Deactivation of Glutaminolysis Sensitizes PIK3CA-Mutated Colorectal Cancer Cells to Aspirin-Induced Growth Inhibition

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
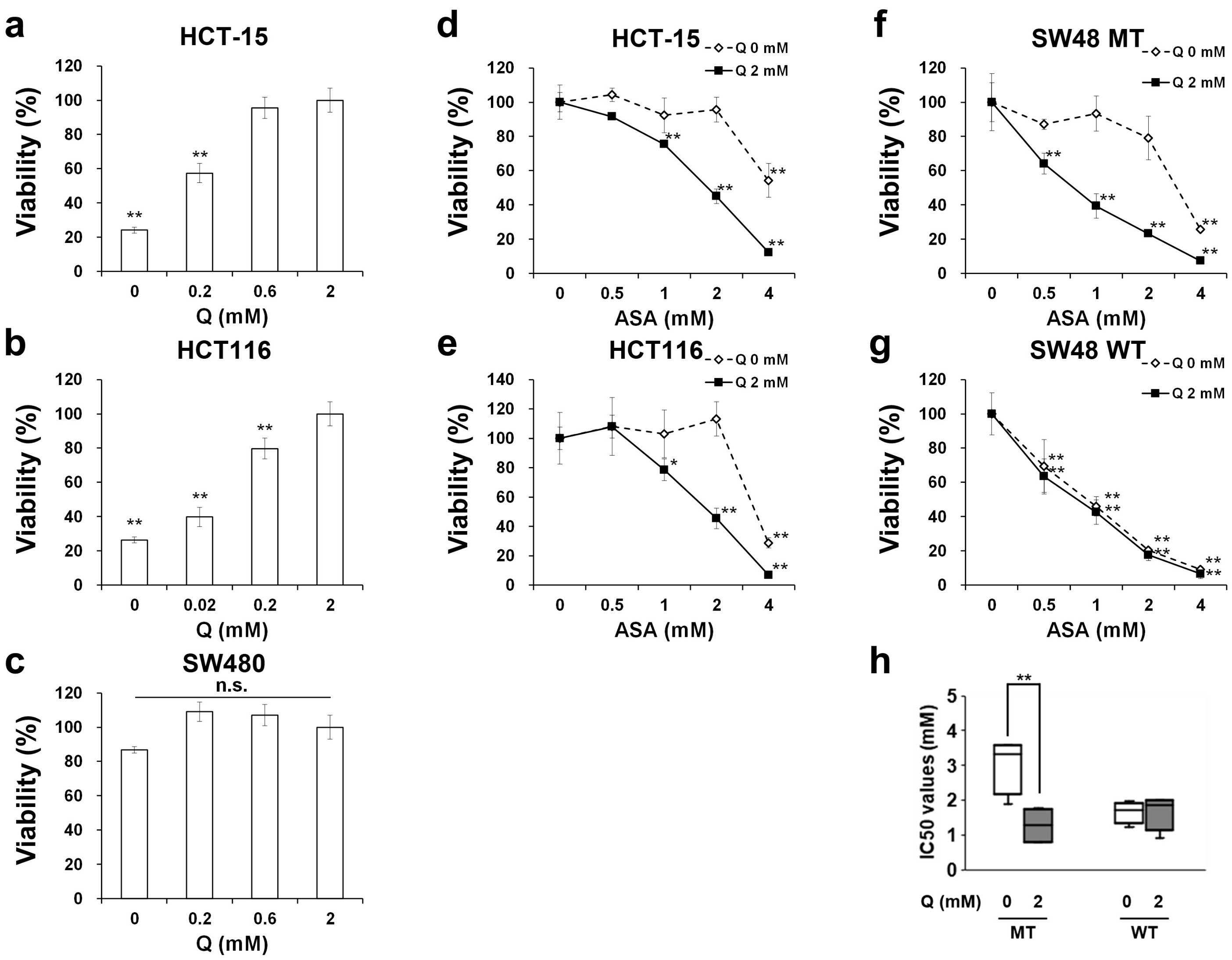
2.1. Aspirin-Mediated Growth Inhibition is Dependent on Glutamine in PIK3CA-Mutated Cells
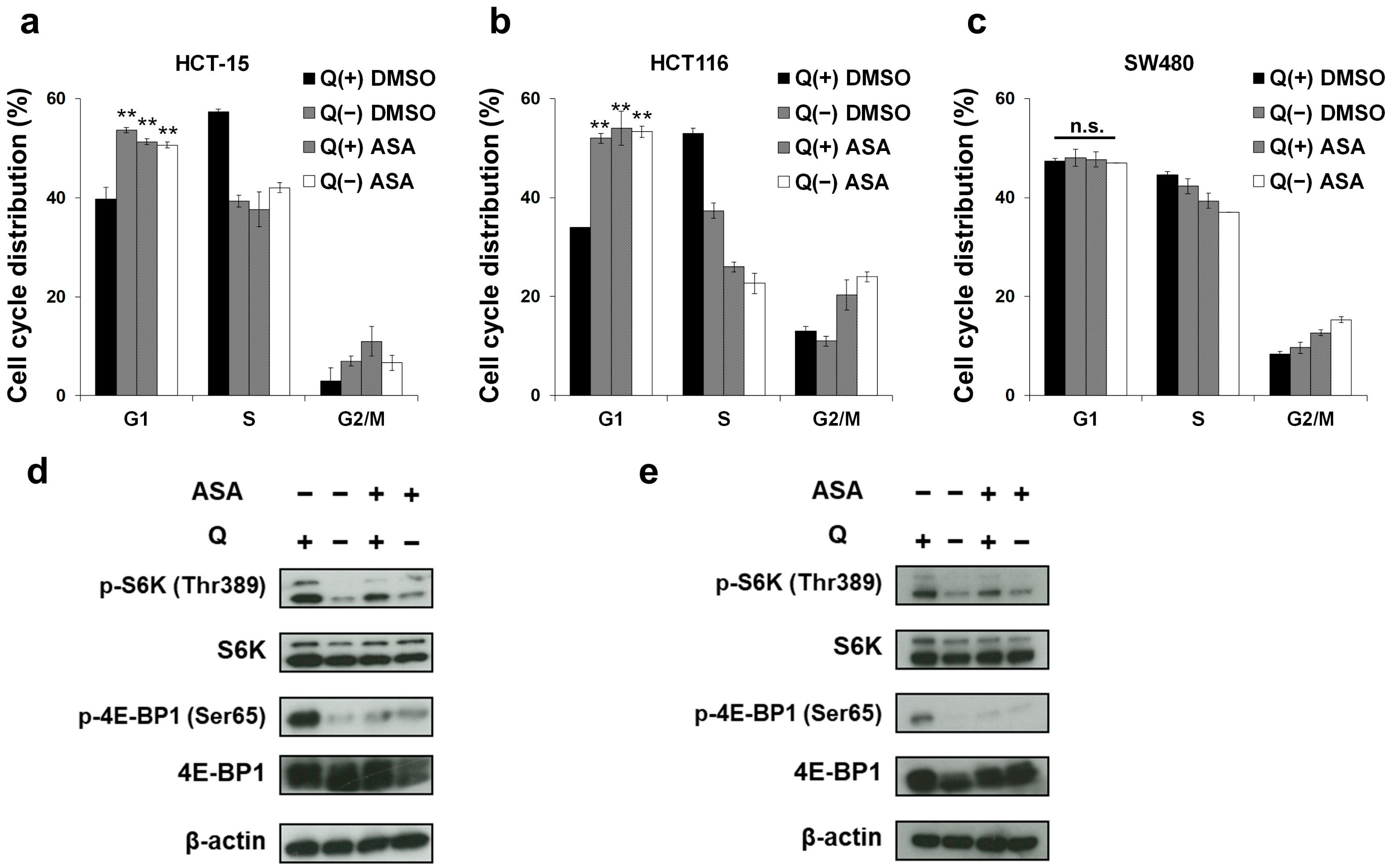
2.2. Glutamine Depletion Mirrors the Effects of Aspirin on the Cell Cycle and the mTOR Pathway in PIK3CA-Mutated Cells
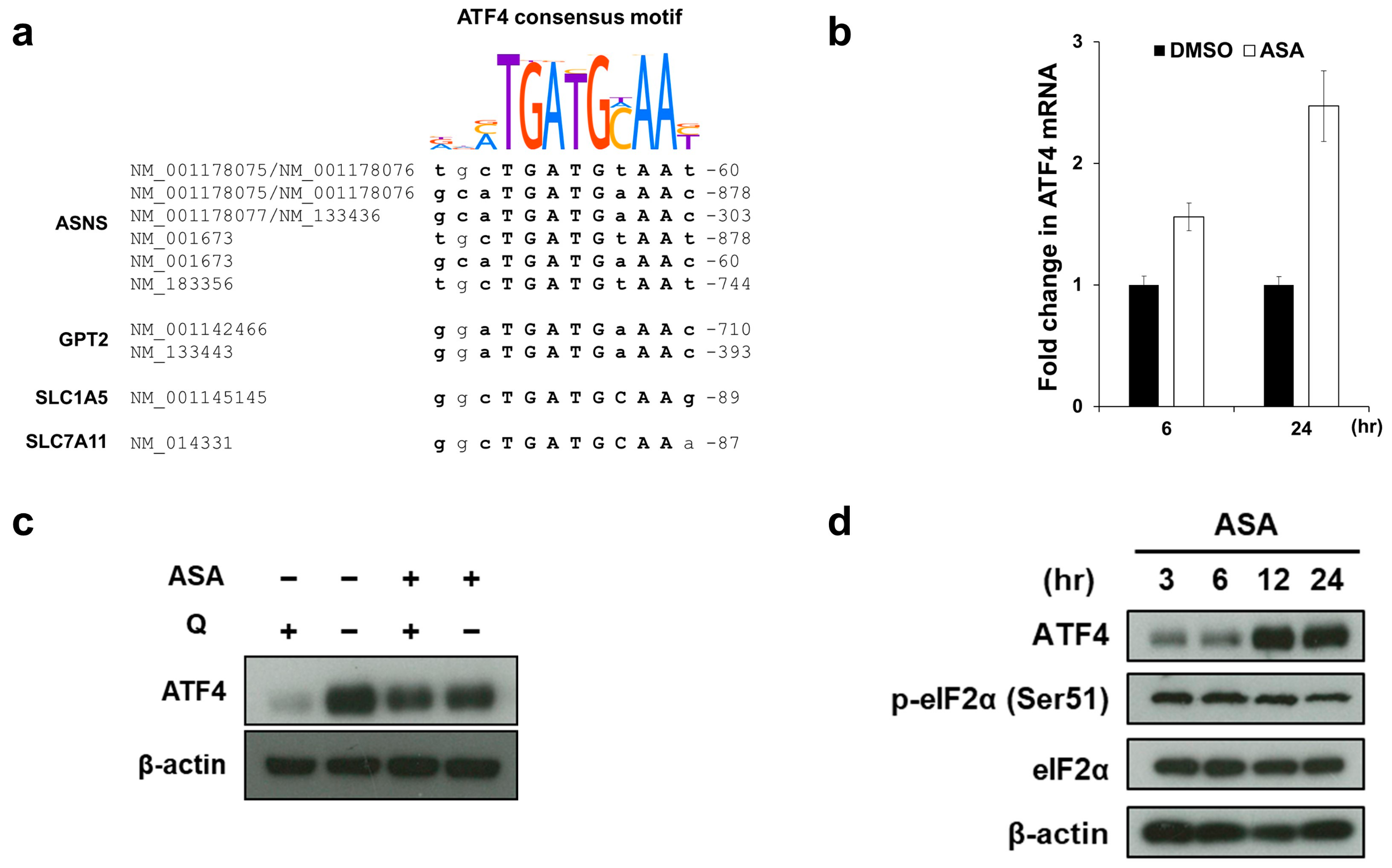
2.3. Aspirin Broadly Upregulates Glutaminolysis-Related Genes
2.4. Aspirin Induces ATF4 Gene Expression via the ER Stress-Independent Mechanism
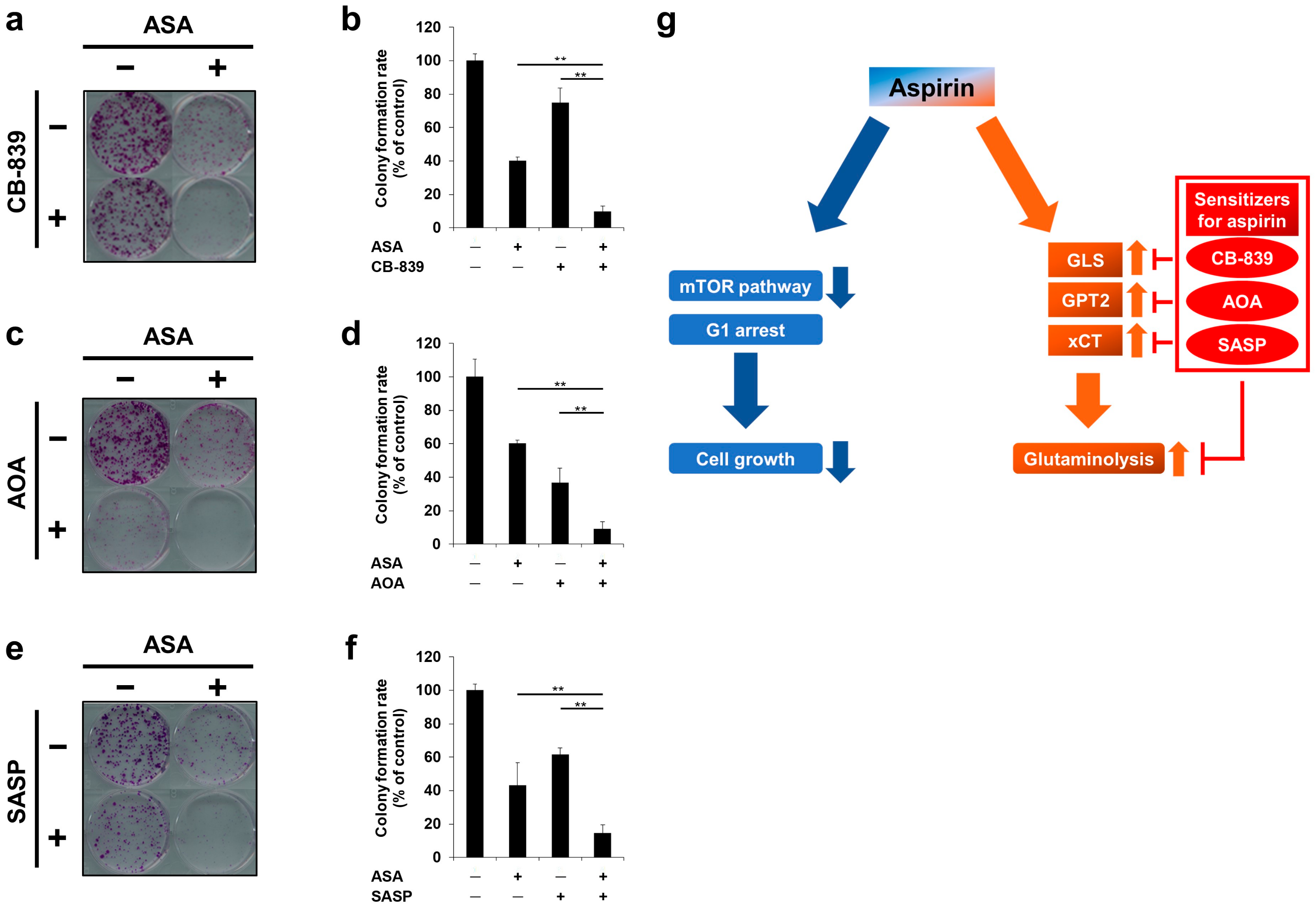
2.5. Glutaminolysis-Targeting Agents Increase the Efficacy of Aspirin in PIK3CA-Mutated Cells
2.6. Cotreatment of Aspirin with Glutaminolysis-Targeting Agents Exhibits Combinatorial Effects in Long-Term Incubation
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture
4.2. Reagents
4.3. Cell Viability Assay
4.4. Cell Cycle Analysis and the Detection of Cell Death
4.5. Protein Isolation and Western Blotting
4.6. RNA Isolation and Real-Time Quantitative Reverse Transcription-PCR
4.7. Microarray Analysis
4.8. RNA Sequencing
4.9. RNA-Seq Data Analysis
4.10. Motif Enrichment Analysis in Promoter Sequences of the Glutaminolysis-Related Genes
4.11. Colony Formation Assay
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Rebecca, S.Y.W. Role of Nonsteroidal Anti-Inflammatory Drugs (NSAIDs) in Cancer Prevention and Cancer Promotion. Adv. Pharmacol. Sci. 2019, 2019, 3418975. [Google Scholar]
- Vallée, A.; Lecarpentier, Y.; Vallée, J.N. Targeting the Canonical WNT/β-Catenin Pathway in Cancer Treatment Using Non-Steroidal Anti-Inflammatory Drugs. Cells 2019, 8, 726. [Google Scholar] [CrossRef] [Green Version]
- Rothwell, P.M.; Fowkes, F.G.R.; Belch, J.F.; Ogawa, H.; Warlow, C.P.; Meade, T.W. Effect of daily aspirin on long-term risk of death due to cancer: Analysis of individual patient data from randomised trials. Lancet 2011, 377, 31–41. [Google Scholar] [CrossRef]
- Rothwell, P.M.; Wilson, M.; Price, J.F.; Belch, J.F.; Meade, T.W.; Mehta, Z. Effect of daily aspirin on risk of cancer metastasis: A study of incident cancers during randomised controlled trials. Lancet 2012, 379, 1591–1601. [Google Scholar] [CrossRef]
- Liao, X.; Lochhead, P.; Nishihara, R.; Morikawa, T.; Kuchiba, A.; Yamauchi, M.; Imamura, Y.; Qian, Z.R.; Baba, Y.; Shima, K.; et al. Aspirin Use, Tumor PIK3CA Mutation, and Colorectal-Cancer Survival. N. Engl. J. Med. 2012, 367, 1596–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domingo, E.; Church, D.N.; Sieber, O.; Ramamoorthy, R.; Yanagisawa, Y.; Johnstone, E.; Davidson, B.; Kerr, D.J.; Tomlinson, I.P.M.; Midgley, R. Evaluation of PIK3CA Mutation As a Predictor of Benefit From Nonsteroidal Anti-Inflammatory Drug Therapy in Colorectal Cancer. J. Clin. Oncol. 2013, 31, 4297–4305. [Google Scholar] [CrossRef] [PubMed]
- Elwood, P.C.; Morgan, G.; Pickering, J.E.; Galante, J.; Weightman, A.L.; Morris, D.; Kelson, M.; Dolwani, S. Aspirin in the Treatment of Cancer: Reductions in Metastatic Spread and in Mortality: A Systematic Review and Meta-Analyses of Published Studies. PLoS ONE 2016, 11, e0152402. [Google Scholar] [CrossRef] [Green Version]
- Liao, X.; Morikawa, T.; Lochhead, P.; Imamura, Y.; Kuchiba, A.; Yamauchi, M.; Nosho, K.; Qian, Z.R.; Nishihara, R.; Meyerhardt, J.A.; et al. Prognostic Role of PIK3CA Mutation in Colorectal Cancer: Cohort Study and Literature Review. Clin. Cancer Res. 2012, 18, 2257–2268. [Google Scholar] [CrossRef] [Green Version]
- Kato, S.; Iida, S.; Higuchi, T.; Ishikawa, T.; Takagi, Y.; Yasuno, M.; Enomoto, M.; Uetake, H.; Sugihara, K. PIK3CA mutation is predictive of poor survival in patients with colorectal cancer. Int. J. Cancer 2007, 121, 1771–1778. [Google Scholar] [CrossRef]
- Cathomas, G. PIK3CA in colorectal cancer. Front. Oncol. 2014, 4, 35. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Shi, Y.-L.; Zhou, K.; Wang, L.-L.; Yan, Z.-X.; Liu, Y.-L.; Xu, L.-L.; Zhao, S.-W.; Chu, H.-L.; Shi, T.-T.; et al. PIK3CA mutations confer resistance to first-line chemotherapy in colorectal cancer. Cell Death Dis. 2018, 9, 739. [Google Scholar] [CrossRef] [PubMed]
- Sartore-Bianchi, A.; Martini, M.; Molinari, F.; Veronese, S.; Nichelatti, M.; Artale, S.; Di Nicolantonio, F.; Saletti, P.; De Dosso, S.; Mazzucchelli, L.; et al. PIK3CA Mutations in Colorectal Cancer Are Associated with Clinical Resistance to EGFR-targeted Monoclonal Antibodies. Cancer Res. 2009, 69, 1851–1857. [Google Scholar] [CrossRef] [Green Version]
- Ilic, N.; Birsoy, K.; Aguirre, A.J.; Kory, N.; Pacold, M.E.; Singh, S.; Moody, S.E.; DeAngelo, J.D.; Spardy, N.A.; Freinkman, E.; et al. PIK3CA mutant tumors depend on oxoglutarate dehydrogenase. Proc. Natl. Acad. Sci. USA 2017, 114, E3434–E3443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, Y.; Samuels, Y.; Li, Q.; Krokowski, D.; Guan, B.-J.; Wang, C.; Jin, Z.; Dong, B.; Cao, B.; Feng, X.; et al. Oncogenic PIK3CA mutations reprogram glutamine metabolism in colorectal cancer. Nat. Commun. 2016, 7, 11971. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Hao, Y.; Wang, Z. Targeting glutamine metabolism in PIK3CA mutant colorectal cancers. Genes Dis. 2016, 3, 241–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jewell, J.L.; Kim, Y.C.; Russell, R.C.; Yu, F.-X.; Park, H.W.; Plouffe, S.W.; Tagliabracci, V.S.; Guan, K.-L. Metabolism. Differential regulation of mTORC1 by leucine and glutamine. Science 2015, 347, 194–198. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.R.; Dorotinsky, C.S.; McGuire, L.J.; Macy, M.L.; Hay, R.J. DLD-1 and HCT-15 Cell Lines Derived Separately from Colorectal Carcinomas Have Totally Different Chromosome Changes But the Same Genetic Origin. Cancer Genet. Cytogenet. 1995, 81, 103–108. [Google Scholar] [CrossRef]
- Ahmed, D.; Eide, P.W.; Eilertsen, I.A.; Danielsen, S.A.; Eknæs, M.; Hektoen, M.; Lind, G.E.; Lothe, R.A. Epigenetic and genetic features of 24 colon cancer cell lines. Oncogenesis 2013, 2, e71. [Google Scholar] [CrossRef]
- Zhu, Y.; Yang, C.; Weng, M.; Zhang, Y.; Yang, C.; Jin, Y.; Yang, W.; He, Y.; Wu, Y.; Zhang, Y.; et al. Identification of TMEM208 and PQLC2 as reference genes for normalizing mRNA expression in colorectal cancer treated with aspirin. Oncotarget 2017, 8, 22759–22771. [Google Scholar] [CrossRef] [Green Version]
- Qing, G.; Li, B.; Vu, A.; Skuli, N.; Walton, Z.E.; Liu, X.; Mayes, P.A.; Wise, D.R.; Thompson, C.B.; Maris, J.M.; et al. ATF4 Regulates MYC-mediated Neuroblastoma Cell Death upon Glutamine Deprivation. Cancer Cell 2012, 22, 631–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gwinn, D.M.; Lee, A.G.; Briones-Martin-del-Campo, M.; Conn, C.S.; Simpson, D.R.; Scott, A.I.; Le, A.; Cowan, T.M.; Ruggero, D.; Sweet-Cordero, E.A. Oncogenic KRAS Regulates Amino Acid Homeostasis and Asparagine Biosynthesis via ATF4 and Alters Sensitivity to L-Asparaginase. Cancer Cell 2018, 33, 91–107.e6. [Google Scholar] [CrossRef] [Green Version]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Jin, L.; Alesi, G.N.; Kang, S. Glutaminolysis as a target for cancer therapy. Oncogene 2016, 35, 3619–3625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drew, D.A.; Cao, Y.; Chan, A.T. Aspirin and colorectal cancer: The promise of precision chemoprevention. Nat. Rev. Cancer 2016, 16, 173–186. [Google Scholar] [CrossRef]
- Saikawa, Y.; Sugiura, T.; Toriumi, F.; Kubota, T.; Suganuma, K.; Isshiki, S.; Otani, Y.; Kumai, K.; Kitajima, M. Cyclooxygenase-2 Gene Induction Causes CDDP Resistance in Colon Cancer Cell Line, HCT-15. Anticancer Res. 2004, 24, 2723–2728. [Google Scholar] [PubMed]
- Semaan, J.; Pinon, A.; Rioux, B.; Hassan, L.; Limami, Y.; Pouget, C.; Fagnere, C.; Sol, V.; Diab-Assaf, M.; Simon, A.; et al. Resistance to 3-HTMC-Induced Apoptosis Through Activation of PI3K/Akt, MEK/ERK, and p38/COX-2/PGE2 Pathways in Human HT-29 and HCT116 Colorectal Cancer Cells. J. Cell. Biochem. 2016, 117, 2875–2885. [Google Scholar] [CrossRef]
- Zumwalt, T.J.; Wodarz, D.; Komarova, N.L.; Toden, S.; Turner, J.; Cardenas, J.; Burn, J.; Chan, A.T.; Boland, C.R.; Goel, A. Aspirin-Induced Chemoprevention and Response Kinetics Are Enhanced by PIK3CA Mutations in Colorectal Cancer Cells. Cancer Prev. Res. (Phila) 2017, 10, 208–218. [Google Scholar] [CrossRef] [Green Version]
- Gu, M.; Nishihara, R.; Chen, Y.; Li, W.; Shi, Y.; Masugi, Y.; Hamada, T.; Kosumi, K.; Liu, L.; da Silva, A.; et al. Aspirin exerts high anti-cancer activity in PIK3CA-mutant colon cancer cells. Oncotarget 2017, 8, 87379–87389. [Google Scholar] [CrossRef] [Green Version]
- Din, F.V.N.; Valanciute, A.; Houde, V.P.; Zibrova, D.; Green, K.A.; Sakamoto, K.; Alessi, D.R.; Dunlop, M.G. Aspirin Inhibits mTOR Signaling, Activates AMP-Activated Protein Kinase, and Induces Autophagy in Colorectal Cancer Cells. Gastroenterology 2012, 142, 1504–1515.e3. [Google Scholar] [CrossRef] [Green Version]
- Henry, W.S.; Laszewski, T.; Tsang, T.; Beca, F.; Beck, A.H.; McAllister, S.S.; Toker, A. Aspirin Suppresses Growth in PI3K-Mutant Breast Cancer by Activating AMPK and Inhibiting mTORC1 Signaling. Cancer Res. 2017, 77, 790–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lien, E.C.; Lyssiotis, C.A.; Juvekar, A.; Hu, H.; Asara, J.M.; Cantley, L.C.; Toker, A. Glutathione biosynthesis is a metabolic vulnerability in PI(3)K/Akt-driven breast cancer. Nat. Cell Biol. 2016, 18, 572–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.; Reyna-Neyra, A.; Philippe, L.; Thoreen, C.C. mTORC1 Balances Cellular Amino Acid Supply with Demand for Protein Synthesis through Post-transcriptional Control of ATF4. Cell Rep. 2017, 19, 1083–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov-Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J.; et al. Antitumor Activity of the Glutaminase Inhibitor CB-839 in Triple-Negative Breast Cancer. Mol. Cancer Ther. 2014, 13, 890–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galan-Cobo, A.; Sitthideatphaiboon, P.; Qu, X.; Poteete, A.; Pisegna, M.A.; Tong, P.; Chen, P.-H.; Boroughs, L.K.; Rodriguez, M.L.M.; Zhang, W.; et al. LKB1 and KEAP1/NRF2 Pathways Cooperatively Promote Metabolic Reprogramming with Enhanced Glutamine Dependence in KRAS-Mutant Lung Adenocarcinoma. Cancer Res. 2019, 79, 3251–3267. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.M.; Dytfeld, D.; Reyes, L.; Robinson, R.M.; Smith, B.; Manevich, Y.; Jakubowiak, A.; Komarnicki, M.; Przybylowicz-Chalecka, A.; Szczepaniak, T.; et al. Glutaminase inhibitor CB-839 synergizes with carfilzomib in resistant multiple myeloma cells. Oncotarget 2017, 8, 35863–35876. [Google Scholar] [CrossRef] [Green Version]
- Gregory, M.A.; Nemkov, T.; Park, H.J.; Zaberezhnyy, V.; Gehrke, S.; Adane, B.; Jordan, C.T.; Hansen, K.C.; D’Alessandro, A.; DeGregori, J. Targeting Glutamine Metabolism and Redox State for Leukemia Therapy. Clin. Cancer Res. 2019, 25, 4079–4090. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.; Sasayama, T.; Irino, Y.; Takata, K.; Nagashima, H.; Satoh, N.; Kyotani, K.; Mizowaki, T.; Imahori, T.; Ejima, Y.; et al. Compensatory glutamine metabolism promotes glioblastoma resistance to mTOR inhibitor treatment. J. Clin. Investig. 2015, 125, 1591–1602. [Google Scholar] [CrossRef]
- Momcilovic, M.; Bailey, S.T.; Lee, J.T.; Fishbein, M.C.; Braas, D.; Go, J.; Graeber, T.G.; Parlati, F.; Demo, S.; Li, R.; et al. The GSK3 Signaling Axis Regulates Adaptive Glutamine Metabolism in Lung Squamous Cell Carcinoma. Cancer Cell 2018, 33, 905–921.e5. [Google Scholar] [CrossRef] [Green Version]
- Otsubo, K.; Nosaki, K.; Imamura, C.K.; Ogata, H.; Fujita, A.; Sakata, S.; Hirai, F.; Toyokawa, G.; Iwama, E.; Harada, T.; et al. Phase I study of salazosulfapyridine in combination with cisplatin and pemetrexed for advanced non-small-cell lung cancer. Cancer Sci. 2017, 108, 1843–1849. [Google Scholar] [CrossRef]
- Li, C.; Chen, H.; Lan, Z.; He, S.; Chen, R.; Wang, F.; Liu, Z.; Li, K.; Cheng, L.; Liu, Y.; et al. mTOR-dependent upregulation of xCT blocks melanin synthesis and promotes tumorigenesis. Cell Death Differ. 2019, 26, 2015–2028. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Hiraoka, Y.; Yamada, K.; Kawasaki, Y.; Hirose, H.; Matsumoto, Y.; Ishikawa, K.; Yasumizu, Y. ikra: RNAseq Pipeline Centered on Salmon. 2019. Available online: https://doi.org/10.5281/zenodo.3352573 (accessed on 20 February 2020).
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krueger, F. Trim Galore. 2015. Available online: https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 20 February 2020).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Ge, S.X.; Son, E.W.; Yao, R. iDEP: An integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinform. 2018, 19, 1–24. [Google Scholar] [CrossRef] [Green Version]
- McLeay, R.C.; Bailey, T.L. Motif Enrichment Analysis: A unified framework and an evaluation on ChIP data. BMC Bioinform. 2010, 11, 165. [Google Scholar] [CrossRef] [Green Version]
- Kulakovskiy, I.V.; Vorontsov, I.E.; Yevshin, I.S.; Sharipov, R.N.; Fedorova, A.D.; Rumynskiy, E.I.; Medvedeva, Y.A.; Magana-Mora, A.; Bajic, V.B.; Papatsenko, D.A.; et al. HOCOMOCO: Towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-Seq analysis. Nucleic Acids Res. 2018, 46, D252–D259. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boku, S.; Watanabe, M.; Sukeno, M.; Yaoi, T.; Hirota, K.; Iizuka-Ohashi, M.; Itoh, K.; Sakai, T. Deactivation of Glutaminolysis Sensitizes PIK3CA-Mutated Colorectal Cancer Cells to Aspirin-Induced Growth Inhibition. Cancers 2020, 12, 1097. https://doi.org/10.3390/cancers12051097
Boku S, Watanabe M, Sukeno M, Yaoi T, Hirota K, Iizuka-Ohashi M, Itoh K, Sakai T. Deactivation of Glutaminolysis Sensitizes PIK3CA-Mutated Colorectal Cancer Cells to Aspirin-Induced Growth Inhibition. Cancers. 2020; 12(5):1097. https://doi.org/10.3390/cancers12051097
Chicago/Turabian StyleBoku, Shogen, Motoki Watanabe, Mamiko Sukeno, Takeshi Yaoi, Kiichi Hirota, Mahiro Iizuka-Ohashi, Kyoko Itoh, and Toshiyuki Sakai. 2020. "Deactivation of Glutaminolysis Sensitizes PIK3CA-Mutated Colorectal Cancer Cells to Aspirin-Induced Growth Inhibition" Cancers 12, no. 5: 1097. https://doi.org/10.3390/cancers12051097
APA StyleBoku, S., Watanabe, M., Sukeno, M., Yaoi, T., Hirota, K., Iizuka-Ohashi, M., Itoh, K., & Sakai, T. (2020). Deactivation of Glutaminolysis Sensitizes PIK3CA-Mutated Colorectal Cancer Cells to Aspirin-Induced Growth Inhibition. Cancers, 12(5), 1097. https://doi.org/10.3390/cancers12051097