Depletion of Trichoplein (TpMs) Causes Chromosome Mis-Segregation, DNA Damage and Chromosome Instability in Cancer Cells

 , , , ,
, , , ,  and
and
Abstract
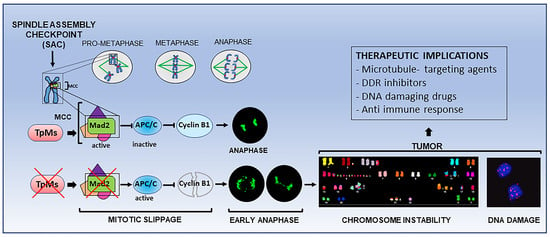
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. TpMs Is Required for Proper Progression through Mitosis and Spindle Checkpoint Activation
2.2. TpMs Depletion Leads to Chromosomal Instability
2.3. TpMs Depletion Induces DNA Damage
2.4. TpMs Depletion Leads to Reduced Expression Level of Mad2 during Mitosis
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Lentivirus and Retrovirus Production, Plasmid Constructs and siRNAs
4.3. Live Cell Imaging
4.4. Spectral Karyotyping
4.5. Immunofluorescence
4.6. Immunoblotting
4.7. Immunoprecipitation
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Thompson, R.; Gatenby, R.; Sidi, S. How cells handle DNA breaks during mitosis: Detection, signaling, repair, and fate choice. Cells 2019, 8, 1049. [Google Scholar] [CrossRef] [PubMed]
- Janssen, A.; Van der Burg, M.; Szuhai, K.; Kops, G.J.P.L.; Medema, R.H. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science 2011, 333, 1895–1898. [Google Scholar] [CrossRef] [PubMed]
- Bakhoum, S.F.; Kabeche, L.; Compton, D.A.; Powell, S.N.; Bastians, H. Mitotic DNA damage response: At the crossroads of structural and numerical cancer chromosome instabilities. Trends Cancer 2017, 3, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Hustedt, N.; Durocher, D. The control of DNA repair by the cell cycle. Nat. Cell Biol. 2016, 19, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, K.S.; Chau, T.; Engebrecht, J. DNA damage response and spindle assembly checkpoint function throughout the cell cycle to ensure genomic integrity. PLoS Genet. 2015, 11. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Davoli, T.; Uno, H.; Wooten, E.C.; Elledge, S.J. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 2017, 355. [Google Scholar] [CrossRef]
- Nishizawa, M.; Izawa, I.; Inoko, A.; Hayashi, Y.; Nagata, K.; Yokoyama, T.; Usukura, J.; Inagaki, M. Identification of trichoplein, a novel keratin filament-binding protein. J. Cell Sci. 2005, 118, 1081–1090. [Google Scholar] [CrossRef]
- Cerqua, C.; Anesti, V.; Pyakurel, A.; Liu, D.; Naon, D.; Wiche, G.; Baffa, R.; Dimmer, K.S.; Scorrano, L. Trichoplein/mitostatin regulates endoplasmic reticulum-mitochondria juxtaposition. EMBO Rep. 2010, 11, 854–860. [Google Scholar] [CrossRef]
- Vecchione, A.; Fassan, M.; Anesti, V.; Morrione, A.; Goldoni, S.; Baldassarre, G.; Byrne, D.; D’Arca, D.; Palazzo, J.P.; Lloyd, J.; et al. MITOSTATIN, a putative tumor suppressor on chromosome 12q24.1, is downregulated in human bladder and breast cancer. Oncogene 2009, 28, 257–269. [Google Scholar] [CrossRef]
- Buraschi, S.; Neill, T.; Iozzo, R.V. Decorin is a devouring proteoglycan: Remodeling of intracellular catabolism via autophagy and mitophagy. Matrix Biol. 2019, 75–76, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Martello, A.; Lauriola, A.; Mellis, D.; Parish, E.; Dawson, J.C.; Imrie, L.; Vidmar, M.; Gammoh, N.; Mitic, T.; Brittan, M.; et al. Trichoplein binds PCM1 and controls endothelial cell function by regulating autophagy. BioRxiv 2019. [Google Scholar] [CrossRef]
- Fassan, M.; D’Arca, D.; Letko, J.; Vecchione, A.; Gardiman, M.P.; McCue, P.; Wildemore, B.; Rugge, M.; Shupp-Byrne, D.; Gomella, L.G.; et al. Mitostatin is down-regulated in human prostate cancer and suppresses the invasive phenotype of prostate cancer cells. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Inoko, A.; Matsuyama, M.; Goto, H.; Ohmuro-Matsuyama, Y.; Hayashi, Y.; Enomoto, M.; Ibi, M.; Urano, T.; Yonemura, S.; Kiyono, T.; et al. Trichoplein and aurora a block aberrant primary cilia assembly in proliferating cells. J. Cell Biol. 2012, 197, 391–405. [Google Scholar] [CrossRef] [PubMed]
- Ibi, M.; Zou, P.; Inoko, A.; Shiromizu, T.; Matsuyama, M.; Hayashi, Y.; Enomoto, M.; Mori, D.; Hirotsune, S.; Kiyono, T.; et al. Trichoplein controls microtubule anchoring at the centrosome by binding to Odf2 and ninein. J. Cell Sci. 2011, 124, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Roschke, A.V.; Stover, K.; Tonon, G.; Schäffer, A.A.; Kirsch, I.R. Stable karyotypes in epithelial cancer cell lines despite high rates of ongoing structural and numerical chromosomal instability. Neoplasia 2002, 4, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Schröck, E.; du Manoir, S.; Veldman, T.; Schoell, B.; Wienberg, J.; Ferguson-Smith, M.A.; Ning, Y.; Ledbetter, D.H.; Bar-Am, I.; Soenksen, D.; et al. Multicolor spectral karyotyping of human chromosomes. Science 1996, 273, 494–497. [Google Scholar] [CrossRef]
- Karpf, A.R.; Matsui, S.I. Genetic disruption of cytosine DNA methyltransferase enzymes induces chromosomal instability in human cancer cells. Cancer Res. 2005, 65, 8635–8639. [Google Scholar] [CrossRef]
- Sharma, A.; Singh, K.; Almasan, A. Histone H2AX phosphorylation: A marker for DNA damage. Methods Mol. Biol. 2012, 920, 613–626. [Google Scholar]
- Rieder, C.L.; Cole, R.W.; Khodjakov, A.; Sluder, G. The checkpoint delaying anaphase in response to chromosome monoorientation is mediated by an inhibitory signal produced by unattached kinetochores. J. Cell Biol. 1995, 130, 941–948. [Google Scholar] [CrossRef]
- Musacchio, A.; Salmon, E.D. The spindle-assembly checkpoint in space and time. Nat. Rev. Mol. Cell Biol. 2007, 8, 379–393. [Google Scholar] [CrossRef]
- Michel, L.; Diaz-Rodriguez, E.; Narayan, G.; Hernando, E.; Murty, V.V.; Benezra, R. Complete loss of the tumor suppressor MAD2 causes premature cyclin B degradation and mitotic failure in human somatic cells. Proc. Natl. Acad. Sci. USA 2004, 101, 4459–4464. [Google Scholar] [CrossRef]
- Michel, L.S.; Liberal, V.; Chatterjee, A.; Kirchwegger, R.; Pasche, B.; Gerald, W.; Dobles, M.; Sorger, P.K.; Murty, V.V.; Benezra, R.; et al. MAD2 haplo-insufficiency causes premature anaphase and chromosome instability in mammalian cells. Nature 2001, 409, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Ittel, A.; Gervais, C.; Galoisy, A.C.; M’Kacher, R.; Mauvieux, L.; Fornecker, L.M. Triradial and quadriradial chromosomes detected in a case of B-cell prolymphocytic leukaemia. Br. J. Haematol. 2017, 179, 704. [Google Scholar] [CrossRef] [PubMed]
- Funk, L.C.; Zasadil, L.M.; Weave, B.A. Living in CIN: Mitotic infidelity and its consequences for tumor promotion and suppression. Dev. Cell 2016, 39, 638–652. [Google Scholar] [CrossRef]
- Crasta, K.; Neil, J.; Ganem, N.J.; Regina Dagher, R.; Lantermann, A.B.; Ivanova, E.V.; Pan, Y.; Nezi, L.; Protopopov, A.; Chowdhury, D.; et al. DNA breaks and chromosome pulverization from errors in mitosis. Nature 2012, 482, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA damage in micronuclei. Nature 2015, 522, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Mai, S. Initiation of telomere-mediated chromosomal rearrangements in cancer. J. Cell Biochem. 2010, 109, 1095–1102. [Google Scholar] [CrossRef]
- Mai, S. The three-dimensional cancer nucleus. Genes Chromosom. Cancer 2019, 58, 462–473. [Google Scholar] [CrossRef]
- Orthwein, A.; Fradet-Turcotte, A.; Noordermeer, S.M.; Canny, M.D.; Brun, C.M.; Strecker, J.; Escribano-Diaz, C.; Durocher, D. Mitosis inhibits DNA double-strand break repair to guard against telomere fusions. Science 2014, 344, 189–193. [Google Scholar] [CrossRef]
- Bakhoum, S.F.; Kabeche, L.; Murnane, J.P.; Zaki, B.I.; Compton, D.A. DNA damage response during mitosis induces whole chromosome mis-segregation. Cancer Discov. 2014, 4, 1281–1289. [Google Scholar] [CrossRef] [PubMed]
- Vossen, M.L.; Alhosawi, H.M.; Aney, K.J.; Burrack, L.S. CaMad2 promotes multiple aspects of genome stability beyond its direct function in chromosome segregation. Genes 2019, 10, 1013. [Google Scholar] [CrossRef] [PubMed]
- Hayward, D.; Alfonso-Perez, T.; Ulrike Gruneberg, U. Orchestration of the spindle assembly checkpoint by CDK1-cyclin B1. FEBS Lett. 2019, 593, 2889–2907. [Google Scholar] [CrossRef]
- Meraldi, P.; Draviam, V.M.; Sorger, P.K. Timing and checkpoints in the regulation of mitotic progression. Dev. Cell 2004, 7, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Howell, B.J.; Hoffman, D.B.; Fang, G.; Murray, A.W.; Salmon, E.D. Visualization of Mad2 dynamics at kinetochores, along spindle fibers, and at spindle poles in living cells. J. Cell Biol. 2000, 150, 1233–1250. [Google Scholar] [CrossRef]
- Li, J.; Dang, N.; Martinez-Lopez, N.; Jowsey, P.A.; Huang, D.; Lightowlers, R.N.; Gao, F.; Huang, J.Y. M2I-1 disrupts the in vivo interaction between CDC20 and MAD2 and increases the sensitivities of cancer cell lines to anti-mitotic drugs via MCL-1s. Cell Div. 2019, 15, 14. [Google Scholar] [CrossRef]
- Haschka, M.; Karbon, G.; Fava, L.L.; Villunger, A. Perturbing mitosis for anti-cancer therapy: Is cell death the only answer? EMBO Rep. 2018, 19. [Google Scholar] [CrossRef]
- Harding, S.M.; Benci, J.L.; Irianto, J.; Discher, D.E.; Minn, A.J.; Greenberg, R.A. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017, 548, 466–470. [Google Scholar] [CrossRef]
- Zasadil, L.M.; Andersen, K.A.; Yeum, D.; Rocque, G.B.; Wilke, L.G.; Tevaarwerk, A.J.; Raines, R.T.; Burkard, M.E.; Weaver, B.A. Cytotoxicity of paclitaxel in breast cancer is due to chromosome missegregation on multipolar spindles. Sci. Transl. Med. 2014, 6, e229. [Google Scholar] [CrossRef]
- Khanna, A. DNA damage in cancer therapeutics: A boon or a curse? Cancer Res. 2015, 75, 2133–2138. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lauriola, A.; Martello, A.; Fantini, S.; Marverti, G.; Zanocco-Marani, T.; Davalli, P.; Guardavaccaro, D.; Mai, S.; Caporali, A.; D’Arca, D. Depletion of Trichoplein (TpMs) Causes Chromosome Mis-Segregation, DNA Damage and Chromosome Instability in Cancer Cells. Cancers 2020, 12, 993. https://doi.org/10.3390/cancers12040993
Lauriola A, Martello A, Fantini S, Marverti G, Zanocco-Marani T, Davalli P, Guardavaccaro D, Mai S, Caporali A, D’Arca D. Depletion of Trichoplein (TpMs) Causes Chromosome Mis-Segregation, DNA Damage and Chromosome Instability in Cancer Cells. Cancers. 2020; 12(4):993. https://doi.org/10.3390/cancers12040993
Chicago/Turabian StyleLauriola, Angela, Andrea Martello, Sebastian Fantini, Gaetano Marverti, Tommaso Zanocco-Marani, Pierpaola Davalli, Daniele Guardavaccaro, Sabine Mai, Andrea Caporali, and Domenico D’Arca. 2020. "Depletion of Trichoplein (TpMs) Causes Chromosome Mis-Segregation, DNA Damage and Chromosome Instability in Cancer Cells" Cancers 12, no. 4: 993. https://doi.org/10.3390/cancers12040993
APA StyleLauriola, A., Martello, A., Fantini, S., Marverti, G., Zanocco-Marani, T., Davalli, P., Guardavaccaro, D., Mai, S., Caporali, A., & D’Arca, D. (2020). Depletion of Trichoplein (TpMs) Causes Chromosome Mis-Segregation, DNA Damage and Chromosome Instability in Cancer Cells. Cancers, 12(4), 993. https://doi.org/10.3390/cancers12040993