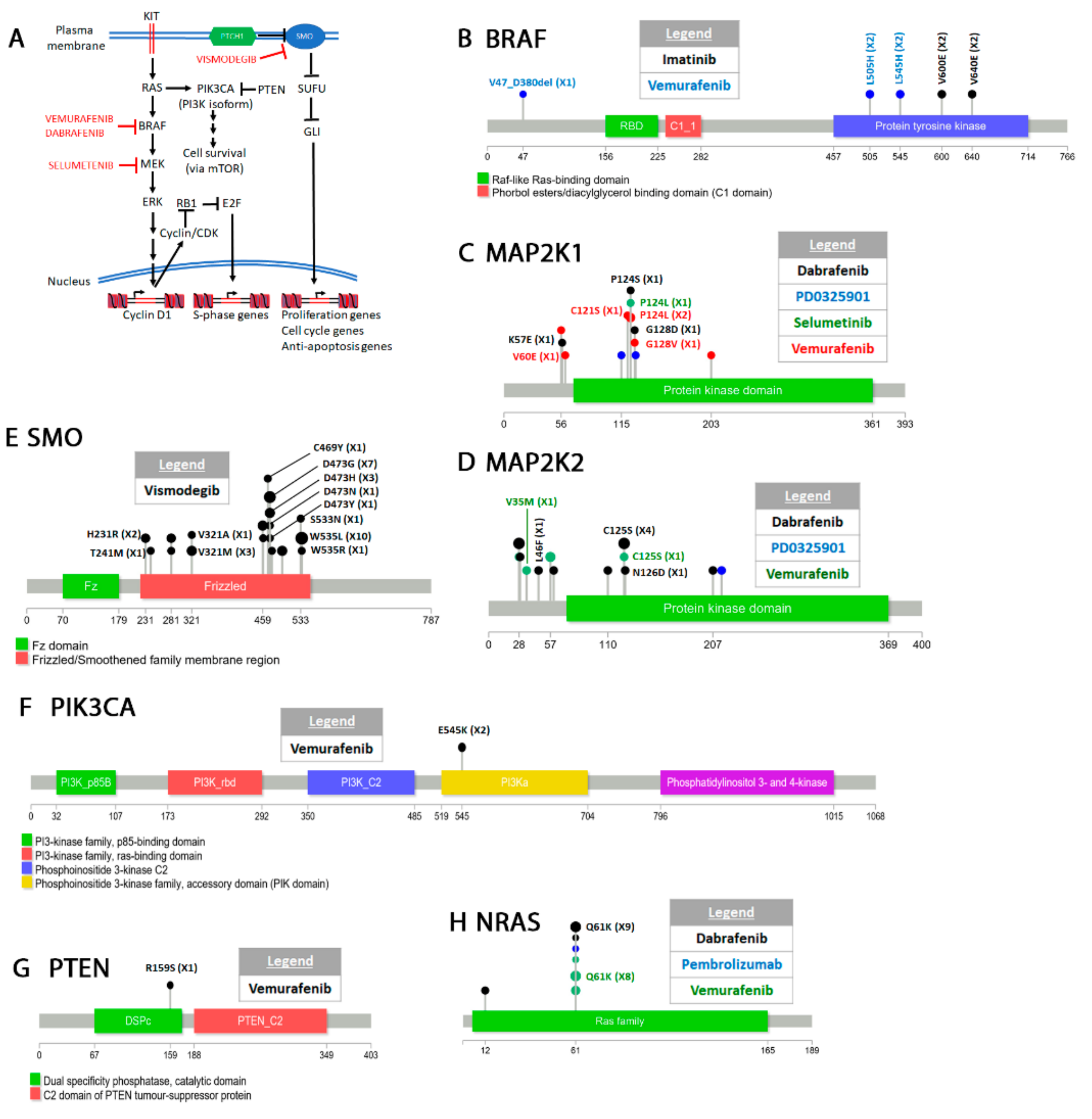{kind=link}
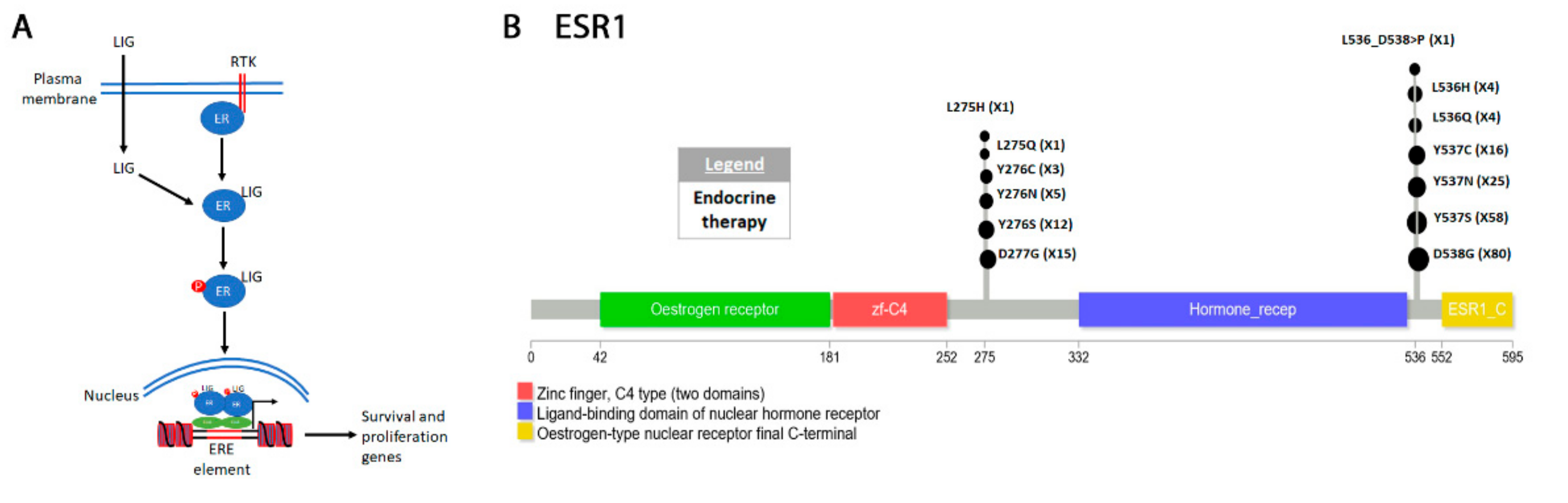{kind=link}
{kind=link}
| Drug Name | Targeted Cancer Type(s) | Gene(s) Acquiring Resistant Mutation(s) | Primary Tissue where Resistant Mutations were Identified | Tumor Type where Resistant Mutations Identified 1 |
|---|---|---|---|---|
| Erlotinib | Non-Small Cell Lung Cancer (NSCLC) (with EGFR activating mutations)2 Pancreatic cancer 3 | EGFR (Epidermal Growth Factor Receptor), MET (Hepatocyte Growth Factor Receptor) | Lung | Adenocarcinoma, non-small cell carcinoma, bronchioloalveolar and acinar adenocarcinoma |
| Gefitinib | NSCLC (with EGFR activating mutations) Breast cancer as well as several other cancers | EGFR, MET | Lung | Adenocarcinoma, non-small cell, squamous cell and pleomorphic carcinoma, mixed adenosquamous carcinoma, bronchioloalveolar, acinar and micropapillary adenocarcinoma |
| Afatinib | NSCLC (with EGFR activating mutations) Breast cancer | EGFR, MET, NF2 (Merlin) | Lung | Adenocarcinoma, non-small cell carcinoma, bronchioloalveolar adenocarcinoma |
| Osimertinib | NSCLC (with EGFR activating mutations and T790M resistant mutation) | EGFR, MET | Lung | Adenocarcinoma, non-small cell carcinoma |
| Olmutinib (HM61713) | NSCLC (with EGFR activating mutations and T790M resistant mutation) | EGFR | Lung | Adenocarcinoma |
| Tesevatinib (XL647) | NSCLC (with EGFR activating mutations and T790M resistant mutation) Polycystic kidney disease Kidney cancer | EGFR | Lung | Adenocarcinoma |
| Capmatinib | NSCLC (with MET activating mutations or MET amplification) | MET | Lung | Adenocarcinoma |
| Alectinib | NSCLC (ALK positive) | ALK (Anaplastic Lymphoma Kinase) | Lung | Adenocarcinoma, non-small cell carcinoma |
| Crizotinib | NSCLC (ALK, ROS1(Proto-oncogene tyrosine-protein kinase ROS) positive) Other ALK positive cancers | ALK, KIT (Proto-oncogene receptor tyrosine kinase), MET | Lung, Soft Tissue | Adenocarcinoma, non-small cell carcinoma, mixed adenosquamous carcinoma, squamous cell carcinoma, NS |
| Ceritinib | NSCLC (ALK positive with resistant mutations to crizotinib) | ALK | Lung | Adenocarcinoma, non-small cell carcinoma |
| Savolitinib | NSCLC Renal and gastric cancers | MET | Lung | Adenocarcinoma |
| Imatinib | Philadelphia chromosome positive leukemia KIT positive GIST (Gastrointestinal stromal tumors) Skin tumors | ABL1(Abelson Murine Leukemia Viral Oncogene Homolog 1), BRAF (serine/threonine-protein kinase B-Raf), KIT, PDGFRA(Platelet Derived Growth Factor Receptor Alpha), CTNNB1 (Beta catenin) AC058822.1 4 | Hematopoietic and lymphoid, Soft Tissue, Skin | Chronic myeloid leukemia, acute lymphoblastic leukemia, blast phase chronic myeloid leukemia, acral lentiginous, epithelioid, spindle, spindle and epithelioid, NS |
| Dasatinib | Philadelphia chromosome positive leukemia (with some imatinib resistant mutations) | ABL1 | Hematopoietic and lymphoid | Acute lymphoblastic leukemia, chronic myeloid leukemia, blast phase chronic myeloid leukemia |
| Nilotinib | Philadelphia chromosome positive leukemia (with some imatinib resistant mutations) GIST (KIT driven tumors) | ABL1, KIT | Hematopoietic and lymphoid, Soft Tissue | Chronic myeloid leukemia, blast phase chronic myeloid leukemia, spindle |
| Bosutinib | Philadelphia chromosome positive leukemia (with some imatinib resistant mutations) | ABL1 | Hematopoietic and lymphoid | Chronic myeloid leukemia, blast phase chronic myeloid leukemia |
| Tyrosine Kinase Inhibitor-NS 5 | NSCLC and leukemias | ABL1, EGFR | Hematopoietic and lymphoid, Lung | Adenocarcinoma, chronic myeloid leukemia, acute lymphoblastic leukemia, non-small cell carcinoma, blast phase chronic myeloid leukemia |
| Ibrutinib | Lymphomas and Chronic lymphocytic leukemia Other B-cell cancers | BTK (Bruton Tyrosine Kinase) | Hematopoietic and lymphoid | Chronic lymphocytic leukemia, lymphoplasmacytic lymphoma, mantle cell lymphoma |
| Quizartinib, Sorafenib | Acute myeloid leukemia Kidney and liver cancers | FLT3 (FMS-like Tyrosine Kinase 3) | Hematopoietic and lymphoid | Acute myeloid leukemia |
| Sunitinib | GISTs (usually imatinib resistant) AML, kidney cancer | FLT3, KIT, PDGFRA | Hematopoietic and lymphoid, Soft Tissue | Acute myeloid leukemia, NS |
| Vemurafenib | Melanoma | BRAF, MAP2K1/2(Mitogen activated protein kinase 1/2), NRAS (Neuroblastoma Ras viral oncogene homolog), PIK3CA(phosphatidylinositol-4,5-bisphosphate 3-kinase alpha), PTEN (Phosphatase and tensin homolog) | Skin, NS | Malignant melanoma, NS |
| Dabrafenib | Melanoma NSCLC with trametinib combination | BRAF, MAP2K1/2, NRAS | Skin, NS | Malignant melanoma |
| Vismodegib | Basal cell carcinoma Gorlin syndrome Small cell lung cancer, other cancers | SMO (Smoothened) | Skin, Central Nervous System, NS | Basal cell carcinoma, NS |
| Selumetinib | Melanoma NSCLC | MAP2K1 | Skin | Malignant melanoma |
| Pembrolizumab | Melanoma NSCLC, other cancers | JAK1/2 (Janus kinase 1/2), NRAS | Skin, NS | Malignant melanoma |
| Endocrine Therapy | Breast cancer | ESR1 (Estrogen receptor alpha) | Breast | ER-positive carcinoma, ductal carcinoma, lobular carcinoma, ductolobular carcinoma |
| Rapamycin | Breast cancer Numerous other diseases Prevention of organ transplant rejection | MTOR (Mammalian target of rapamycin) | Breast | NS |
| PD0325901 | Breast, GISTs Various other cancers | MAP2K1/2 | Breast, Large Intestine | Adenocarcinoma, NS |
| Androgen Ablation | Prostate cancer | Androgen Receptor | Prostate | Adenocarcinoma, NS |
| Abiraterone | Castration resistant prostate cancer | Androgen Receptor | Prostate | Adenocarcinoma, NS |
| Ketoconazole, LHRH (Luteinizing hormone releasing hormone) | Castration resistant prostate cancer | Androgen Receptor | Prostate | NS |
| Enzalutamide | Castration resistant prostate cancer | Androgen Receptor | Prostate | Adenocarcinoma, NS |
| Flutamide | Castration resistant prostate cancer | Androgen Receptor | Prostate | NS |
| Infigratinib (BGJ398) | Biliary tract cancer with sorafenib resistant mutations | FGFR2 (Fibroblast growth factor receptor 2) | Biliary Tract | Cholangiocarcinoma |
| PF-04217903 | Kidney tumor | MET | Kidney | Papillary renal cell carcinoma |
| Everolimus | Thyroid cancer Other cancers | MTOR | Thyroid | Anaplastic carcinoma |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamid, A.B.; Petreaca, R.C. Secondary Resistant Mutations to Small Molecule Inhibitors in Cancer Cells. Cancers 2020, 12, 927. https://doi.org/10.3390/cancers12040927
Hamid AB, Petreaca RC. Secondary Resistant Mutations to Small Molecule Inhibitors in Cancer Cells. Cancers. 2020; 12(4):927. https://doi.org/10.3390/cancers12040927
Chicago/Turabian StyleHamid, Abdulaziz B., and Ruben C. Petreaca. 2020. "Secondary Resistant Mutations to Small Molecule Inhibitors in Cancer Cells" Cancers 12, no. 4: 927. https://doi.org/10.3390/cancers12040927