TESC Promotes TGF-?/EGFR-FOXM1-Mediated Tumor Progression in Cholangiocarcinoma

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
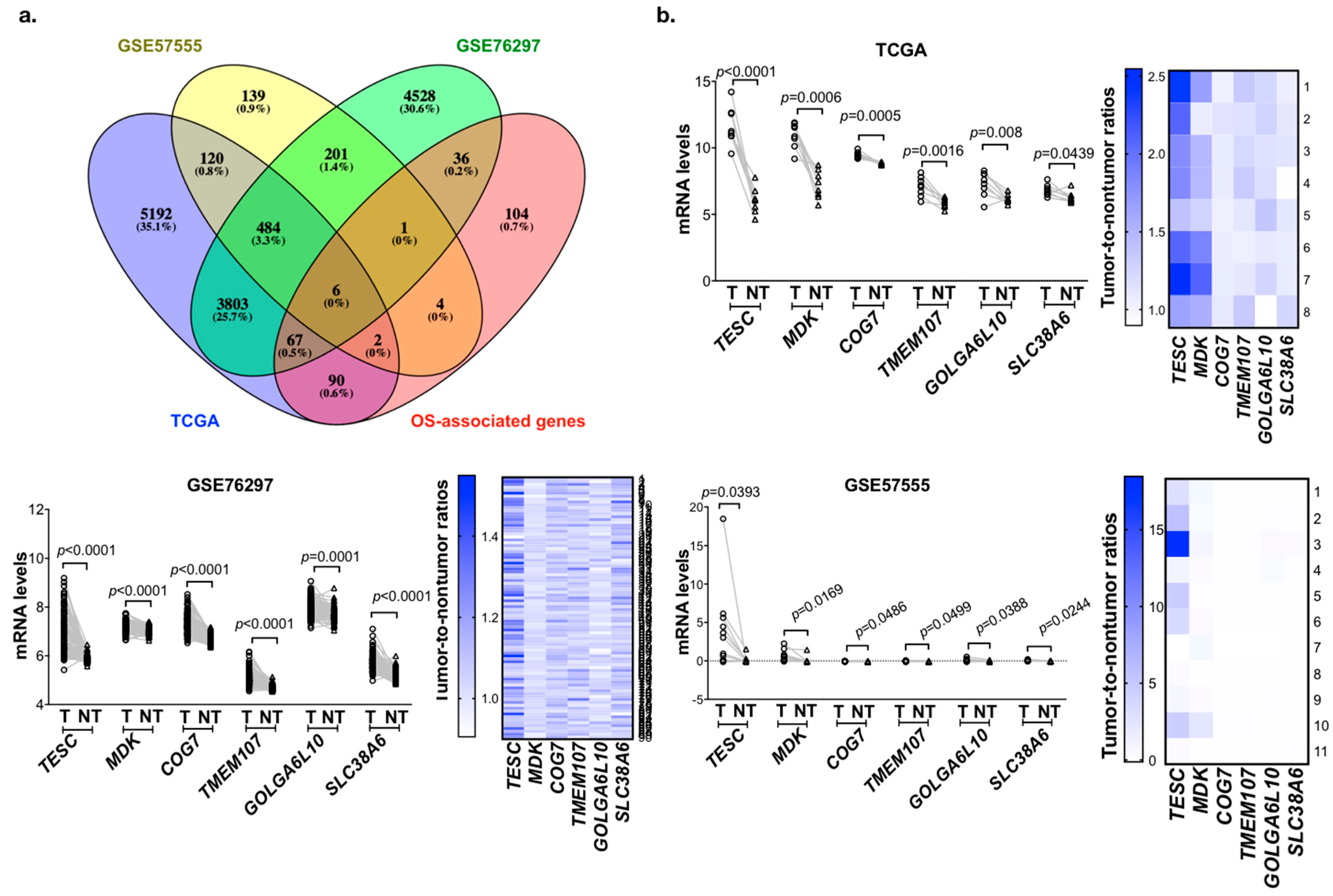
2.1. Identification of Genes Upregulated and Associated with Overall Survival (OS) in Cholangiocarcinoma
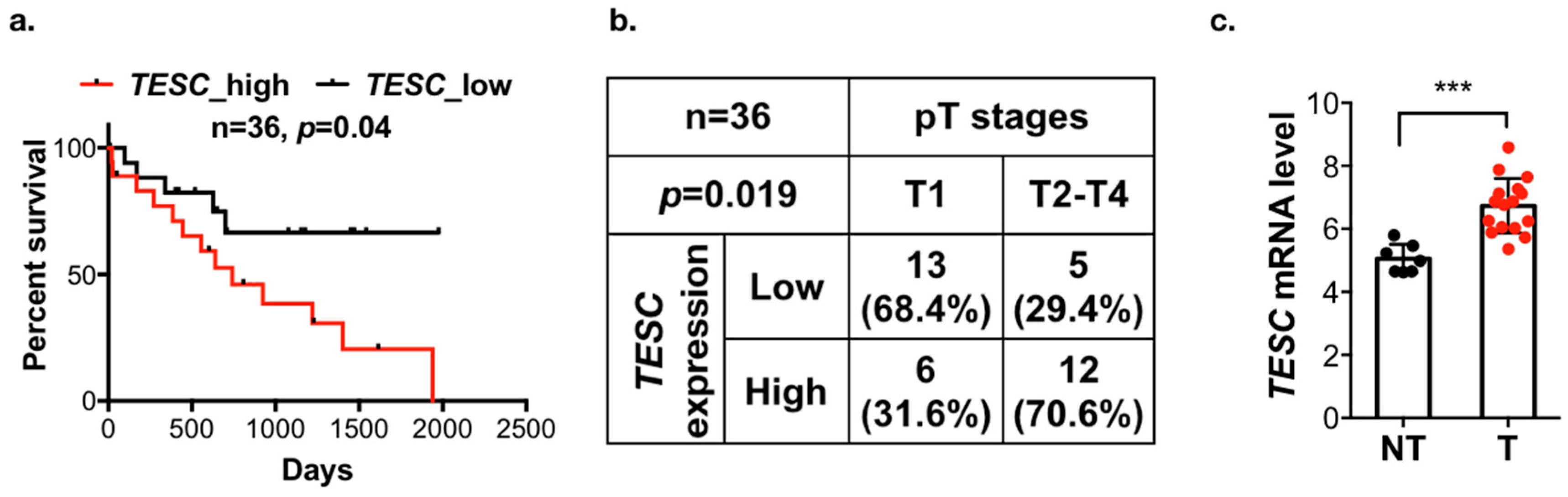
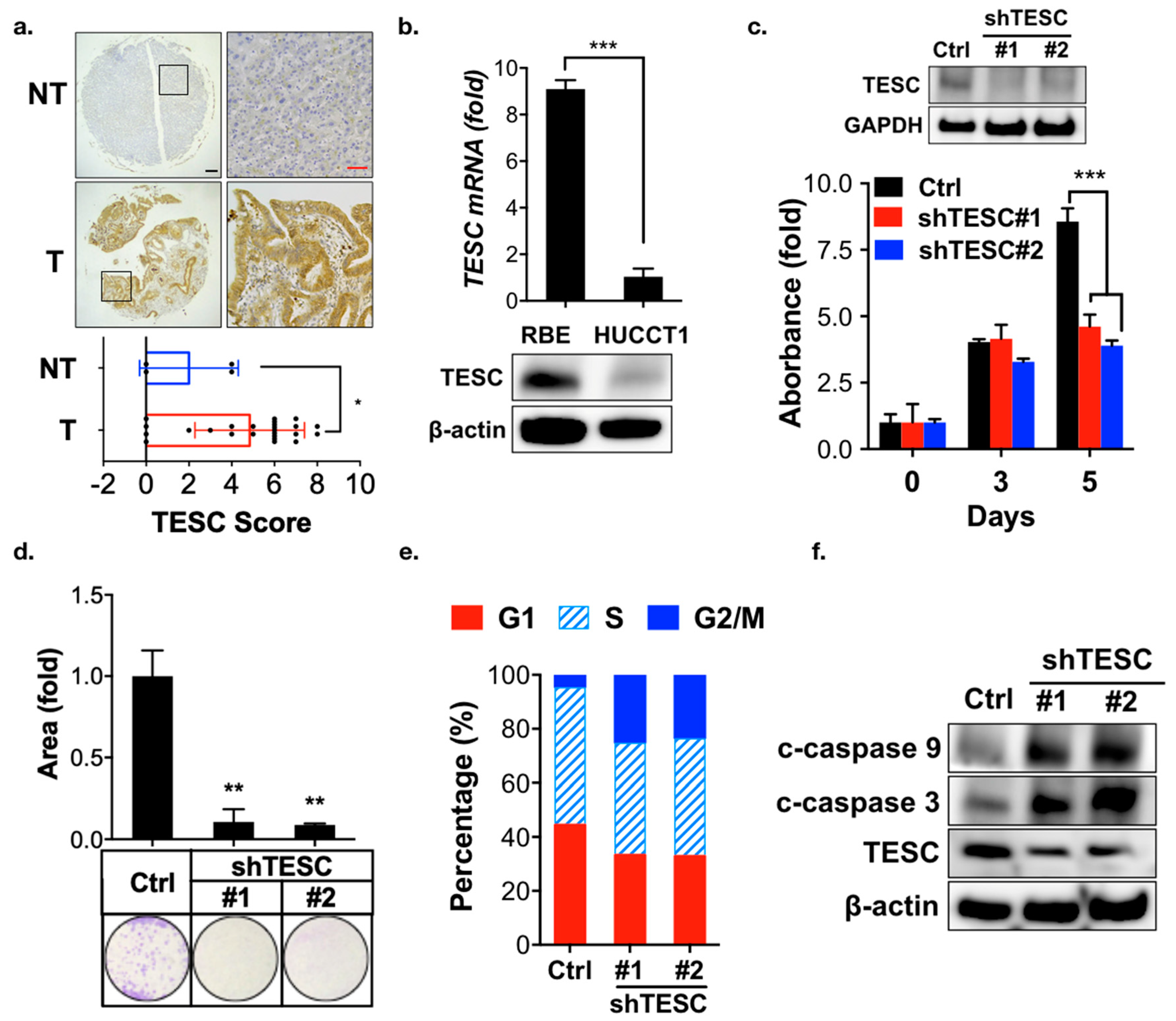
2.2. Participation of TESC in Cholangiocarcinoma
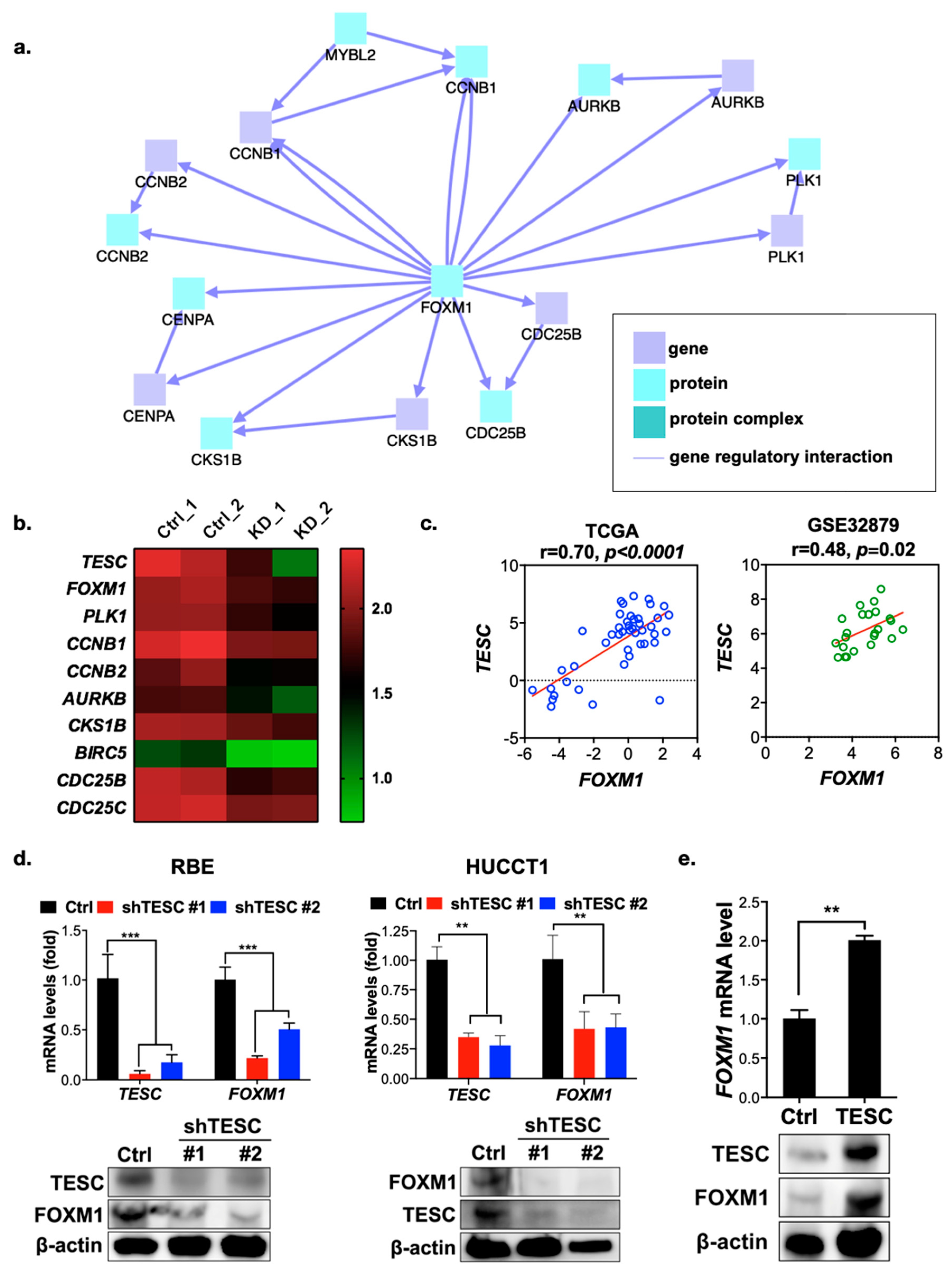
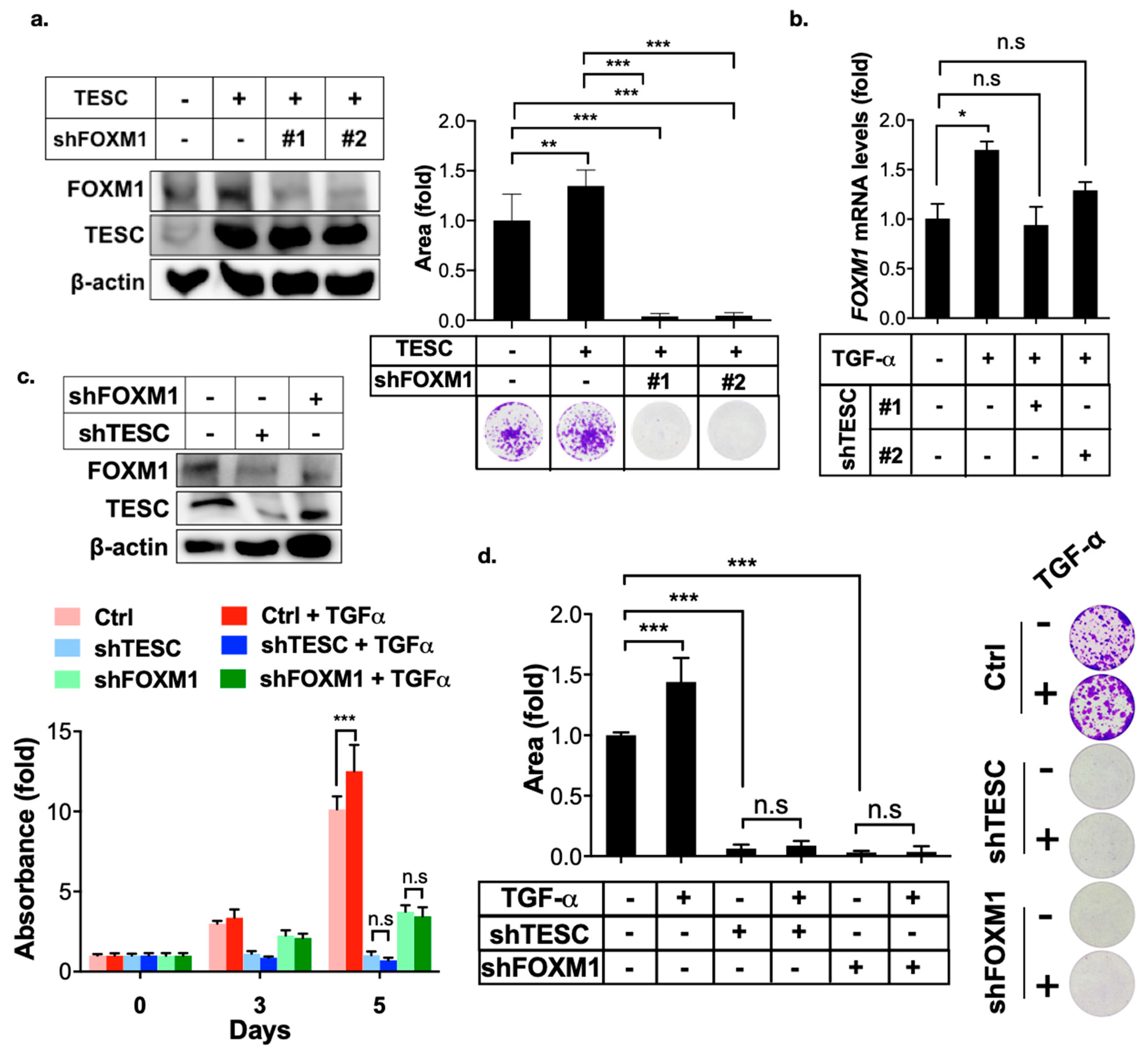
2.3. TESC Regulates G2/M Phase Through FOXM1
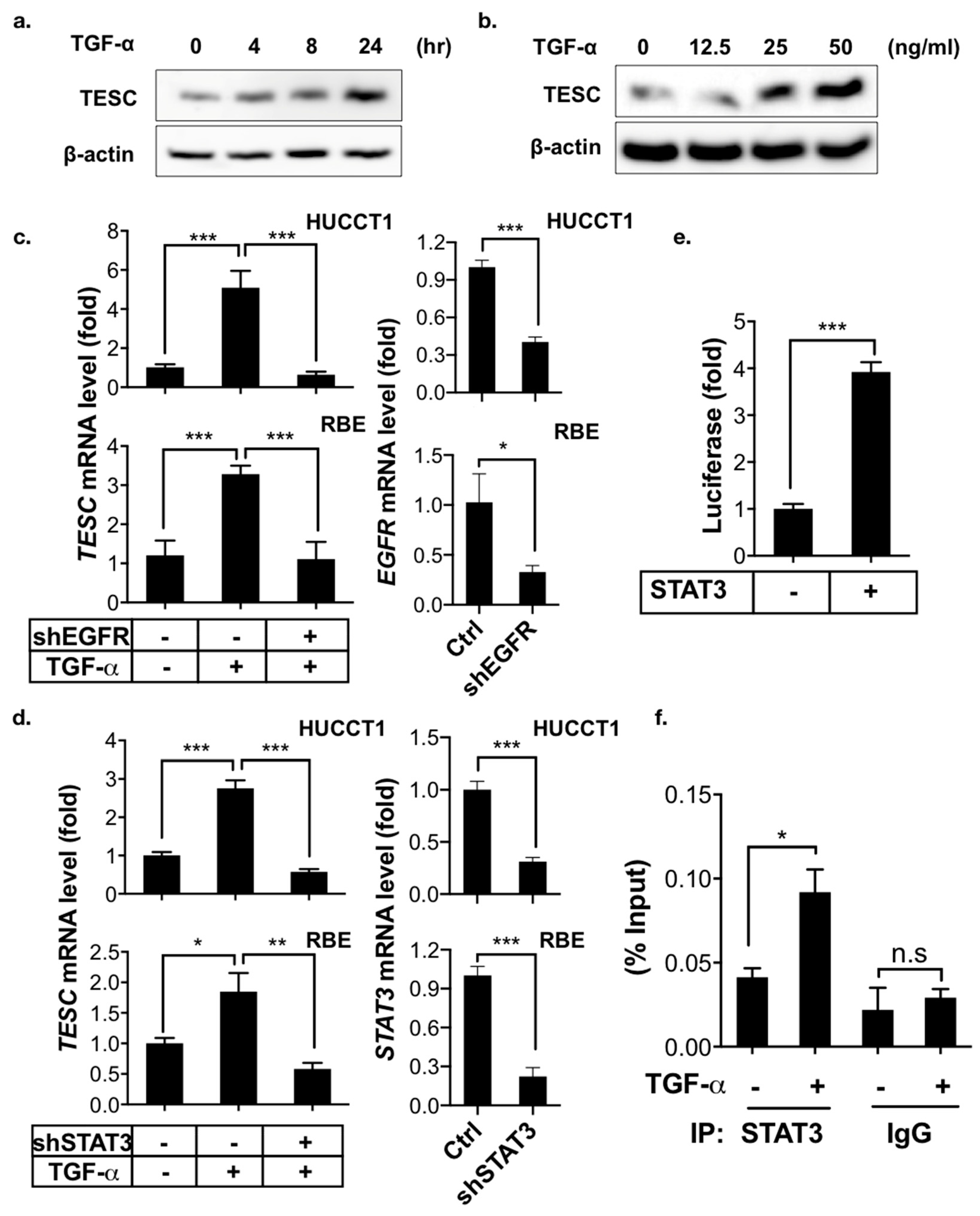
2.4. TESC Is Induced by TGF-α/STAT3 Signaling
2.5. TESC Enhances TGF-α-Induced Cell Proliferation Via FOXM1
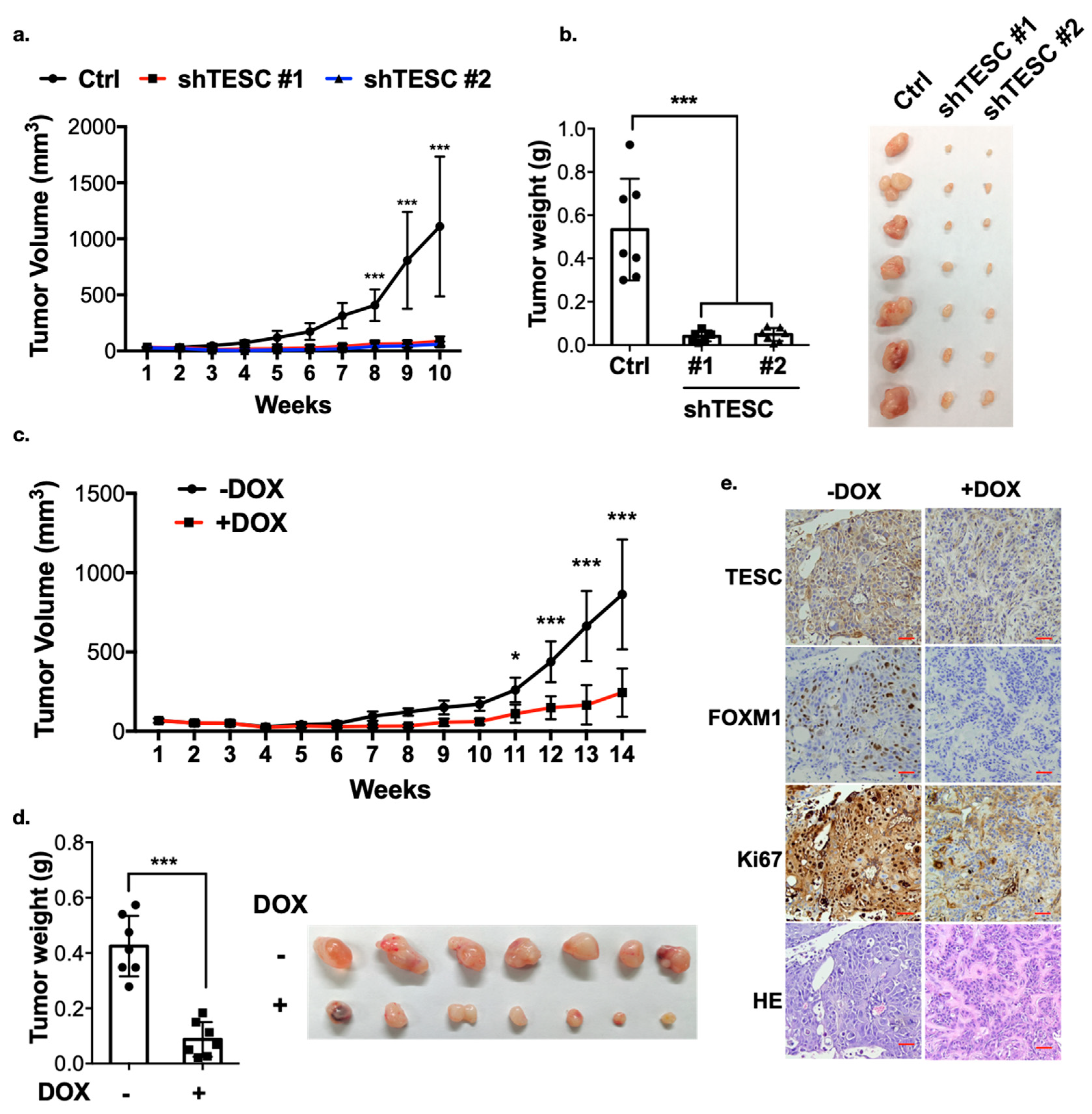
2.6. TESC Mediates ICC Tumor Growth In Vivo
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Reverse Transcription-Quantitative PCR
4.4. MTT Assay
4.5. Clonogenic Assay
4.6. Luciferase Reporter Assay
4.7. Chromatin Immunoprecipitation (ChIP)
4.8. Co-Immunoprecipitation Assay and Immunoblotting
4.9. Plasmid Construction
4.10. Xenograft Tumorigenicity Assay
4.11. Immunohistochemistry
4.12. Cell Cycle Analysis
4.13. Wound Healing Assay
4.14. Patients and Tissue Samples
4.15. Next-Generation Sequencing and Data Analysis
4.16. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alvaro, D. The challenge of cholangiocarcinoma diagnosis: The turning point is in extracellular vesicles? Hepatology 2017, 66, 1029–1031. [Google Scholar] [CrossRef]
- Park, H.M.; Yun, S.P.; Lee, E.C.; Lee, S.D.; Han, S.S.; Kim, S.H.; Park, S.J. Outcomes for Patients with Recurrent Intrahepatic Cholangiocarcinoma After Surgery. Ann. Surg. Oncol. 2016, 23, 4392–4400. [Google Scholar] [CrossRef] [PubMed]
- Lewis, H.L.; Rahnemai-Azar, A.A.; Dillhoff, M.; Schmidt, C.R.; Pawlik, T.M. Current Management of Perihilar Cholangiocarcinoma and Future Perspectives. Chirurgia (Bucur) 2017, 112, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, M.; Yamashita, Y.I.; Imai, K.; Umezaki, N.; Yamao, T.; Okabe, H.; Nakagawa, S.; Hashimoto, D.; Chikamoto, A.; Ishiko, T.; et al. Predictors of Cure of Intrahepatic Cholangiocarcinoma after Hepatic Resection. Anticancer Res. 2017, 37, 6971–6975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bridgewater, J.; Galle, P.R.; Khan, S.A.; Llovet, J.M.; Park, J.W.; Patel, T.; Pawlik, T.M.; Gores, G.J. Guidelines for the diagnosis and management of intrahepatic cholangiocarcinoma. J. Hepatol. 2014, 60, 1268–1289. [Google Scholar] [CrossRef] [Green Version]
- Perera, E.M.; Martin, H.; Seeherunvong, T.; Kos, L.; Hughes, I.A.; Hawkins, J.R.; Berkovitz, G.D. Tescalcin, a Novel Gene Encoding a Putative EF-Hand Ca2+-Binding Protein, Col9a3, and Renin Are Expressed in the Mouse Testis during the Early Stages of Gonadal Differentiation. Endocrinology 2001, 142, 455–463. [Google Scholar] [CrossRef]
- Mailänder, J.; Müller-Esterl, W.; Dedio, J. Human homolog of mouse tescalcin associates with Na+/H+ exchanger type-1. FEBS Lett. 2001, 507, 331–335. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Liu, Y.; Kay, C.M.; Müller-Esterl, W.; Fliegel, L. The Na+/H+ Exchanger Cytoplasmic Tail: Structure, Function, and Interactions with Tescalcin. Biochemistry 2003, 42, 7448–7456. [Google Scholar] [CrossRef]
- Levay, K.; Slepak, V.Z. Tescalcin is an essential factor in megakaryocytic differentiation associated with Ets family gene expression. J. Clin. Investig. 2007, 117, 2672–2683. [Google Scholar] [CrossRef] [Green Version]
- Port, M.; Boltze, C.; Wang, Y.; Roper, B.; Meineke, V.; Abend, M. A radiation-induced gene signature distinguishes post-Chernobyl from sporadic papillary thyroid cancers. Radiat. Res. 2007, 168, 639–649. [Google Scholar] [CrossRef] [Green Version]
- Stein, L.; Rothschild, J.; Luce, J.; Cowell, J.K.; Thomas, G.; Bogdanova, T.I.; Tronko, M.D.; Hawthorn, L. Copy Number and Gene Expression Alterations in Radiation-Induced Papillary Thyroid Carcinoma from Chernobyl Pediatric Patients. Thyroid 2009, 20, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.H.; Han, S.R.; Kim, J.T.; Lee, S.J.; Yeom, Y.I.; Min, J.K.; Lee, C.H.; Kim, J.W.; Yoon, S.R.; Yoon, D.Y.; et al. The EF-hand calcium-binding protein tescalcin is a potential oncotarget in colorectal cancer. Oncotarget 2014, 5, 2149–2160. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Kang, Y.H.; Oh, B.M.; Uhm, T.G.; Park, S.Y.; Kim, T.W.; Han, S.R.; Lee, S.J.; Lee, Y.; Lee, H.G. Tescalcin expression contributes to invasive and metastatic activity in colorectal cancer. Tumour Biol. 2016, 37, 13843–13853. [Google Scholar] [CrossRef] [PubMed]
- Luo, A.J.; Tan, J.; He, L.Y.; Jiang, X.Z.; Jiang, Z.Q.; Zeng, Q.; Yao, K.; Xue, J. Suppression of Tescalcin inhibits growth and metastasis in renal cell carcinoma via downregulating NHE1 and NF-kB signaling. Exp. Mol. Pathol. 2019, 107, 110–117. [Google Scholar] [CrossRef]
- Dictor, M.; Ehinger, M.; Mertens, F.; Akervall, J.; Wennerberg, J. Abnormal cell cycle regulation in malignancy. Am. J. Clin. Pathol. 1999, 112, S40–S52. [Google Scholar]
- Sherr, C.J. Cancer cell cycles. Science 1996, 274, 1672–1677. [Google Scholar] [CrossRef] [Green Version]
- Leung, T.W.C.; Lin, S.S.W.; Tsang, A.C.C.; Tong, C.S.W.; Ching, J.C.Y.; Leung, W.Y.; Gimlich, R.; Wong, G.G.; Yao, K.-M. Over-expression of FoxM1 stimulates cyclin B1 expression. FEBS Lett. 2001, 507, 59–66. [Google Scholar] [CrossRef] [Green Version]
- Laoukili, J.; Kooistra, M.R.; Bras, A.; Kauw, J.; Kerkhoven, R.M.; Morrison, A.; Clevers, H.; Medema, R.H. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat. Cell Biol. 2005, 7, 126–136. [Google Scholar] [CrossRef]
- Wang, I.C.; Chen, Y.-J.; Hughes, D.; Petrovic, V.; Major, M.L.; Park, H.J.; Tan, Y.; Ackerson, T.; Costa, R.H. Forkhead Box M1 Regulates the Transcriptional Network of Genes Essential for Mitotic Progression and Genes Encoding the SCF (Skp2-Cks1) Ubiquitin Ligase. Mol. Cell. Biol. 2005, 25, 10875–10894. [Google Scholar] [CrossRef] [Green Version]
- Park, H.J.; Costa, R.H.; Lau, L.F.; Tyner, A.L.; Raychaudhuri, P. Anaphase-Promoting Complex/Cyclosome-Cdh1-Mediated Proteolysis of the Forkhead Box M1 Transcription Factor Is Critical for Regulated Entry into S Phase. Mol. Cell. Biol. 2008, 28, 5162–5171. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.S.; Miao, R.C.; Wan, Y.; Zhang, L.Q.; Qu, K.; Liu, C. FoxM1 as a novel therapeutic target for cancer drug therapy. APJCP 2015, 16, 23–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.S.H.; Chang, H.W.; Lin, I.H.; Chien, L.N.; Wu, M.J.; Liu, Y.R.; Chu, P.G.; Xie, G.; Dong, F.; Jia, W.; et al. Long-term Proton Pump Inhibitor Administration Caused Physiological and Microbiota Changes in Rats. Sci. Rep. 2020, 10, 866. [Google Scholar] [CrossRef] [PubMed]
- Werneburg, N.W.; Yoon, J.-H.; Higuchi, H.; Gores, G.J. Bile acids activate EGF receptor via a TGF-α-dependent mechanism in human cholangiocyte cell lines. Am. J. Physiol.-Gastrointest. Liver Physiol. 2003, 285, G31–G36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Xu, H.; Zhou, Z.; Tian, Y.; Cao, X.; Cheng, G.; Liu, Q. Blocking of the EGFR-STAT3 signaling pathway through afatinib treatment inhibited the intrahepatic cholangiocarcinoma. Exp. Ther. Med. 2018, 15, 4995–5000. [Google Scholar] [CrossRef]
- Stoll, S.W.; Stuart, P.E.; Swindell, W.R.; Tsoi, L.C.; Li, B.; Gandarillas, A.; Lambert, S.; Johnston, A.; Nair, R.P.; Elder, J.T. The EGF receptor ligand amphiregulin controls cell division via FoxM1. Oncogene 2016, 35, 2075–2086. [Google Scholar] [CrossRef] [Green Version]
- Chang-Panesso, M.; Kadyrov, F.F.; Lalli, M.; Wu, H.; Ikeda, S.; Kefaloyianni, E.; Abdelmageed, M.M.; Herrlich, A.; Kobayashi, A.; Humphreys, B.D. FOXM1 drives proximal tubule proliferation during repair from acute ischemic kidney injury. J. Clin. Investig. 2019, 129, 5501–5517. [Google Scholar] [CrossRef]
- Di Sole, F.; Vadnagara, K.; Moe, O.W.; Babich, V. Calcineurin homologous protein: A multifunctional Ca2+-binding protein family. Am. J. Physiol. Renal Physiol. 2012, 303, F165–F179. [Google Scholar] [CrossRef] [Green Version]
- Kolobynina, K.G.; Solovyova, V.V.; Levay, K.; Rizvanov, A.A.; Slepak, V.Z. Emerging roles of the single EF-hand Ca2+ sensor tescalcin in the regulation of gene expression, cell growth and differentiation. J. Cell Sci. 2016, 129, 3533–3540. [Google Scholar] [CrossRef] [Green Version]
- Barroso, M.R.; Bernd, K.K.; DeWitt, N.D.; Chang, A.; Mills, K.; Sztul, E.S. A novel Ca2+-binding protein, p22, is required for constitutive membrane traffic. J. Biol. Chem. 1996, 271, 10183–10187. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Barber, D.L. A calcineurin homologous protein inhibits GTPase-stimulated Na-H exchange. Proc. Natl. Acad. Sci. USA 1996, 93, 12631–12636. [Google Scholar] [CrossRef] [Green Version]
- Inoue, H.; Nakamura, Y.; Nagita, M.; Takai, T.; Masuda, M.; Nakamura, N.; Kanazawa, H. Calcineurin homologous protein isoform 2 (CHP2), Na+/H+ exchangers-binding protein, is expressed in intestinal epithelium. Biol. Pharm. Bull. 2003, 26, 148–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.D.; Zhang, X.; Li, R.; Wang, Y.D.; Wang, Y.L.; Han, K.J.; Qian, X.P.; Yang, C.G.; Liu, P.; Wei, Q.; et al. CHP2 activates the calcineurin/nuclear factor of activated T cells signaling pathway and enhances the oncogenic potential of HEK293 cells. J. Biol. Chem. 2008, 283, 32660–32668. [Google Scholar] [CrossRef] [Green Version]
- Pang, T.; Wakabayashi, S.; Shigekawa, M. Expression of calcineurin B homologous protein 2 protects serum deprivation-induced cell death by serum-independent activation of Na+/H+ exchanger. J. Biol. Chem. 2002, 277, 43771–43777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Q.; Kong, B.; Yang, X.; Cui, B.; Wei, Y.; Yang, Q. Overexpression of CHP2 enhances tumor cell growth, invasion and metastasis in ovarian cancer. In Vivo 2007, 21, 593–598. [Google Scholar] [PubMed]
- Hammam, A.A.; Eissa, H.H.; El Masry, M.R.; Mahmoud, S. CHP2 gene expression and quantitation in Egyptian patients with acute leukemia. Meta Gene 2014, 2, 323–331. [Google Scholar] [CrossRef]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Xue, S.; Yang, J.J. Calciomics: Integrative studies of Ca2+-binding proteins and their interactomes in biological systems. Metall. Integr. Biometal Sci. 2013, 5, 29–42. [Google Scholar] [CrossRef] [Green Version]
- Ukarapong, S.; Bao, Y.; Perera, E.M.; Berkovitz, G.D. Megakaryocyte development is normal in mice with targeted disruption of Tescalcin. Exp. Cell Res. 2012, 318, 662–669. [Google Scholar] [CrossRef]
- Fan, J.; Xing, Y.; Wen, X.; Jia, R.; Ni, H.; He, J.; Ding, X.; Pan, H.; Qian, G.; Ge, S.; et al. Long non-coding RNA ROR decoys gene-specific histone methylation to promote tumorigenesis. Genome Biol. 2015, 16, 139. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.H.; Gwak, G.Y.; Lee, H.S.; Bronk, S.F.; Werneburg, N.W.; Gores, G.J. Enhanced epidermal growth factor receptor activation in human cholangiocarcinoma cells. J. Hepatol. 2004, 41, 808–814. [Google Scholar] [CrossRef]
- Yoshikawa, D.; Ojima, H.; Iwasaki, M.; Hiraoka, N.; Kosuge, T.; Kasai, S.; Hirohashi, S.; Shibata, T. Clinicopathological and prognostic significance of EGFR, VEGF, and HER2 expression in cholangiocarcinoma. Br. J. Cancer 2008, 98, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Choi, S.I.; Kim, R.K.; Cho, E.W.; Kim, I.G. Tescalcin/c-Src/IGF1Rbeta-mediated STAT3 activation enhances cancer stemness and radioresistant properties through ALDH1. Sci. Rep. 2018, 8, 10711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Park, S.H.; Chang, H.M.; Kim, J.S.; Choi, H.J.; Lee, M.A.; Jang, J.S.; Jeung, H.C.; Kang, J.H.; Lee, H.W.; et al. Gemcitabine and oxaliplatin with or without erlotinib in advanced biliary-tract cancer: A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2012, 13, 181–188. [Google Scholar] [CrossRef]
- Hsieh, C.H.; Chou, Y.T.; Kuo, M.H.; Tsai, H.P.; Chang, J.L.; Wu, C.W. A targetable HB-EGF-CITED4 axis controls oncogenesis in lung cancer. Oncogene 2017, 36, 2946–2956. [Google Scholar] [CrossRef]
- Chou, Y.T.; Lee, C.C.; Hsiao, S.H.; Lin, S.E.; Lin, S.C.; Chung, C.H.; Chung, C.H.; Kao, Y.R.; Wang, Y.H.; Chen, C.T.; et al. The emerging role of SOX2 in cell proliferation and survival and its crosstalk with oncogenic signaling in lung cancer. Stem Cells 2013, 31, 2607–2619. [Google Scholar] [CrossRef]
- Chou, Y.T.; Lin, H.H.; Lien, Y.C.; Wang, Y.H.; Hong, C.F.; Kao, Y.R.; Lin, S.C.; Chang, Y.C.; Lin, S.Y.; Chen, S.J.; et al. EGFR promotes lung tumorigenesis by activating miR-7 through a Ras/ERK/Myc pathway that targets the Ets2 transcriptional repressor ERF. Cancer Res. 2010, 70, 8822–8831. [Google Scholar] [CrossRef] [Green Version]
- Chou, Y.T.; Hsieh, C.H.; Chiou, S.H.; Hsu, C.F.; Kao, Y.R.; Lee, C.C.; Chung, C.H.; Wang, Y.H.; Hsu, H.S.; Pang, S.T.; et al. CITED2 functions as a molecular switch of cytokine-induced proliferation and quiescence. Cell Death Differ. 2012, 19, 2015–2028. [Google Scholar] [CrossRef] [Green Version]
- Oishi, N.; Kumar, M.R.; Roessler, S.; Ji, J.; Forgues, M.; Budhu, A.; Zhao, X.; Andersen, J.B.; Ye, Q.H.; Jia, H.L.; et al. Transcriptomic profiling reveals hepatic stem-like gene signatures and interplay of miR-200c and epithelial-mesenchymal transition in intrahepatic cholangiocarcinoma. Hepatology 2012, 56, 1792–1803. [Google Scholar] [CrossRef]
- Chaisaingmongkol, J.; Budhu, A.; Dang, H.; Rabibhadana, S.; Pupacdi, B.; Kwon, S.M.; Forgues, M.; Pomyen, Y.; Bhudhisawasdi, V.; Lertprasertsuke, N.; et al. Common Molecular Subtypes among Asian Hepatocellular Carcinoma and Cholangiocarcinoma. Cancer Cell 2017, 32, 57–70 e53. [Google Scholar] [CrossRef] [Green Version]
- Murakami, Y.; Kubo, S.; Tamori, A.; Itami, S.; Kawamura, E.; Iwaisako, K.; Ikeda, K.; Kawada, N.; Ochiya, T.; Taguchi, Y.H. Comprehensive analysis of transcriptome and metabolome analysis in Intrahepatic Cholangiocarcinoma and Hepatocellular Carcinoma. Sci. Rep. 2015, 5, 16294. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GSEA. Available online: http://www.broadinstitute.org/gsea (accessed on 20 May 2019).
- ConsensusPathDB. Available online: http://cpdb.molgen.mpg.de (accessed on 15 May 2019).







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsieh, C.-H.; Chu, C.-Y.; Lin, S.-E.; Yang, Y.-C.S.H.; Chang, H.-S.; Yen, Y. TESC Promotes TGF-?/EGFR-FOXM1-Mediated Tumor Progression in Cholangiocarcinoma. Cancers 2020, 12, 1105. https://doi.org/10.3390/cancers12051105
Hsieh C-H, Chu C-Y, Lin S-E, Yang Y-CSH, Chang H-S, Yen Y. TESC Promotes TGF-?/EGFR-FOXM1-Mediated Tumor Progression in Cholangiocarcinoma. Cancers. 2020; 12(5):1105. https://doi.org/10.3390/cancers12051105
Chicago/Turabian StyleHsieh, Cheng-Han, Cheng-Ying Chu, Sey-En Lin, Yu-Chen S.H. Yang, Hung-Shu Chang, and Yun Yen. 2020. "TESC Promotes TGF-?/EGFR-FOXM1-Mediated Tumor Progression in Cholangiocarcinoma" Cancers 12, no. 5: 1105. https://doi.org/10.3390/cancers12051105
APA StyleHsieh, C.-H., Chu, C.-Y., Lin, S.-E., Yang, Y.-C. S. H., Chang, H.-S., & Yen, Y. (2020). TESC Promotes TGF-?/EGFR-FOXM1-Mediated Tumor Progression in Cholangiocarcinoma. Cancers, 12(5), 1105. https://doi.org/10.3390/cancers12051105