Nelfinavir Inhibits the TCF11/Nrf1-Mediated Proteasome Recovery Pathway in Multiple Myeloma

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
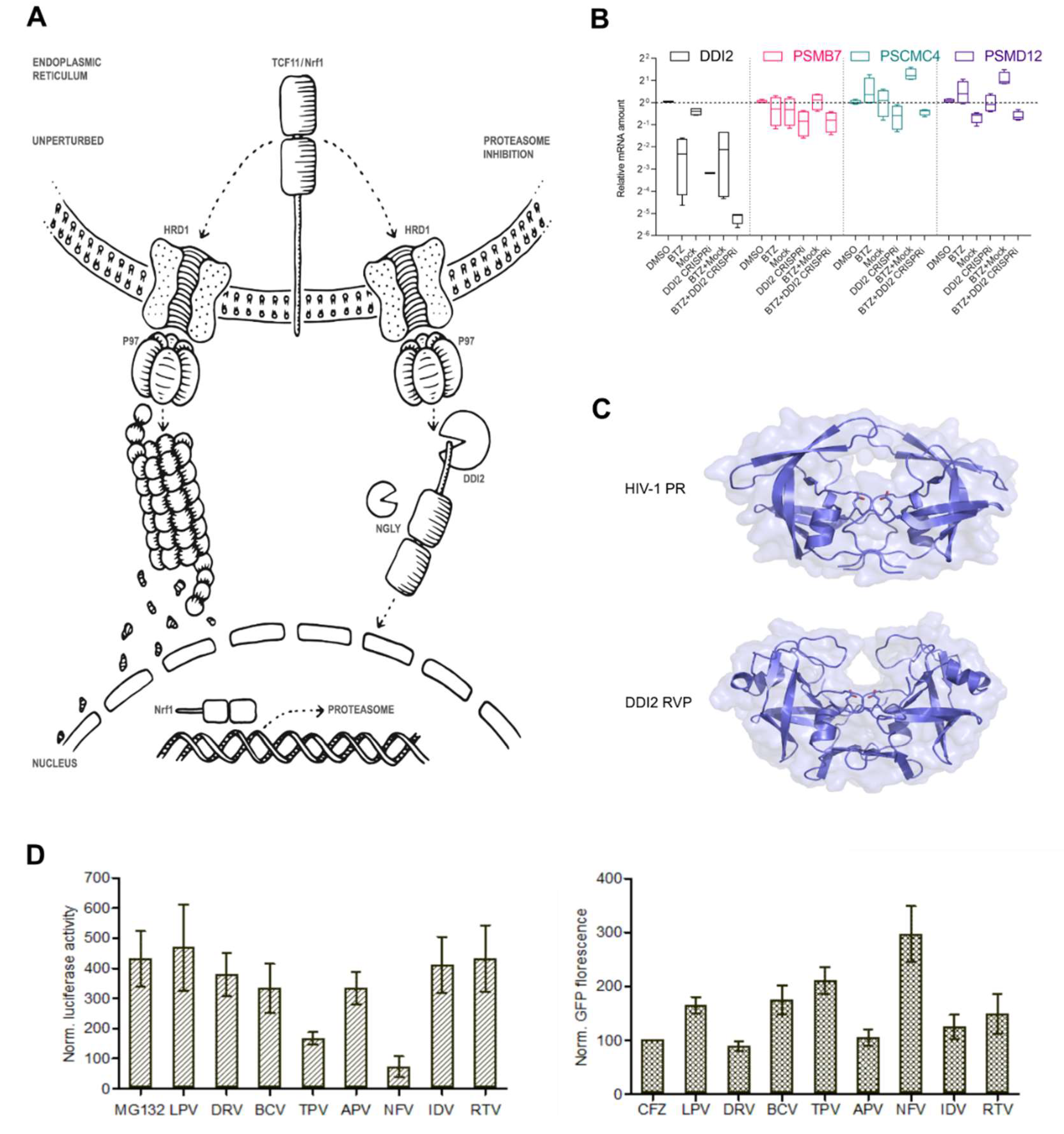
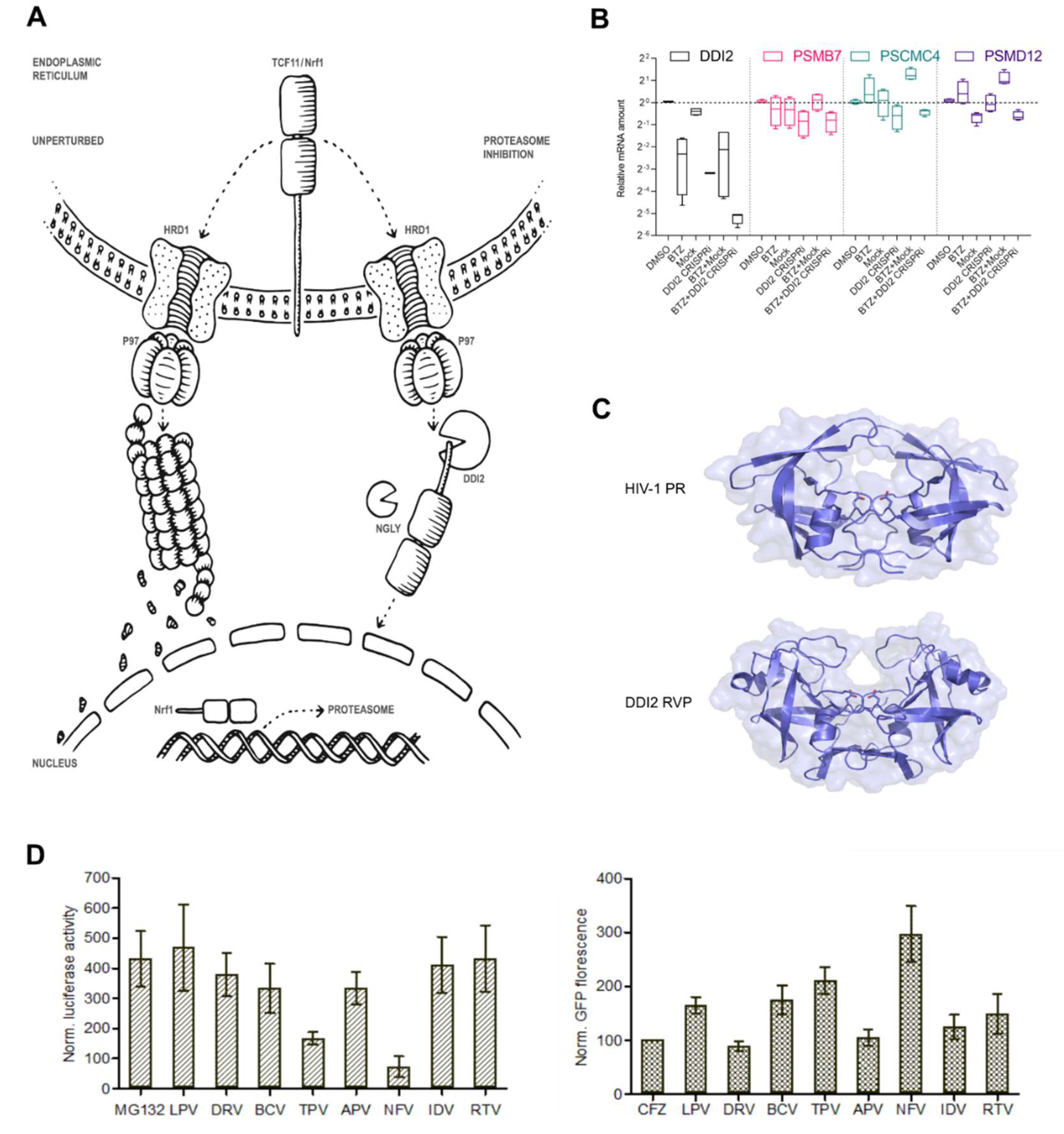
2.1. Efficient Proteasome Re-Synthesis in MM Is Dependent on DDI2 and Can Be Attenuated by HIV PI Nelfinavir
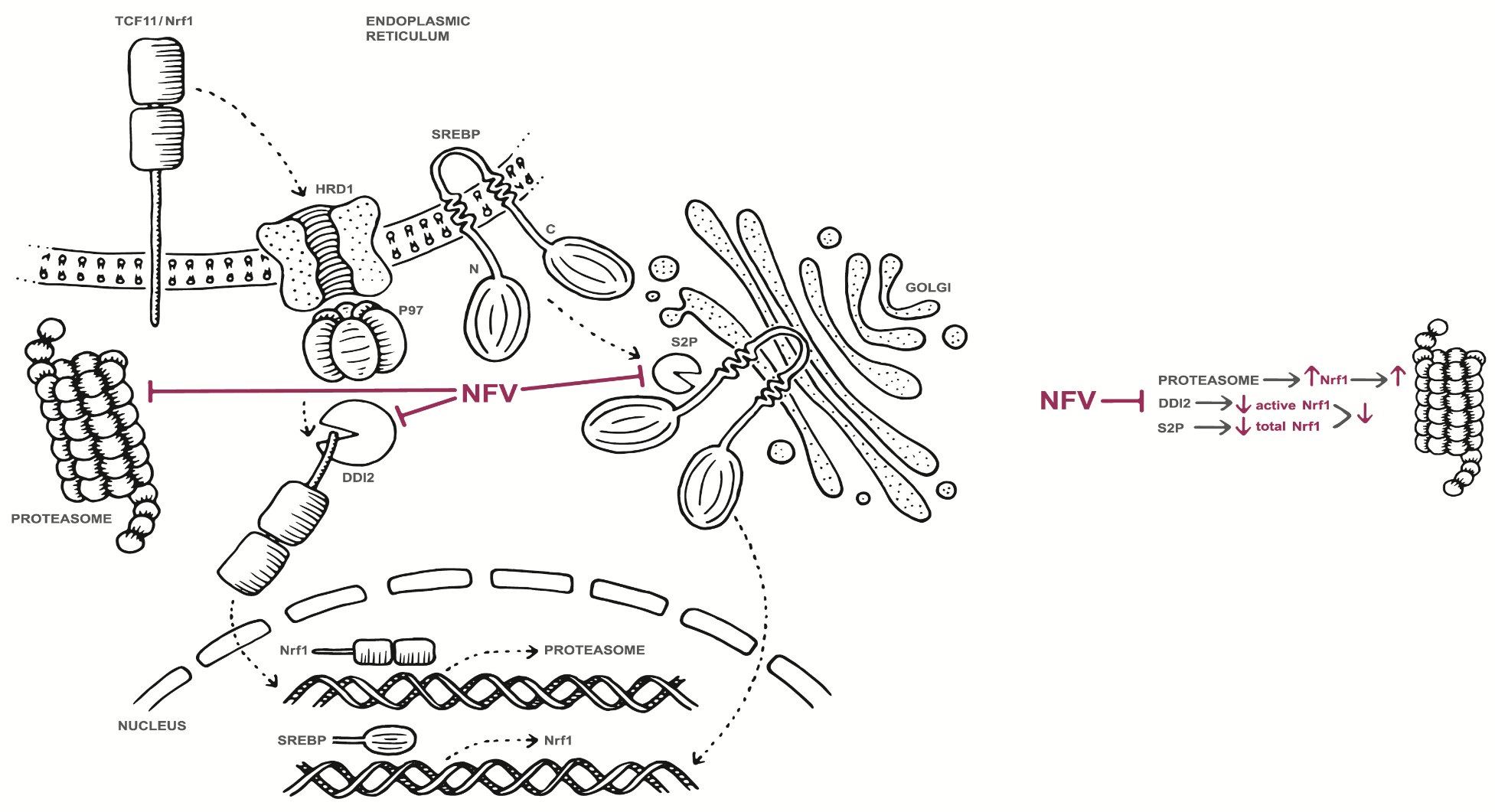
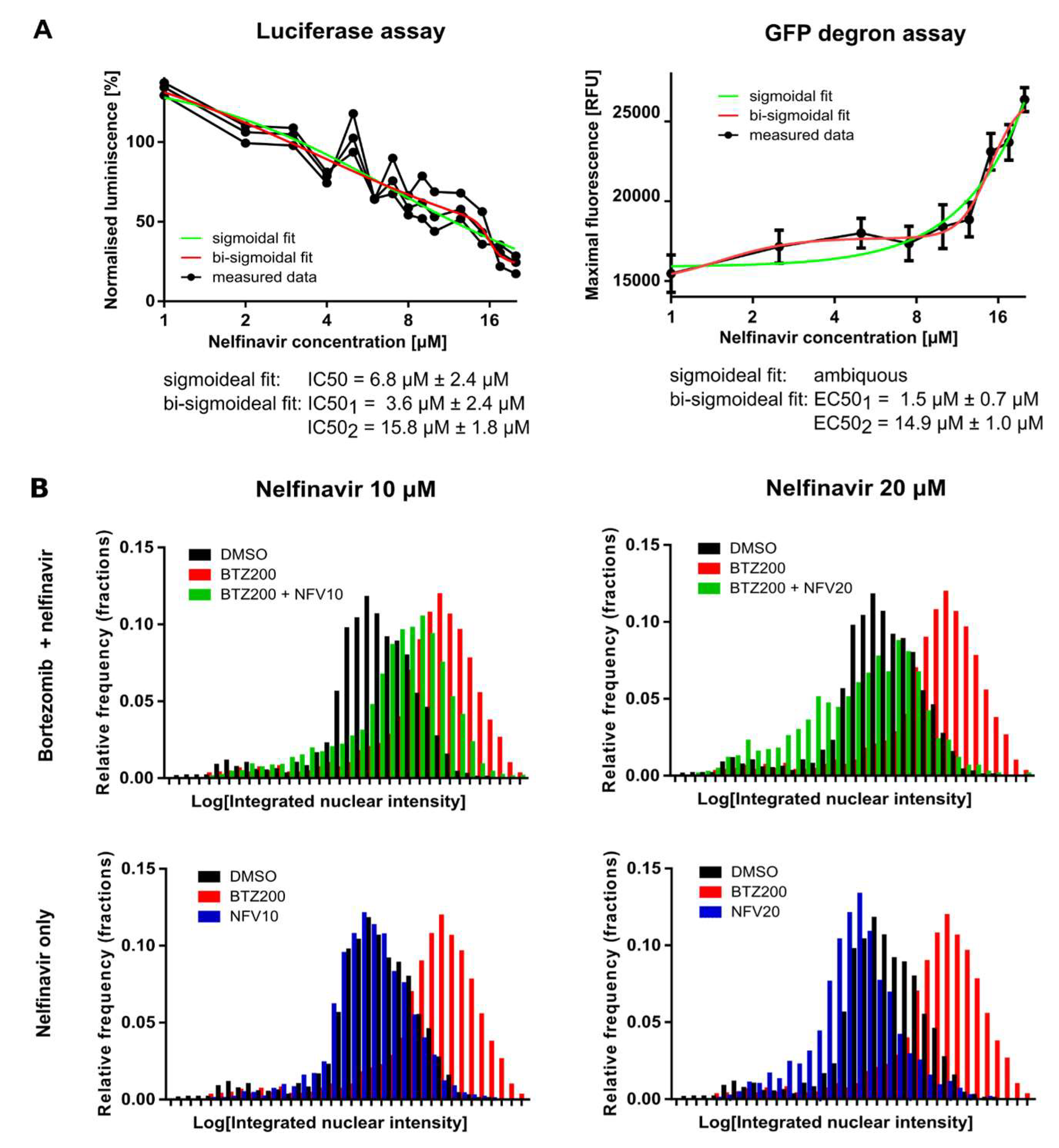
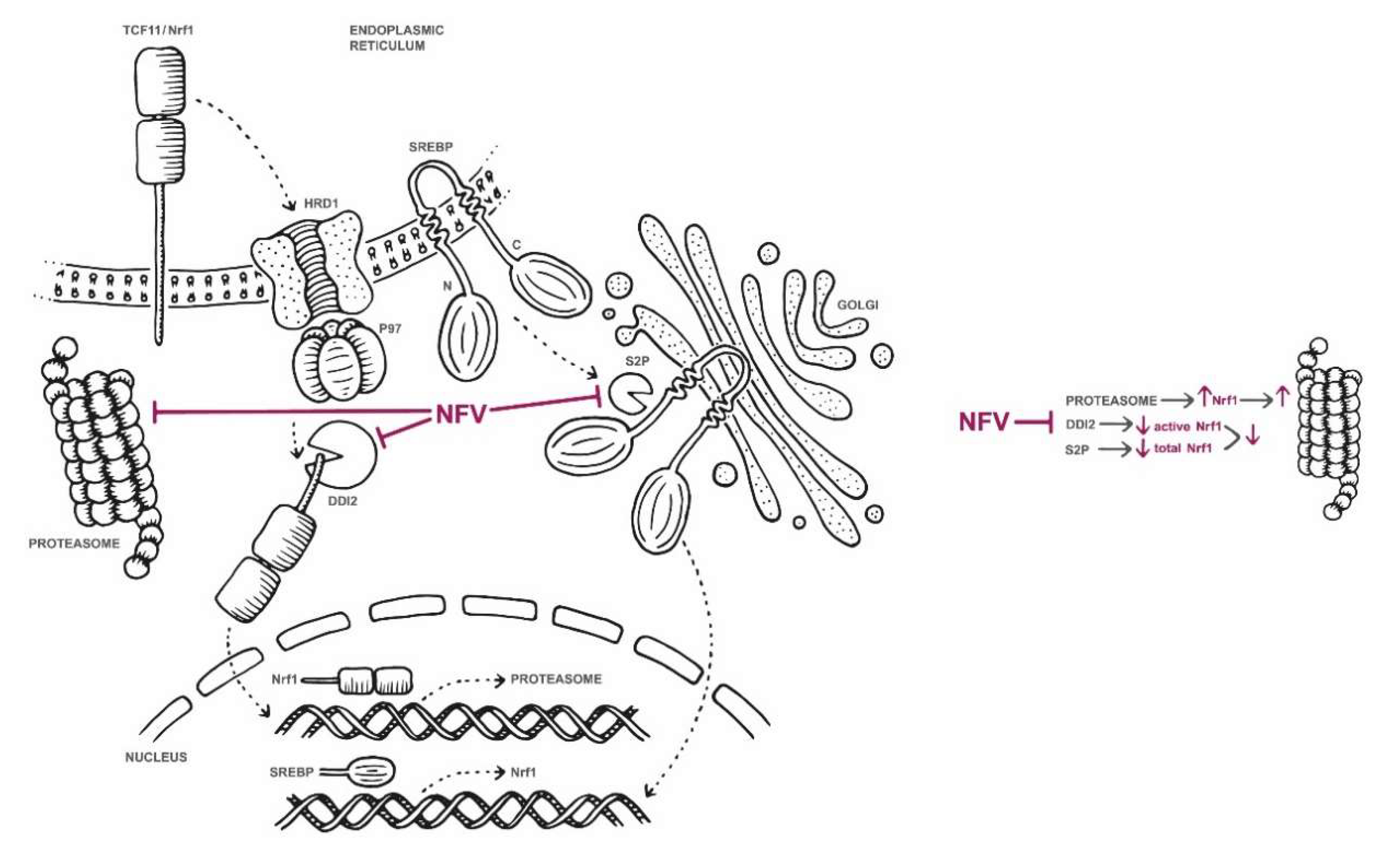
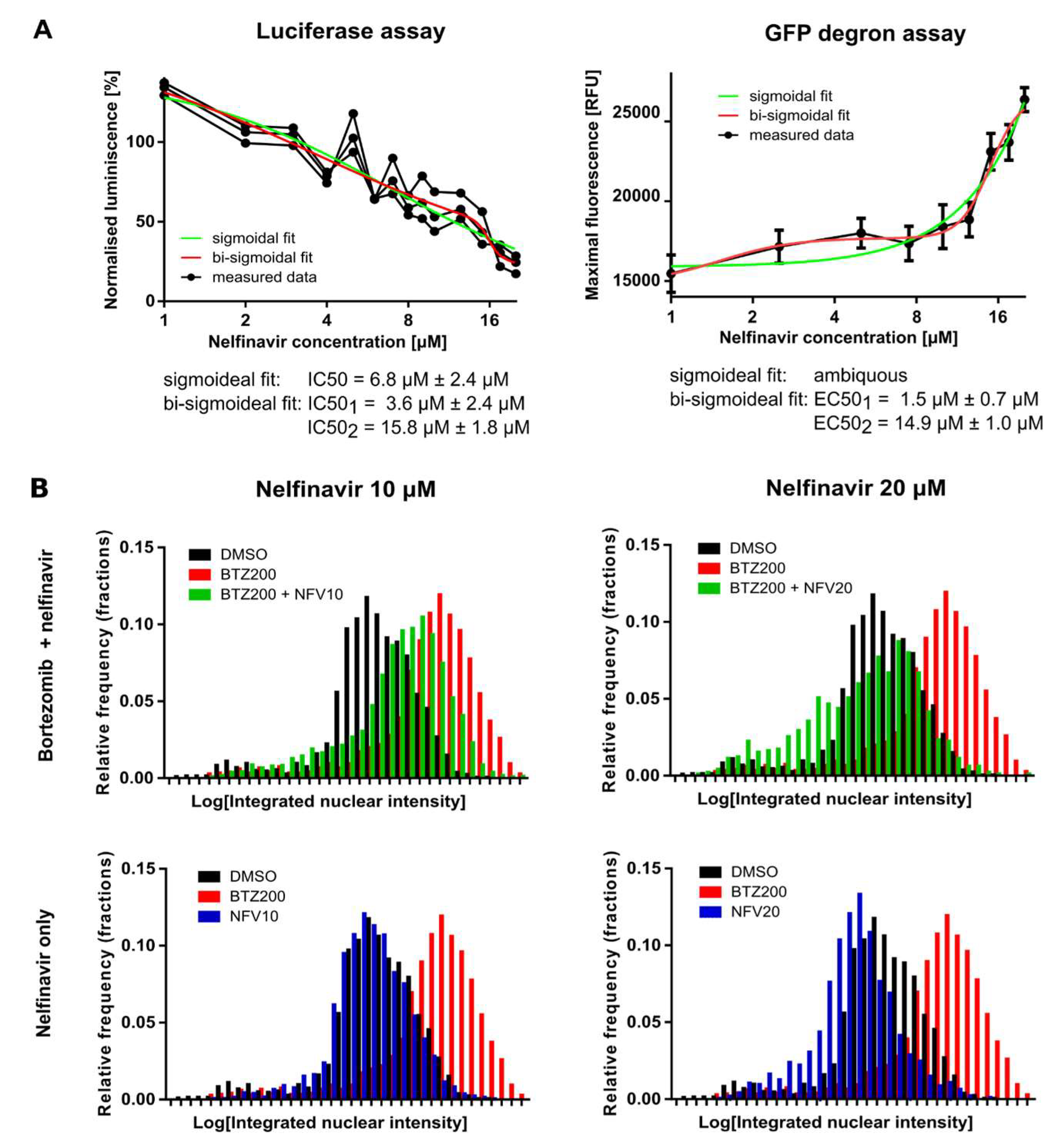
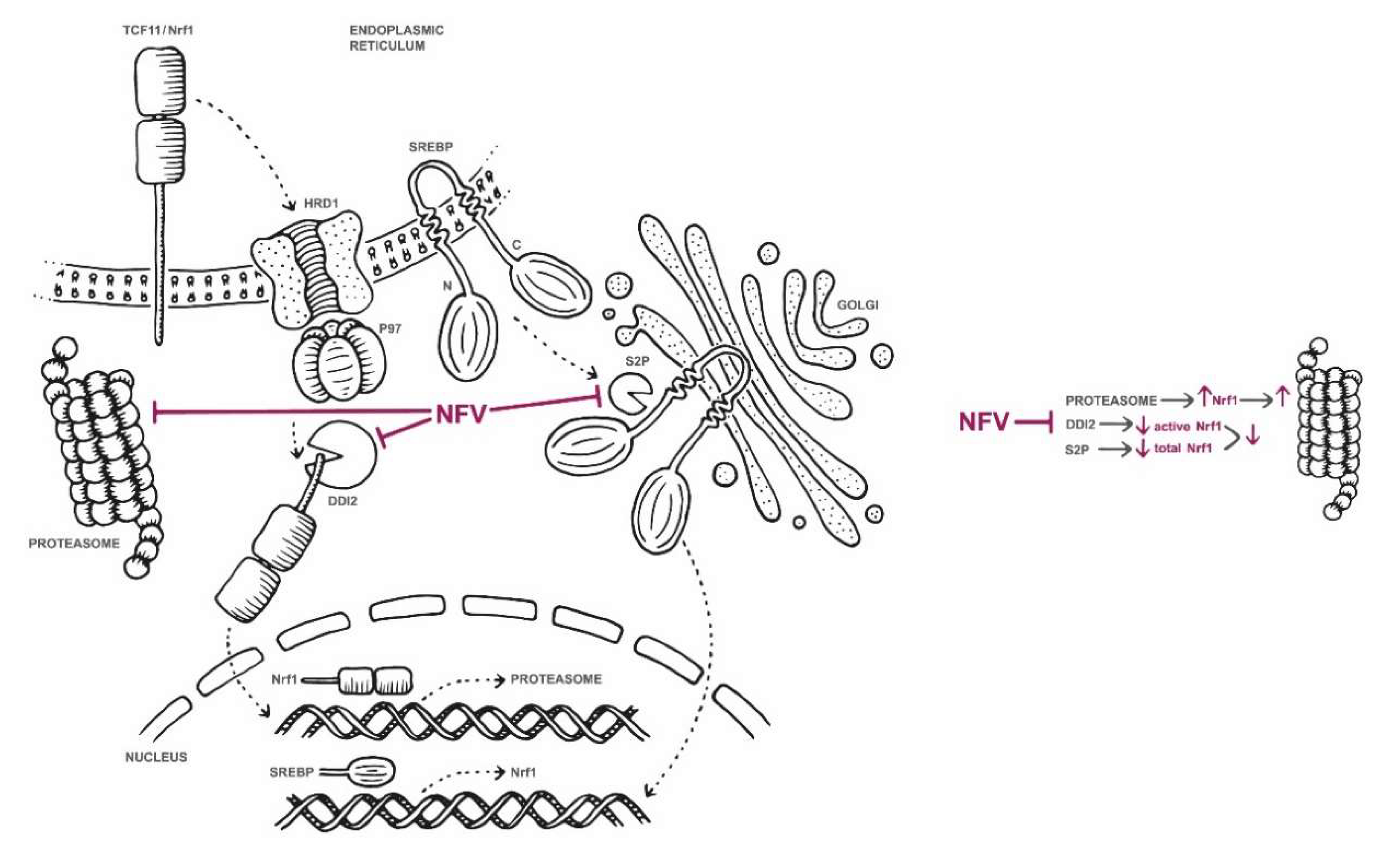
2.2. Nelfinavir Inhibits the TCF11/Nrf1 Pathway by a Dual Mode of Action and Decreases the TCF11/Nrf1 Levels in the Nucleus
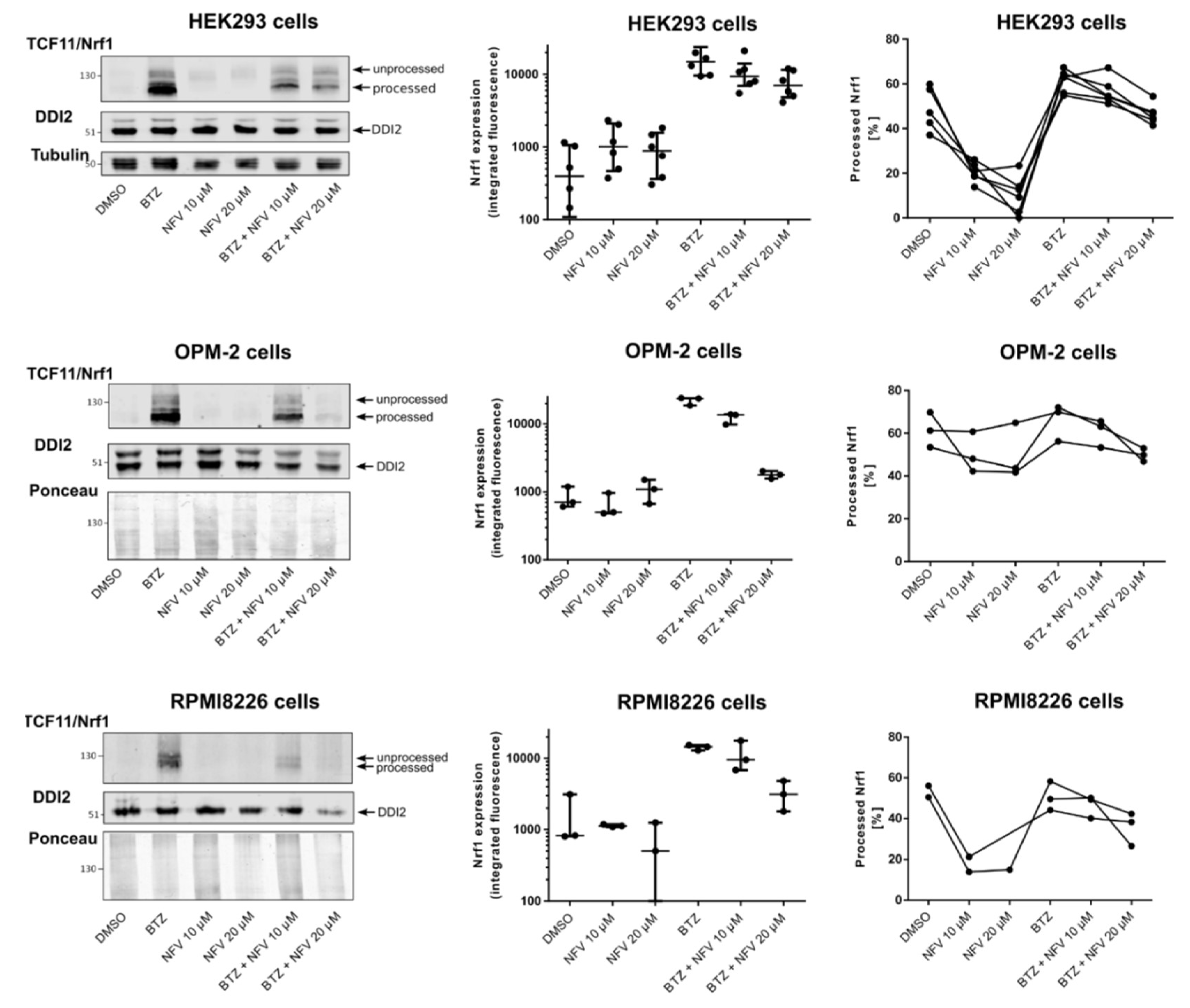
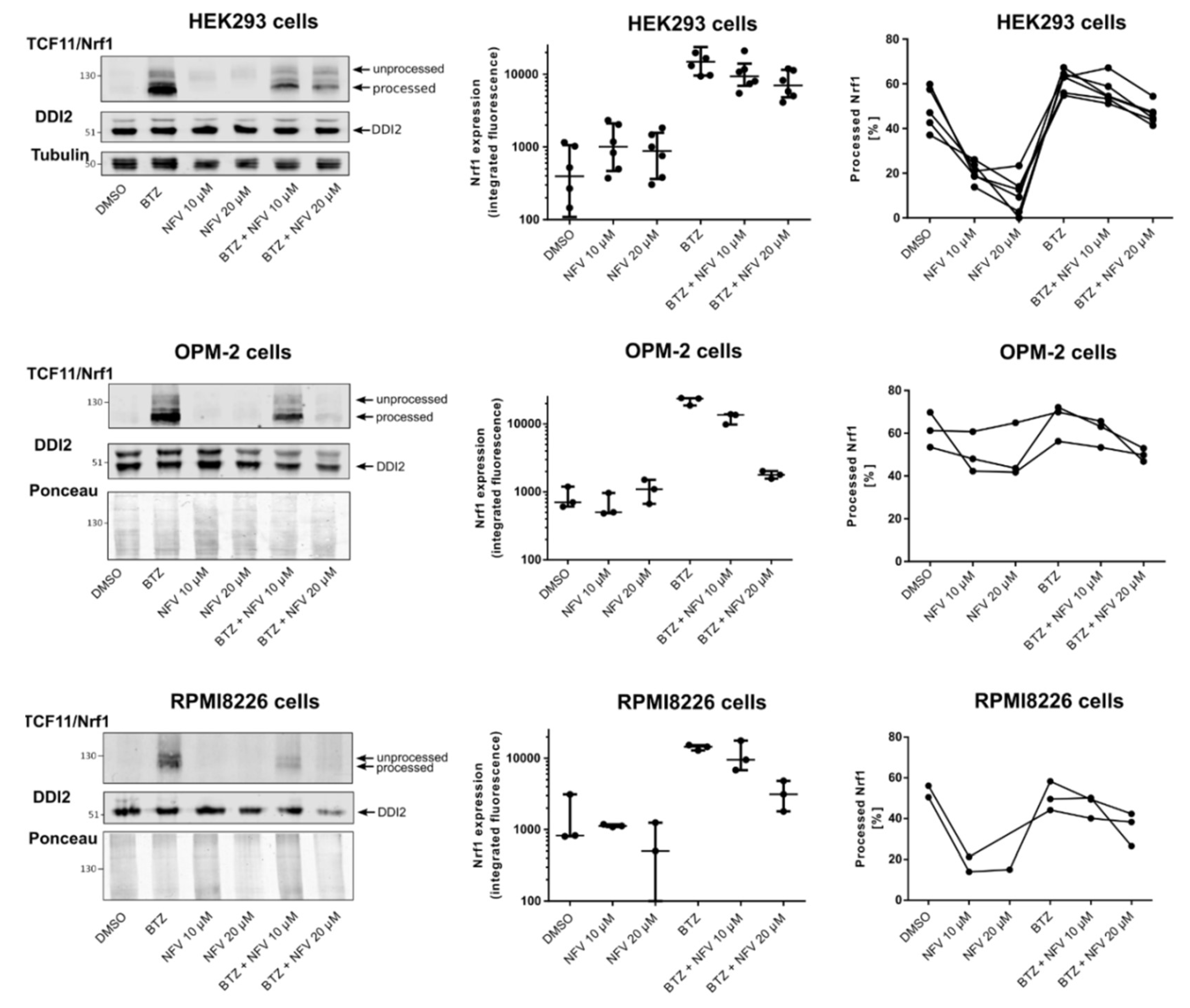
2.3. Nelfinavir Inhibits Proteolytic Processing of TCF11/Nrf1 in MM Cells
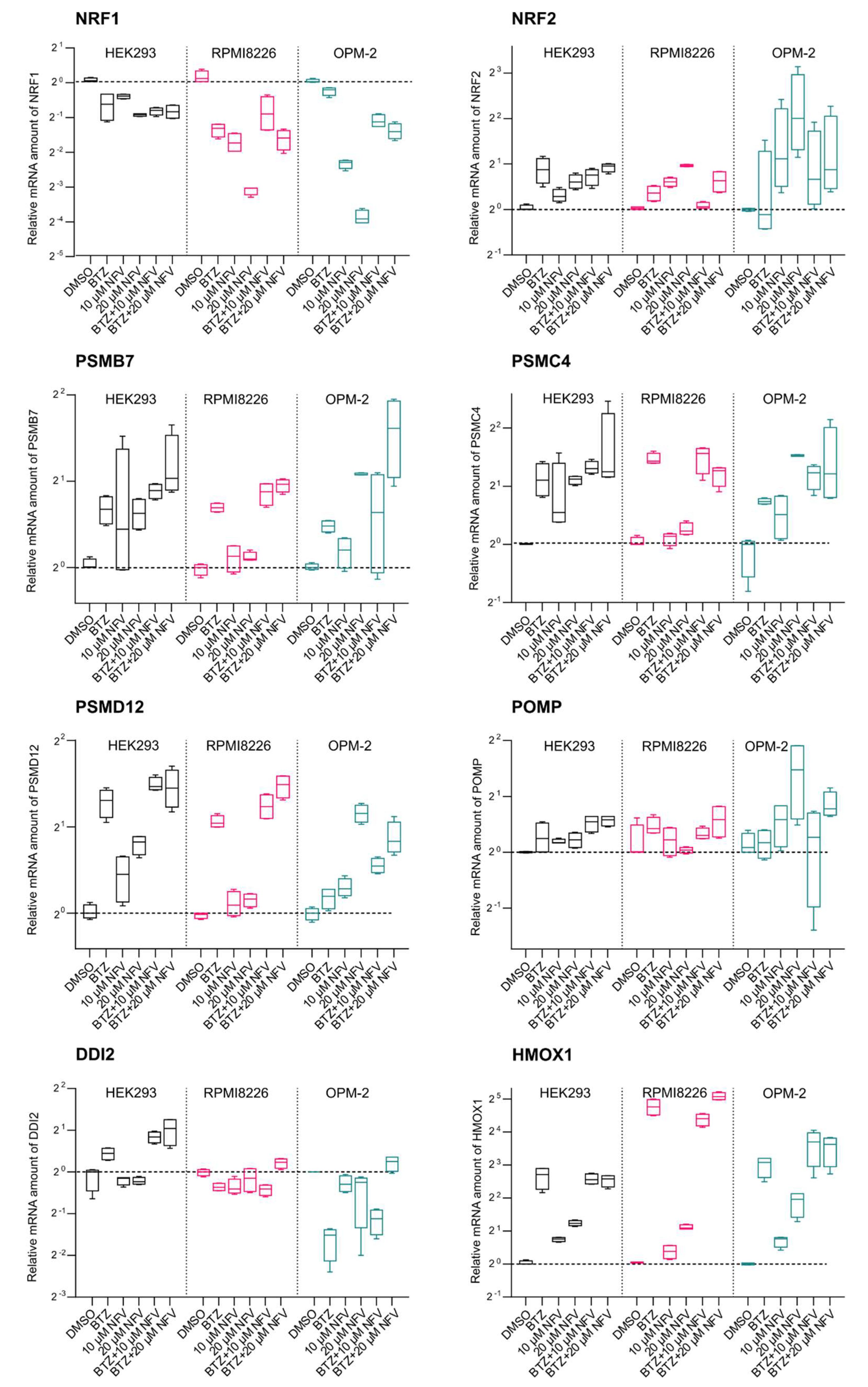
2.4. Nelfinavir Decreases TCF11/Nrf1 Gene Expression and Activates Nrf2
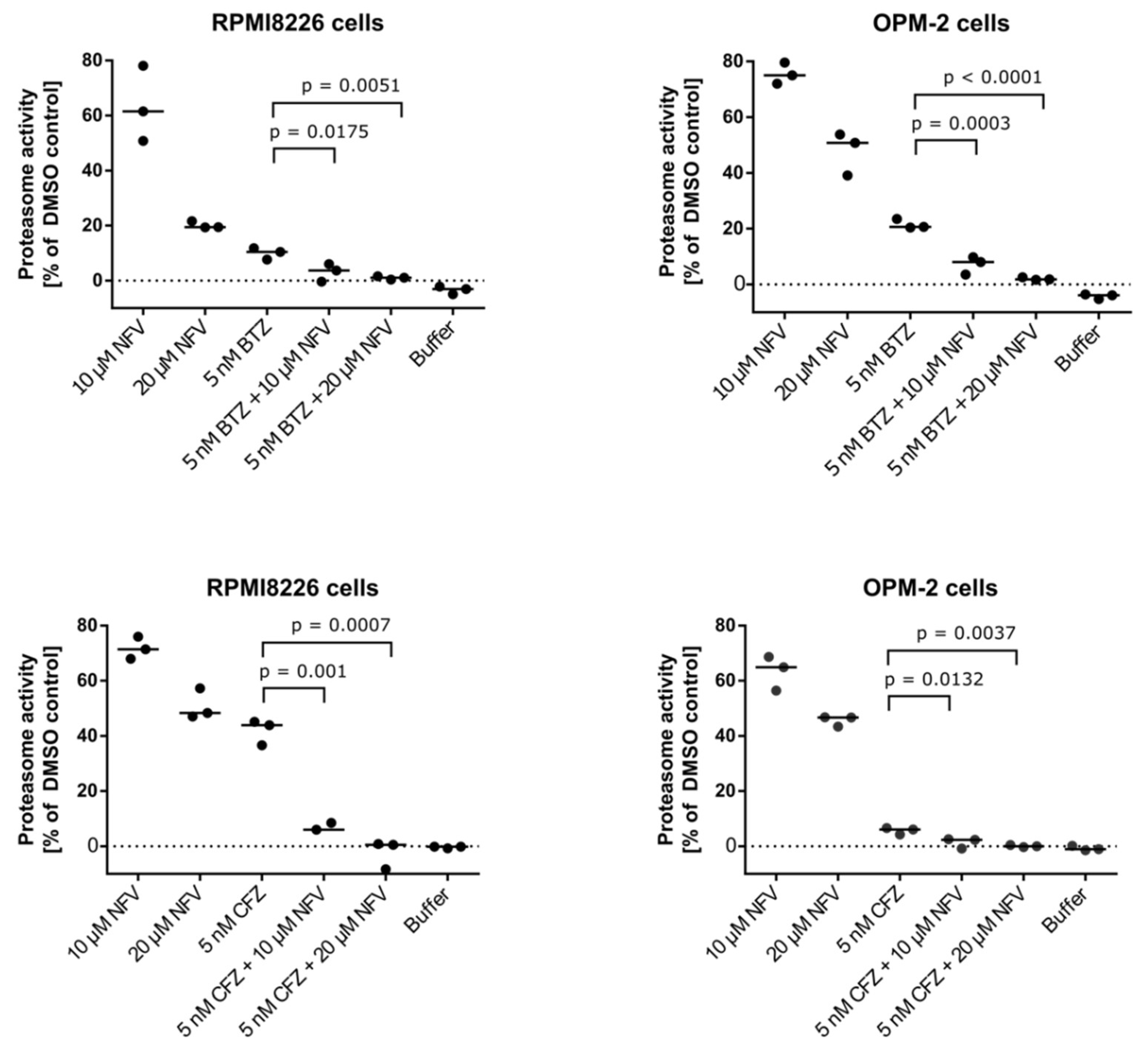
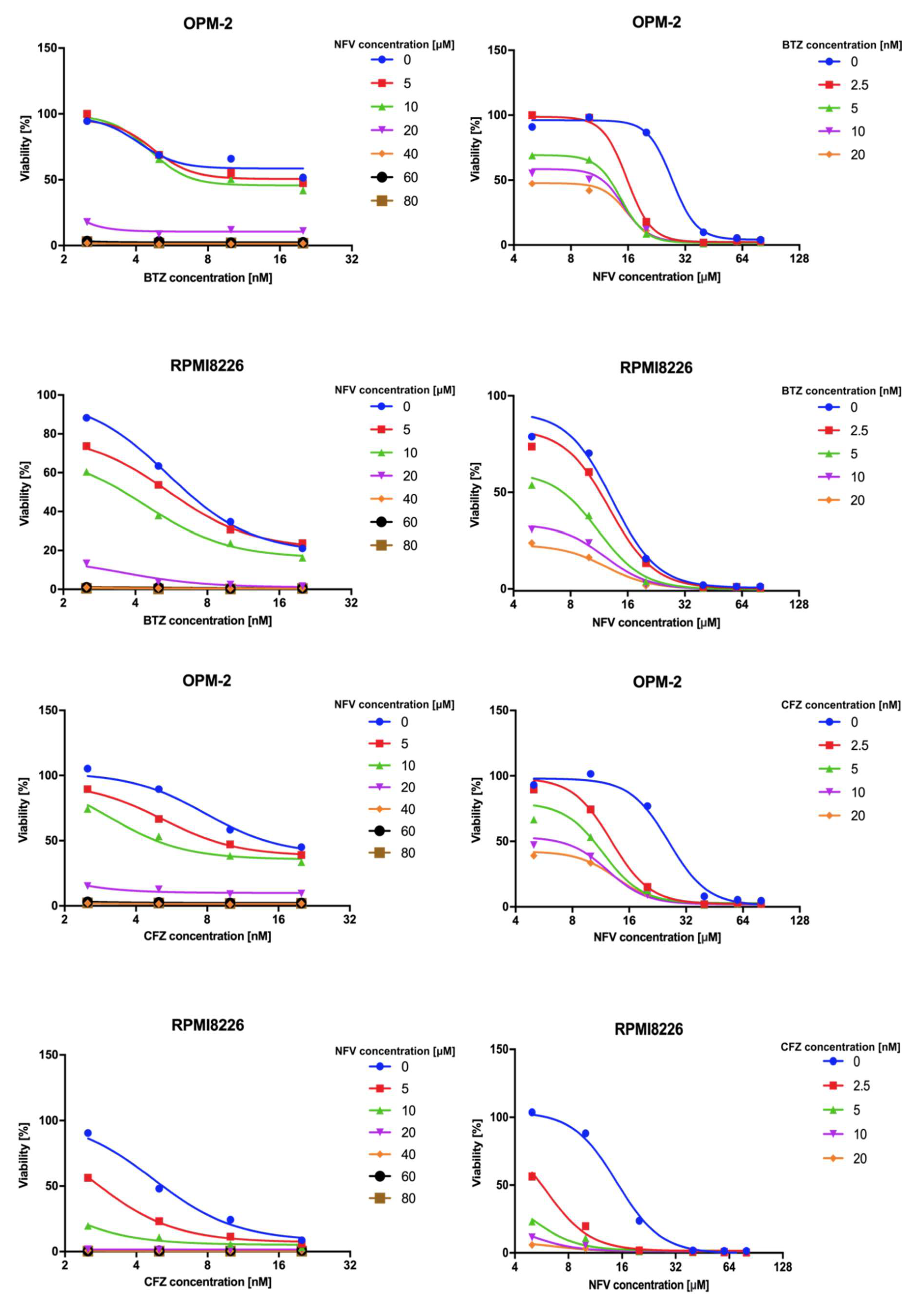
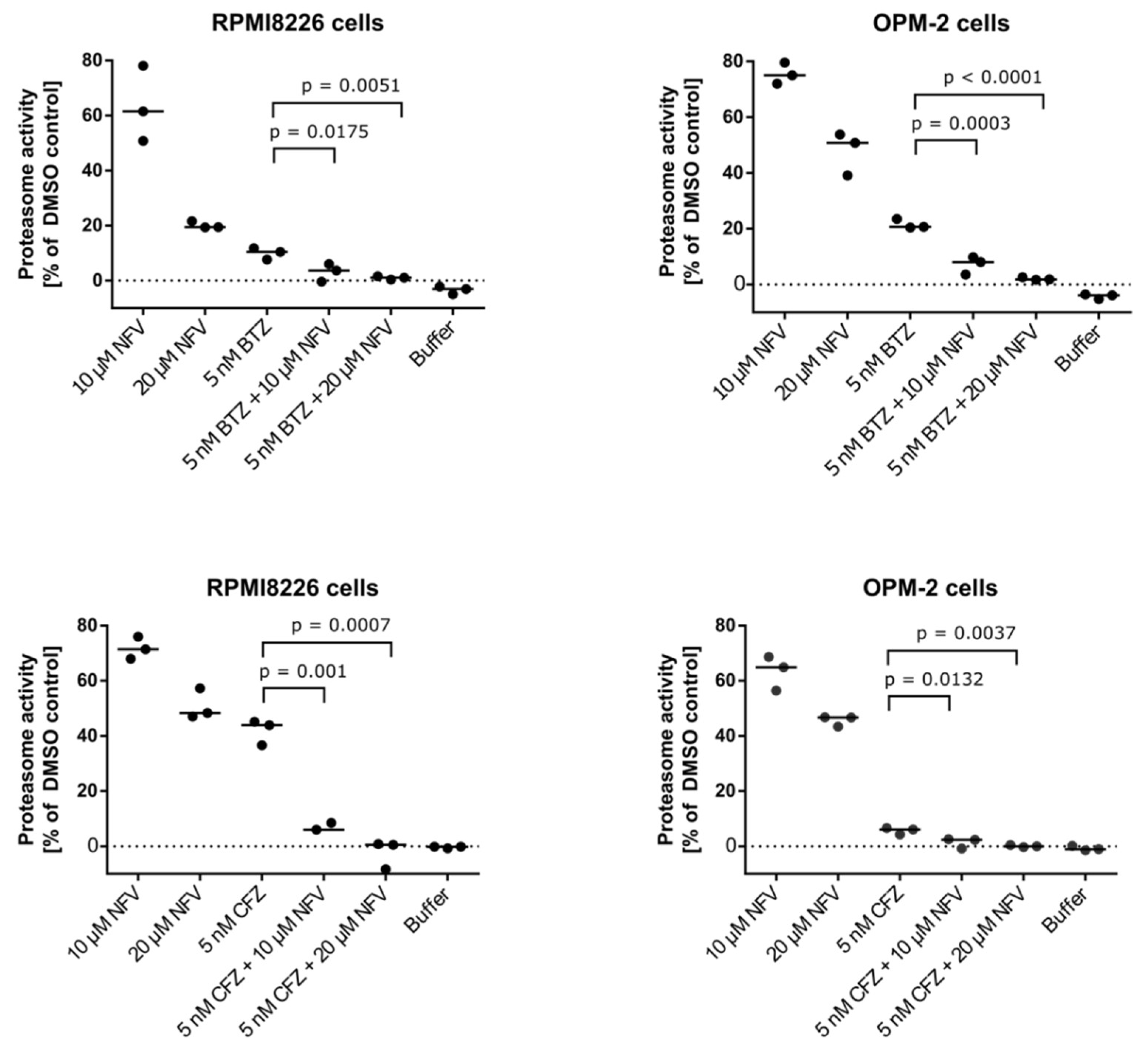
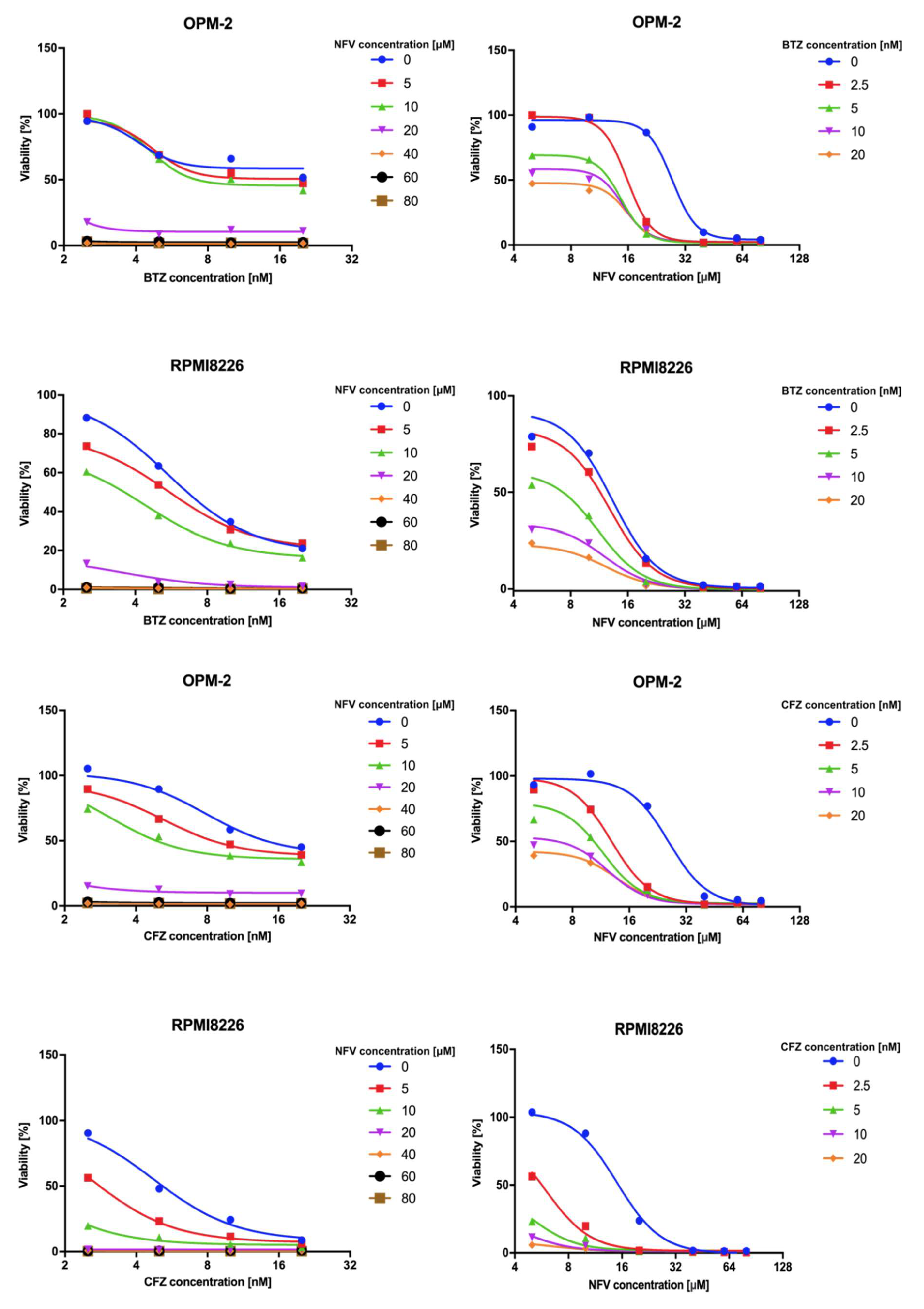
2.5. Nelfinavir Sensitizes MM Cells to BTZ and CFZ Treatment
3. Discussion
4. Materials and Methods
4.1. Constructs
4.2. Cells and Inhibitors
4.3. Lentiviral Transductions
4.4. Luciferase Assays
4.5. N-end Rule GFP Reporter Assays
4.6. Quantitative Reverse Transcription PCR
4.7. Immunoblot Analysis
4.8. Proteasome Activity Assay and Cell Viability Assay
4.9. High-Throughput Confocal Imaging
4.10. Statistical Evaluation and Regression
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Neznanov, N.; Komarov, A.P.; Neznanova, L.; Stanhope-Baker, P.; Gudkov, A.V. Proteotoxic stress targeted therapy (PSTT): Induction of protein misfolding enhances the antitumor effect of the proteasome inhibitor bortezomib. Oncotarget 2011, 2, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obeng, E.A.; Carlson, L.M.; Gutman, D.M.; Harrington, W.J., Jr.; Lee, K.P.; Boise, L.H. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006, 107, 4907–4916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, J. The proteasome: A suitable antineoplastic target. Nat. Rev. Cancer 2004, 4, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Kumar, S. Multiple Myeloma: Diagnosis and Treatment. Mayo Clin. Proc. 2016, 91, 101–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radhakrishnan, S.K.; Lee, C.S.; Young, P.; Beskow, A.; Chan, J.Y.; Deshaies, R.J. Transcription Factor Nrf1 Mediates the Proteasome Recovery Pathway after Proteasome Inhibition in Mammalian Cells. Mol. Cell 2010, 38, 17–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steffen, J.; Seeger, M.; Koch, A.; Kruger, E. Proteasomal degradation is transcriptionally controlled by TCF11 via an ERAD-dependent feedback loop. Mol. Cell 2010, 40, 147–158. [Google Scholar] [CrossRef]
- Radhakrishnan, S.K.; den Besten, W.; Deshaies, R.J. p97-dependent retrotranslocation and proteolytic processing govern formation of active Nrf1 upon proteasome inhibition. Elife 2014, 3, e01856. [Google Scholar] [CrossRef]
- Meiners, S.; Heyken, D.; Weller, A.; Ludwig, A.; Stangl, K.; Kloetzel, P.M.; Kruger, E. Inhibition of proteasome activity induces concerted expression of proteasome genes and de novo formation of Mammalian proteasomes. J. Biol. Chem. 2003, 278, 21517–21525. [Google Scholar] [CrossRef] [Green Version]
- Vangala, J.R.; Sotzny, F.; Kruger, E.; Deshaies, R.J.; Radhakrishnan, S.K. Nrf1 can be processed and activated in a proteasome-independent manner. Curr. Biol. 2016, 26, R834–R835. [Google Scholar] [CrossRef] [Green Version]
- Koizumi, S.; Irie, T.; Hirayama, S.; Sakurai, Y.; Yashiroda, H.; Naguro, I.; Ichijo, H.; Hamazaki, J.; Murata, S. The aspartyl protease DDI2 activates Nrf1 to compensate for proteasome dysfunction. Elife 2016, 5. [Google Scholar] [CrossRef]
- Northrop, A.; Vangala, J.R.; Feygin, A.; Radhakrishnan, S.K. Disabling the Protease DDI2 Attenuates the Transcriptional Activity of NRF1 and Potentiates Proteasome Inhibitor Cytotoxicity. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siva, M.; Svoboda, M.; Veverka, V.; Trempe, J.F.; Hofmann, K.; Kozisek, M.; Hexnerova, R.; Sedlak, F.; Belza, J.; Brynda, J.; et al. Human DNA-Damage-Inducible 2 Protein Is Structurally and Functionally Distinct from Its Yeast Ortholog. Sci. Rep. 2016, 6, 30443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, R.E.; Powell, D.J.; Berry, C. HIV proteinase inhibitors target the Ddi1-like protein of Leishmania parasites. FASEB J. 2011, 25, 1729–1736. [Google Scholar] [CrossRef] [PubMed]
- Gills, J.J.; Lopiccolo, J.; Tsurutani, J.; Shoemaker, R.H.; Best, C.J.; Abu-Asab, M.S.; Borojerdi, J.; Warfel, N.A.; Gardner, E.R.; Danish, M.; et al. Nelfinavir, A lead HIV protease inhibitor, is a broad-spectrum, anticancer agent that induces endoplasmic reticulum stress, autophagy, and apoptosis in vitro and in vivo. Clin. Cancer Res. 2007, 13, 5183–5194. [Google Scholar] [CrossRef] [Green Version]
- Bono, C.; Karlin, L.; Harel, S.; Mouly, E.; Labaume, S.; Galicier, L.; Apcher, S.; Sauvageon, H.; Fermand, J.P.; Bories, J.C.; et al. The human immunodeficiency virus-1 protease inhibitor nelfinavir impairs proteasome activity and inhibits the proliferation of multiple myeloma cells in vitro and in vivo. Haematologica 2012, 97, 1101–1109. [Google Scholar] [CrossRef]
- Brüning, A.; Rahmeh, M.; Gingelmaier, A.; Friese, K. The mitochondria-independent cytotoxic effect of nelfinavir on leukemia cells can be enhanced by sorafenib-mediated mcl-1 downregulation and mitochondrial membrane destabilization. Mol. Cancer 2010, 9. [Google Scholar] [CrossRef]
- Driessen, C.; Kraus, M.; Joerger, M.; Rosing, H.; Bader, J.; Hitz, F.; Berset, C.; Xyrafas, A.; Hawle, H.; Berthod, G.; et al. Treatment with the HIV protease inhibitor nelfinavir triggers the unfolded protein response and may overcome proteasome inhibitor resistance of multiple myeloma in combination with bortezomib: A phase I trial (SAKK 65/08). Haematologica 2016, 101, 346–355. [Google Scholar] [CrossRef]
- Driessen, C.; Muller, R.; Novak, U.; Cantoni, N.; Betticher, D.; Mach, N.; Rufer, A.; Mey, U.; Samaras, P.; Ribi, K.; et al. Promising activity of nelfinavir-bortezomib-dexamethasone in proteasome inhibitor-refractory multiple myeloma. Blood 2018, 132, 2097–2100. [Google Scholar] [CrossRef]
- Guan, M.; Fousek, K.; Jiang, C.; Guo, S.; Synold, T.; Xi, B.; Shih, C.C.; Chow, W.A. Nelfinavir induces liposarcoma apoptosis through inhibition of regulated intramembrane proteolysis of SREBP-1 and ATF6. Clin. Cancer Res. 2011, 17, 1796–1806. [Google Scholar] [CrossRef] [Green Version]
- Guan, M.; Su, L.; Yuan, Y.C.; Li, H.; Chow, W.A. Nelfinavir and nelfinavir analogs block site-2 protease cleavage to inhibit castration-resistant prostate cancer. Sci. Rep. 2015, 5, 9698. [Google Scholar] [CrossRef]
- Gupta, A.K.; Cerniglia, G.J.; Mick, R.; McKenna, W.G.; Muschel, R.J. HIV protease inhibitors block Akt signaling and radiosensitize tumor cells both in vitro and in vivo. Cancer Res. 2005, 65, 8256–8265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plastaras, J.P.; Vapiwala, N.; Ahmed, M.S.; Gudonis, D.; Cerniglia, G.J.; Feldman, M.D.; Frank, I.; Gupta, A.K. Validation and toxicity of PI3K/Akt pathway inhibition by HIV protease inhibitors in humans. Cancer Biol. Ther. 2008, 7, 628–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Ikezoe, T.; Nishioka, C.; Bandobashi, K.; Takeuchi, T.; Adachi, Y.; Kobayashi, M.; Takeuchi, S.; Koeffler, H.P.; Taguchi, H. NFV, an HIV-1 protease inhibitor, induces growth arrest, reduced Akt signalling, apoptosis and docetaxel sensitisation in NSCLC cell lines. Br. J. Cancer 2006, 95, 1653–1662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraus, M.; Bader, J.; Overkleeft, H.; Driessen, C. Nelfinavir augments proteasome inhibition by bortezomib in myeloma cells and overcomes bortezomib and carfilzomib resistance. Blood Cancer J. 2013, 3, e103. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.K.; Li, B.; Cerniglia, G.J.; Ahmed, M.S.; Hahn, S.M.; Maity, A. The HIV protease inhibitor nelfinavir downregulates Akt phosphorylation by inhibiting proteasomal activity and inducing the unfolded protein response. Neoplasia 2007, 9, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Vangala, J.R.; Radhakrishnan, S.K. Nrf1-mediated transcriptional regulation of the proteasome requires a functional TIP60 complex. J. Biol. Chem. 2019, 294, 2036–2045. [Google Scholar] [CrossRef] [Green Version]
- Dantuma, N.P.; Lindsten, K.; Glas, R.; Jellne, M.; Masucci, M.G. Short-lived green fluorescent proteins for quantifying ubiquitin/proteasome-dependent proteolysis in living cells. Nat. Biotechnol. 2000, 18, 538–543. [Google Scholar] [CrossRef]
- Heaslet, H.; Rosenfeld, R.; Giffin, M.; Lin, Y.C.; Tam, K.; Torbett, B.E.; Elder, J.H.; McRee, D.E.; Stout, C.D. Conformational flexibility in the flap domains of ligand-free HIV protease. Acta Crystallogr. D Biol. Crystallogr. 2007, 63, 866–875. [Google Scholar] [CrossRef]
- McQuin, C.; Goodman, A.; Chernyshev, V.; Kamentsky, L.; Cimini, B.A.; Karhohs, K.W.; Doan, M.; Ding, L.; Rafelski, S.M.; Thirstrup, D.; et al. CellProfiler 3.0: Next-generation image processing for biology. PLoS Biol. 2018, 16, e2005970. [Google Scholar] [CrossRef] [Green Version]
- Kawabata, S.; Gills, J.J.; Mercado-Matos, J.R.; Lopiccolo, J.; Wilson, W., 3rd; Hollander, M.C.; Dennis, P.A. Synergistic effects of nelfinavir and bortezomib on proteotoxic death of NSCLC and multiple myeloma cells. Cell Death Dis. 2012, 3, e353. [Google Scholar] [CrossRef]
- Tomlin, F.M.; Gerling-Driessen, U.I.M.; Liu, Y.C.; Flynn, R.A.; Vangala, J.R.; Lentz, C.S.; Clauder-Muenster, S.; Jakob, P.; Mueller, W.F.; Ordonez-Rueda, D.; et al. Inhibition of NGLY1 Inactivates the Transcription Factor Nrf1 and Potentiates Proteasome Inhibitor Cytotoxicity. ACS Cent. Sci. 2017, 3, 1143–1155. [Google Scholar] [CrossRef] [PubMed]
- Onchieku, N.M.; Mogire, R.; Ndung′u, L.; Mwitari, P.; Kimani, F.; Matoke-Muhia, D.; Kiboi, D.; Magoma, G. Deciphering the targets of retroviral protease inhibitors in Plasmodium berghei. PLoS ONE 2018, 13, e0201556. [Google Scholar] [CrossRef] [PubMed]
- Yip, M.C.J.; Bodnar, N.O.; Rapoport, T.A. Ddi1 is a ubiquitin-dependent protease. Proc. Natl. Acad. Sci. USA 2020. [Google Scholar] [CrossRef] [Green Version]
- Hitz, F.; Kraus, M.; Pabst, T.; Hess, D.; Besse, L.; Silzle, T.; Novak, U.; Seipel, K.; Rondeau, S.; Studeli, S.; et al. Nelfinavir and lenalidomide/dexamethasone in patients with lenalidomide-refractory multiple myeloma. A phase I/II Trial (SAKK 39/10). Blood Cancer J. 2019, 9, 70. [Google Scholar] [CrossRef] [PubMed]
- Soprano, M.; Sorriento, D.; Rusciano, M.R.; Maione, A.S.; Limite, G.; Forestieri, P.; D′Angelo, D.; D′Alessio, M.; Campiglia, P.; Formisano, P.; et al. Oxidative Stress Mediates the Antiproliferative Effects of Nelfinavir in Breast Cancer Cells. PLoS ONE 2016, 11, e0155970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruning, A.; Burger, P.; Vogel, M.; Rahmeh, M.; Gingelmaiers, A.; Friese, K.; Lenhard, M.; Burges, A. Nelfinavir induces the unfolded protein response in ovarian cancer cells, resulting in ER vacuolization, cell cycle retardation and apoptosis. Cancer Biol. Ther. 2009, 8, 226–232. [Google Scholar] [CrossRef]
- Ben-Romano, R.; Rudich, A.; Etzion, S.; Potashnik, R.; Kagan, E.; Greenbaum, U.; Bashan, N. Nelfinavir induces adipocyte insulin resistance through the induction of oxidative stress: Differential protective effect of antioxidant agents. Antivir. Ther. 2006, 11, 1051–1060. [Google Scholar]
- Zhang, Y.; Manning, B.D. mTORC1 signaling activates NRF1 to increase cellular proteasome levels. Cell Cycle 2015, 14, 2011–2017. [Google Scholar] [CrossRef] [Green Version]
- Besse, A.; Stolze, S.C.; Rasche, L.; Weinhold, N.; Morgan, G.J.; Kraus, M.; Bader, J.; Overkleeft, H.S.; Besse, L.; Driessen, C. Carfilzomib resistance due to ABCB1/MDR1 overexpression is overcome by nelfinavir and lopinavir in multiple myeloma. Leukemia 2018, 32, 391–401. [Google Scholar] [CrossRef] [Green Version]
- Dyer, B.W.; Ferrer, F.A.; Klinedinst, D.K.; Rodriguez, R. A noncommercial dual luciferase enzyme assay system for reporter gene analysis. Anal. Biochem. 2000, 282, 158–161. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fassmannová, D.; Sedlák, F.; Sedláček, J.; Špička, I.; Grantz Šašková, K. Nelfinavir Inhibits the TCF11/Nrf1-Mediated Proteasome Recovery Pathway in Multiple Myeloma. Cancers 2020, 12, 1065. https://doi.org/10.3390/cancers12051065
Fassmannová D, Sedlák F, Sedláček J, Špička I, Grantz Šašková K. Nelfinavir Inhibits the TCF11/Nrf1-Mediated Proteasome Recovery Pathway in Multiple Myeloma. Cancers. 2020; 12(5):1065. https://doi.org/10.3390/cancers12051065
Chicago/Turabian StyleFassmannová, Dominika, František Sedlák, Jindřich Sedláček, Ivan Špička, and Klára Grantz Šašková. 2020. "Nelfinavir Inhibits the TCF11/Nrf1-Mediated Proteasome Recovery Pathway in Multiple Myeloma" Cancers 12, no. 5: 1065. https://doi.org/10.3390/cancers12051065
APA StyleFassmannová, D., Sedlák, F., Sedláček, J., Špička, I., & Grantz Šašková, K. (2020). Nelfinavir Inhibits the TCF11/Nrf1-Mediated Proteasome Recovery Pathway in Multiple Myeloma. Cancers, 12(5), 1065. https://doi.org/10.3390/cancers12051065