Inducing Angiogenesis, a Key Step in Cancer Vascularization, and Treatment Approaches



Abstract
:1. Introduction
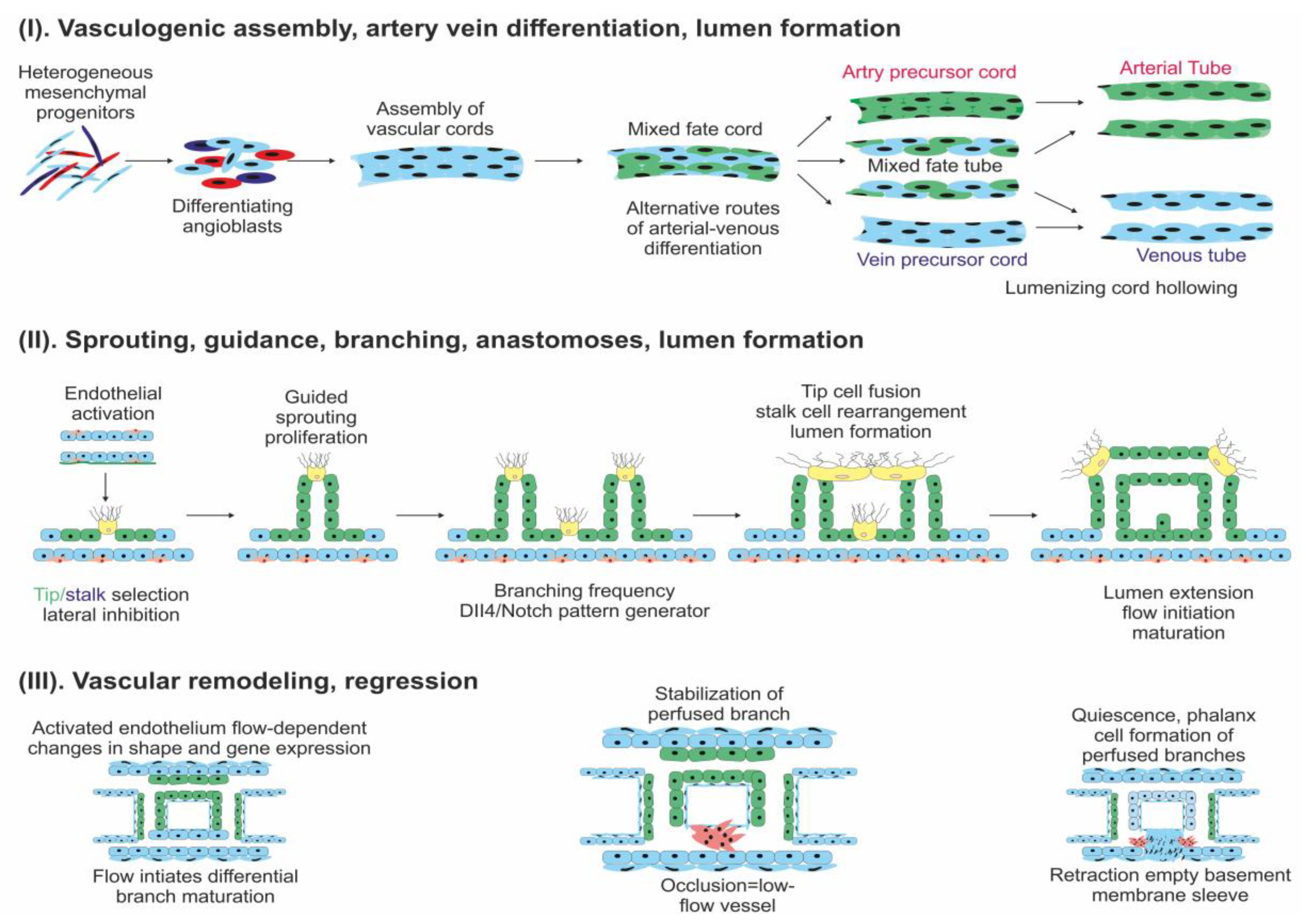
2. Angiogenesis in Normal Tissue
3. Angiogenesis in Cancer, a Literature Review
4. Pro- and Anti-Angiogenic Factors
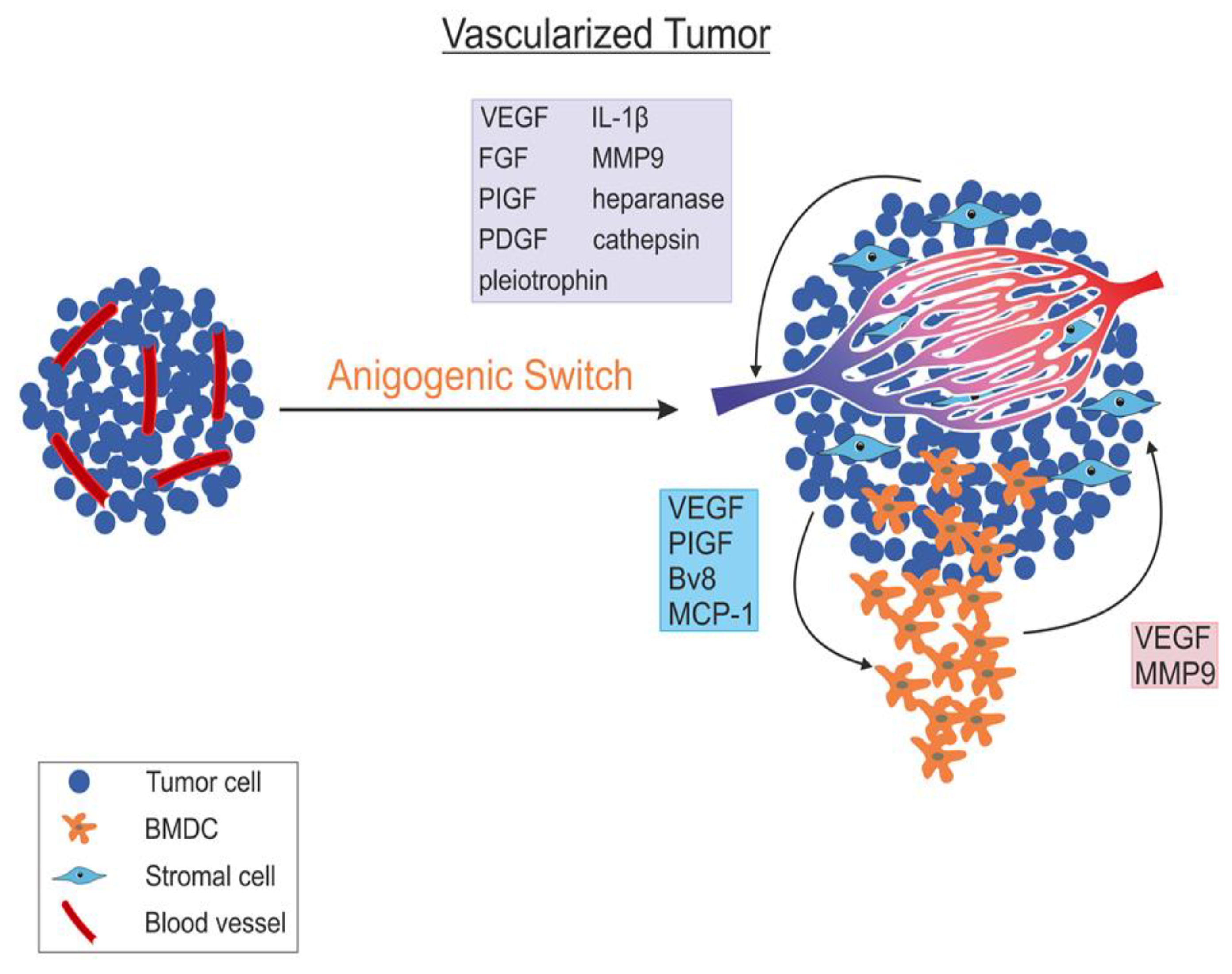
5. Angiogenic Switch
6. Tumour Vasculature Modulation as a Therapeutic Option
Vascular Promotion Therapy
7. Immune Modulation
8. Anti-Angiogenic Therapy
9. Novel and Future Approaches to Modify Angiogenesis as Anti-Cancer Option
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Ang | Angiopoietin |
| CRC | Colorectal Cancer |
| EC | Endothelial cells |
| ECM | Extra-cellular matrix |
| EPC | Endothelial progenitor cells |
| FGF | Fibroblast growth factor |
| FOLFIRI | Fluorouracil, Leucovorin and Irinotecan |
| FOXP3 | Forkhead box protein P3 |
| HCC | Hepatocellular Carcinoma |
| NSCLC | Non-small Cell Lung Cancer |
| PDGF | Platelet-derived growth factor |
| PFS | Progression free survival |
| PlGF | Placenta growth factor |
| TAFs | Tumour associated fibroblasts |
| TK | Tyrosine kinases |
| TME | Tumour micro-environment |
| TSP1 | Thrombospondin1 |
| VEGF | Vascular endothelial growth factor |
| ZA | Zoledronic acid |
References
- Suh, D.Y. Understanding angiogenesis and its clinical applications. Ann. Clin. Lab. Sci. 2000, 30, 227–238. [Google Scholar] [PubMed]
- Nishida, N.; Yano, H.; Nishida, T.; Kamura, T.; Kojiro, M. Angiogenesis in cancer. Vasc. Health Risk Manag. 2006, 2, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Ravi, R.; Mookerjee, B.; Bhujwalla, Z.M.; Sutter, C.H.; Artemov, D.; Zeng, Q.; Dillehay, L.E.; Madan, A.; Semenza, G.L.; Bedi, A. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha. Genes. Dev. 2000, 14, 34–44. [Google Scholar] [PubMed]
- McCoy, R.J.; O’Brien, F.J. Influence of shear stress in perfusion bioreactor cultures for the development of three-dimensional bone tissue constructs: A review. Tissue. Eng. Part B Rev. 2010, 16, 587–601. [Google Scholar] [CrossRef]
- Carmeliet, P. Angiogenesis in health and disease. Nat. Med. 2003, 9, 653–660. [Google Scholar] [CrossRef]
- Kuehbacher, A.; Urbich, C.; Zeiher, A.M.; Dimmeler, S. Role of Dicer and Drosha for endothelial microRNA expression and angiogenesis. Circ. Res. 2007, 101, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Chan, Y.C.; Roy, S.; Khanna, S.; Sen, C.K. Downregulation of endothelial microRNA-200b supports cutaneous wound angiogenesis by desilencing GATA binding protein 2 and vascular endothelial growth factor receptor 2. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1372–1382. [Google Scholar] [CrossRef] [Green Version]
- Chan, Y.C.; Khanna, S.; Roy, S.; Sen, C.K. miR-200b targets Ets-1 and is down-regulated by hypoxia to induce angiogenic response of endothelial cells. J. Biol. Chem. 2011, 286, 2047–2056. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Q.; Cui, X.; Zhang, D.; Yang, Y.; Yan, X.; Liu, M.; Niang, B.; Aziz, F.; Liu, S.; Yan, Q.; et al. miR-200b inhibits proliferation and metastasis of breast cancer by targeting fucosyltransferase IV and alpha1,3-fucosylated glycans. Oncogenesis 2017, 6, 358. [Google Scholar] [CrossRef] [Green Version]
- Hua, Z.; Lv, Q.; Ye, W.; Wong, C.K.; Cai, G.; Gu, D.; Ji, Y.; Zhao, C.; Wang, J.; Yang, B.B.; et al. MiRNA-directed regulation of VEGF and other angiogenic factors under hypoxia. PLoS ONE 2006, 1, 116. [Google Scholar] [CrossRef] [Green Version]
- Tsai, Y.H.; Wu, M.F.; Wu, Y.H.; Chang, S.J.; Lin, S.F.; Sharp, T.V.; Wang, H.W. The M type K15 protein of Kaposi’s sarcoma-associated herpesvirus regulates microRNA expression via its SH2-binding motif to induce cell migration and invasion. J. Virol. 2009, 83, 622–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doganov, N.; Negentsov, N. Clinical trial of the laksafer preparation on patients following gynecological operations. Akush. Ginekol. 1989, 28, 47–51. [Google Scholar]
- Chen, Y.; Gorski, D.H. Regulation of angiogenesis through a microRNA (miR-130a) that down-regulates antiangiogenic homeobox genes GAX and HOXA5. Blood 2008, 111, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Wurdinger, T.; Tannous, B.A.; Saydam, O.; Skog, J.; Grau, S.; Soutschek, J.; Weissleder, R.; Breakefield, X.O.; Krichevsky, A.M. miR-296 regulates growth factor receptor overexpression in angiogenic endothelial cells. Cancer Cell 2008, 14, 382–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byatt, G.; Dalrymple-Alford, J.C. Both anteromedial and anteroventral thalamic lesions impair radial-maze learning in rats. Behav. Neurosci. 1996, 110, 1335–1348. [Google Scholar] [CrossRef]
- Pulkkinen, K.; Malm, T.; Turunen, M.; Koistinaho, J.; Yla-Herttuala, S. Hypoxia induces microRNA miR-210 in vitro and in vivo ephrin-A3 and neuronal pentraxin 1 are potentially regulated by miR-210. FEBS Lett. 2008, 582, 2397–2401. [Google Scholar] [CrossRef] [Green Version]
- Ventura, A.; Young, A.G.; Winslow, M.M.; Lintault, L.; Meissner, A.; Erkeland, S.J.; Newman, J.; Bronson, R.T.; Crowley, D.; Stone, J.R.; et al. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell 2008, 132, 875–886. [Google Scholar] [CrossRef] [Green Version]
- Baeriswyl, V.; Christofori, G. The angiogenic switch in carcinogenesis. Semin. Cancer Biol. 2009, 19, 329–337. [Google Scholar] [CrossRef]
- Saggar, J.K.; Yu, M.; Tan, Q.; Tannock, I.F. The tumor microenvironment and strategies to improve drug distribution. Front. Oncol. 2013, 3, 154. [Google Scholar] [CrossRef] [Green Version]
- Bridges, E.; Harris, A.L. Vascular-promoting therapy reduced tumor growth and progression by improving chemotherapy efficacy. Cancer Cell 2015, 27, 7–9. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Retrospective: Judah Folkman (1933–2008). Science 2008, 319, 1055. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Kubo, H.; Fujiwara, T.; Jussila, L.; Hashi, H.; Ogawa, M.; Shimizu, K.; Awane, M.; Sakai, Y.; Takabayashi, A.; Alitalo, K.; et al. Involvement of vascular endothelial growth factor receptor-3 in maintenance of integrity of endothelial cell lining during tumor angiogenesis. Blood 2000, 96, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Goel, H.L.; Mercurio, A.M. VEGF targets the tumour cell. Nat. Rev. Cancer 2013, 13, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Fantozzi, A.; Gruber, D.C.; Pisarsky, L.; Heck, C.; Kunita, A.; Yilmaz, M.; Meyer-Schaller, N.; Cornille, K.; Hopfer, U.; Bentires-Alj, M.; et al. VEGF-mediated angiogenesis links EMT-induced cancer stemness to tumor initiation. Cancer Res. 2014, 74, 1566–1575. [Google Scholar] [CrossRef] [Green Version]
- Ghersi, G. Roles of molecules involved in epithelial/mesenchymal transition during angiogenesis. Front. Biosci. 2008, 13, 2335–2355. [Google Scholar] [CrossRef] [Green Version]
- Polyak, K.; Weinberg, R.A. Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat. Rev. Cancer 2009, 9, 265–273. [Google Scholar] [CrossRef]
- Galdiero, M.R.; Bonavita, E.; Barajon, I.; Garlanda, C.; Mantovani, A.; Jaillon, S. Tumor associated macrophages and neutrophils in cancer. Immunobiology 2013, 218, 1402–1410. [Google Scholar] [CrossRef]
- Friesel, R.E.; Maciag, T. Molecular mechanisms of angiogenesis: Fibroblast growth factor signal transduction. FASEB J. 1995, 9, 919–925. [Google Scholar] [CrossRef]
- Augustin, H.G.; Koh, G.Y.; Thurston, G.; Alitalo, K. Control of vascular morphogenesis and homeostasis through the angiopoietin-Tie system. Nat. Rev. Mol. Cell Biol. 2009, 10, 165–177. [Google Scholar] [CrossRef]
- Good, D.J.; Polverini, P.J.; Rastinejad, F.; Le Beau, M.M.; Lemons, R.S.; Frazier, W.A.; Bouck, N.P. A tumor suppressor-dependent inhibitor of angiogenesis is immunologically and functionally indistinguishable from a fragment of thrombospondin. Proc. Natl. Acad. Sci. USA 1990, 87, 6624–6628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Reilly, M.S.; Boehm, T.; Shing, Y.; Fukai, N.; Vasios, G.; Lane, W.S.; Flynn, E.; Birkhead, J.R.; Olsen, B.R.; Folkman, J. Endostatin: An endogenous inhibitor of angiogenesis and tumor growth. Cell 1997, 88, 277–285. [Google Scholar] [CrossRef] [Green Version]
- O’Reilly, M.S.; Holmgren, L.; Shing, Y.; Chen, C.; Rosenthal, R.A.; Moses, M.; Lane, W.S.; Cao, Y.; Sage, E.H.; Folkman, J. Angiostatin: A novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell 1994, 79, 315–328. [Google Scholar] [CrossRef]
- Pugh, C.W.; Ratcliffe, P.J. Regulation of angiogenesis by hypoxia: Role of the HIF system. Nat. Med. 2003, 9, 677–684. [Google Scholar] [CrossRef]
- North, S.; Moenner, M.; Bikfalvi, A. Recent developments in the regulation of the angiogenic switch by cellular stress factors in tumors. Cancer Lett. 2005, 218, 1–14. [Google Scholar] [CrossRef]
- Folkman, J.; Watson, K.; Ingber, D.; Hanahan, D. Induction of angiogenesis during the transition from hyperplasia to neoplasia. Nature 1989, 339, 58–61. [Google Scholar] [CrossRef]
- Gannon, G.; Mandriota, S.J.; Cui, L.; Baetens, D.; Pepper, M.S.; Christofori, G. Overexpression of vascular endothelial growth factor-A165 enhances tumor angiogenesis but not metastasis during beta-cell carcinogenesis. Cancer Res. 2002, 62, 603–608. [Google Scholar]
- O’Reilly, T.; Lane, H.A.; Wood, J.M.; Schnell, C.; Littlewood-Evans, A.; Brueggen, J.; McSheehy, P.M. Everolimus and PTK/ZK show synergistic growth inhibition in the orthotopic BL16/BL6 murine melanoma model. Cancer Chemother. Pharmacol. 2011, 67, 193–200. [Google Scholar] [CrossRef]
- Vajkoczy, P.; Menger, M.D.; Vollmar, B.; Schilling, L.; Schmiedek, P.; Hirth, K.P.; Ullrich, A.; Fong, T.A. Inhibition of tumor growth, angiogenesis, and microcirculation by the novel Flk-1 inhibitor SU5416 as assessed by intravital multi-fluorescence videomicroscopy. Neoplasia 1999, 1, 31–41. [Google Scholar] [CrossRef] [Green Version]
- Bergers, G.; Javaherian, K.; Lo, K.M.; Folkman, J.; Hanahan, D. Effects of angiogenesis inhibitors on multistage carcinogenesis in mice. Science 1999, 284, 808–812. [Google Scholar] [CrossRef] [Green Version]
- Bergers, G.; Song, S. The role of pericytes in blood-vessel formation and maintenance. Neuro. Oncol. 2005, 7, 452–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollard, J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef]
- Zumsteg, A.; Christofori, G. Corrupt policemen: Inflammatory cells promote tumor angiogenesis. Curr. Opin. Oncol. 2009, 21, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Bertolini, F.; Shaked, Y.; Mancuso, P.; Kerbel, R.S. The multifaceted circulating endothelial cell in cancer: Towards marker and target identification. Nat. Rev. Cancer 2006, 6, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Bailey, A.S.; Willenbring, H.; Jiang, S.; Anderson, D.A.; Schroeder, D.A.; Wong, M.H.; Grompe, M.; Fleming, W.H. Myeloid lineage progenitors give rise to vascular endothelium. Proc. Natl. Acad. Sci. USA 2006, 103, 13156–13161. [Google Scholar] [CrossRef] [Green Version]
- Nolan, D.J.; Ciarrocchi, A.; Mellick, A.S.; Jaggi, J.S.; Bambino, K.; Gupta, S.; Heikamp, E.; McDevitt, M.R.; Scheinberg, D.A.; Benezra, R.; et al. Bone marrow-derived endothelial progenitor cells are a major determinant of nascent tumor neovascularization. Genes. Dev. 2007, 21, 1546–1558. [Google Scholar] [CrossRef] [Green Version]
- Kuczynski, E.A.; Vermeulen, P.B.; Pezzella, F.; Kerbel, R.S.; Reynolds, A.R. Vessel co-option in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 469–493. [Google Scholar] [CrossRef]
- Kurzrock, R.; Stewart, D.J. Exploring the Benefit/Risk Associated with Antiangiogenic Agents for the Treatment of Non-Small Cell Lung Cancer Patients. Clin. Cancer Res. 2017, 23, 1137–1148. [Google Scholar] [CrossRef] [Green Version]
- Khasraw, M.; Ameratunga, M.; Grommes, C. Bevacizumab for the treatment of high-grade glioma: An update after phase III trials. Expert Opin. Biol. Ther. 2014, 14, 729–740. [Google Scholar] [CrossRef]
- Vasudev, N.S.; Reynolds, A.R. Anti-angiogenic therapy for cancer: Current progress, unresolved questions and future directions. Angiogenesis 2014, 17, 471–494. [Google Scholar] [CrossRef] [Green Version]
- Leenders, W.P.; Kusters, B.; de Waal, R.M. Vessel co-option: How tumors obtain blood supply in the absence of sprouting angiogenesis. Endothelium 2002, 9, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Winkler, F. Hostile takeover: How tumours hijack pre-existing vascular environments to thrive. J. Pathol. 2017, 242, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Szabo, V.; Bugyik, E.; Dezso, K.; Ecker, N.; Nagy, P.; Timar, J.; Tovari, J.; Laszlo, V.; Bridgeman, V.L.; Wan, E.; et al. Mechanism of tumour vascularization in experimental lung metastases. J. Pathol. 2015, 235, 384–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuczynski, E.A.; Yin, M.; Bar-Zion, A.; Lee, C.R.; Butz, H.; Man, S.; Daley, F.; Vermeulen, P.B.; Yousef, G.M.; Foster, F.S.; et al. Co-option of Liver Vessels and Not Sprouting Angiogenesis Drives Acquired Sorafenib Resistance in Hepatocellular Carcinoma. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raymaekers, K.; Stegen, S.; van Gastel, N.; Carmeliet, G. The vasculature: A vessel for bone metastasis. Bonekey Rep. 2015, 4, 742. [Google Scholar] [CrossRef] [Green Version]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.Y.; et al. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 2013, 15, 807–817. [Google Scholar] [CrossRef]
- Baker, G.J.; Yadav, V.N.; Motsch, S.; Koschmann, C.; Calinescu, A.A.; Mineharu, Y.; Camelo-Piragua, S.I.; Orringer, D.; Bannykh, S.; Nichols, W.S.; et al. Mechanisms of glioma formation: Iterative perivascular glioma growth and invasion leads to tumor progression, VEGF-independent vascularization, and resistance to antiangiogenic therapy. Neoplasia 2014, 16, 543–561. [Google Scholar] [CrossRef] [Green Version]
- Terayama, N.; Terada, T.; Nakanuma, Y. Histologic growth patterns of metastatic carcinomas of the liver. Jpn. J. Clin. Oncol. 1996, 26, 24–29. [Google Scholar] [CrossRef] [Green Version]
- Kanai, T.; Hirohashi, S.; Upton, M.P.; Noguchi, M.; Kishi, K.; Makuuchi, M.; Yamasaki, S.; Hasegawa, H.; Takayasu, K.; Moriyama, N.; et al. Pathology of small hepatocellular carcinoma. A proposal for a new gross classification. Cancer 1987, 60, 810–819. [Google Scholar] [CrossRef]
- Frentzas, S.; Simoneau, E.; Bridgeman, V.L.; Vermeulen, P.B.; Foo, S.; Kostaras, E.; Nathan, M.; Wotherspoon, A.; Gao, Z.H.; Shi, Y.; et al. Vessel co-option mediates resistance to anti-angiogenic therapy in liver metastases. Nat. Med. 2016, 22, 1294–1302. [Google Scholar] [CrossRef] [Green Version]
- Bridgeman, V.L.; Vermeulen, P.B.; Foo, S.; Bilecz, A.; Daley, F.; Kostaras, E.; Nathan, M.R.; Wan, E.; Frentzas, S.; Schweiger, T.; et al. Vessel co-option is common in human lung metastases and mediates resistance to anti-angiogenic therapy in preclinical lung metastasis models. J. Pathol. 2017, 241, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Guerin, E.; Man, S.; Xu, P.; Kerbel, R.S. A model of postsurgical advanced metastatic breast cancer more accurately replicates the clinical efficacy of antiangiogenic drugs. Cancer Res. 2013, 73, 2743–2748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, P.P.; Demircioglu, F.; Ghazaly, E.; Alrawashdeh, W.; Stratford, M.R.; Scudamore, C.L.; Cereser, B.; Crnogorac-Jurcevic, T.; McDonald, S.; Elia, G.; et al. Dual-action combination therapy enhances angiogenesis while reducing tumor growth and spread. Cancer Cell 2015, 27, 123–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynolds, A.R.; Hart, I.R.; Watson, A.R.; Welti, J.C.; Silva, R.G.; Robinson, S.D.; Da Violante, G.; Gourlaouen, M.; Salih, M.; Jones, M.C.; et al. Stimulation of tumor growth and angiogenesis by low concentrations of RGD-mimetic integrin inhibitors. Nat. Med. 2009, 15, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Giraudo, E.; Inoue, M.; Hanahan, D. An amino-bisphosphonate targets MMP-9-expressing macrophages and angiogenesis to impair cervical carcinogenesis. J. Clin. Invest. 2004, 114, 623–633. [Google Scholar] [CrossRef]
- Santini, D.; Vincenzi, B.; Dicuonzo, G.; Avvisati, G.; Massacesi, C.; Battistoni, F.; Gavasci, M.; Rocci, L.; Tirindelli, M.C.; Altomare, V.; et al. Zoledronic acid induces significant and long-lasting modifications of circulating angiogenic factors in cancer patients. Clin. Cancer Res. 2003, 9, 2893–2897. [Google Scholar]
- Nozawa, H.; Chiu, C.; Hanahan, D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 12493–12498. [Google Scholar] [CrossRef] [Green Version]
- Bertagnolli, M.M.; Eagle, C.J.; Zauber, A.G.; Redston, M.; Solomon, S.D.; Kim, K.; Tang, J.; Rosenstein, R.B.; Wittes, J.; Corle, D.; et al. Celecoxib for the prevention of sporadic colorectal adenomas. N. Engl. J. Med. 2006, 355, 873–884. [Google Scholar] [CrossRef]
- Aragon-Ching, J.B.; Li, H.; Gardner, E.R.; Figg, W.D. Thalidomide analogues as anticancer drugs. Recent. Pat. Anticancer. Drug Discov. 2007, 2, 167–174. [Google Scholar] [CrossRef]
- Al-Abd, A.M.; Alamoudi, A.J.; Abdel-Naim, A.B.; Neamatallah, T.A.; Ashour, O.M. Anti-angiogenic agents for the treatment of solid tumors: Potential pathways, therapy and current strategies—A review. J. Adv. Res. 2017, 8, 591–605. [Google Scholar] [CrossRef]
- Meadows, K.L.; Hurwitz, H.I. Anti-VEGF therapies in the clinic. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartnett, M.E.; Martiniuk, D.; Byfield, G.; Geisen, P.; Zeng, G.; Bautch, V.L. Neutralizing VEGF decreases tortuosity and alters endothelial cell division orientation in arterioles and veins in a rat model of ROP: Relevance to plus disease. Invest. Ophthalmol. Vis. Sci. 2008, 49, 3107–3114. [Google Scholar] [CrossRef] [PubMed]
- Goffin, J.R.; Straume, O.; Chappuis, P.O.; Brunet, J.S.; Begin, L.R.; Hamel, N.; Wong, N.; Akslen, L.A.; Foulkes, W.D. Glomeruloid microvascular proliferation is associated with p53 expression, germline BRCA1 mutations and an adverse outcome following breast cancer. Br. J. Cancer 2003, 89, 1031–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sitohy, B.; Chang, S.; Sciuto, T.E.; Masse, E.; Shen, M.; Kang, P.M.; Jaminet, S.C.; Benjamin, L.E.; Bhatt, R.S.; Dvorak, A.M.; et al. Early Actions of Anti-Vascular Endothelial Growth Factor/Vascular Endothelial Growth Factor Receptor Drugs on Angiogenic Blood Vessels. Am. J. Pathol. 2017, 187, 2337–2347. [Google Scholar] [CrossRef] [Green Version]
- Gordon, M.S.; Margolin, K.; Talpaz, M.; Sledge, G.W., Jr.; Holmgren, E.; Benjamin, R.; Stalter, S.; Shak, S.; Adelman, D. Phase I safety and pharmacokinetic study of recombinant human anti-vascular endothelial growth factor in patients with advanced cancer. J. Clin. Oncol. 2001, 19, 843–850. [Google Scholar] [CrossRef]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef] [Green Version]
- Ilhan-Mutlu, A.; Osswald, M.; Liao, Y.; Gommel, M.; Reck, M.; Miles, D.; Mariani, P.; Gianni, L.; Lutiger, B.; Nendel, V.; et al. Bevacizumab Prevents Brain Metastases Formation in Lung Adenocarcinoma. Mol. Cancer Ther. 2016, 15, 702–710. [Google Scholar] [CrossRef] [Green Version]
- Montero, A.J.; Escobar, M.; Lopes, G.; Gluck, S.; Vogel, C. Bevacizumab in the treatment of metastatic breast cancer: Friend or foe? Curr. Oncol. Rep. 2012, 14, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Rosen, L.S.; Jacobs, I.A.; Burkes, R.L. Bevacizumab in Colorectal Cancer: Current Role in Treatment and the Potential of Biosimilars. Target. Oncol. 2017, 12, 599–610. [Google Scholar] [CrossRef]
- Saltz, L.B.; Clarke, S.; Diaz-Rubio, E.; Scheithauer, W.; Figer, A.; Wong, R.; Koski, S.; Lichinitser, M.; Yang, T.S.; Rivera, F.; et al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: A randomized phase III study. J. Clin. Oncol. 2008, 26, 2013–2019. [Google Scholar] [CrossRef] [Green Version]
- Van Cutsem, E.; Cervantes, A.; Adam, R.; Sobrero, A.; Van Krieken, J.H.; Aderka, D.; Aranda Aguilar, E.; Bardelli, A.; Benson, A.; Bodoky, G.; et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann. Oncol. 2016, 27, 1386–1422. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Ryoo, B.Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.Y.; Breder, V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Pembrolizumab As Second-Line Therapy in Patients With Advanced Hepatocellular Carcinoma in KEYNOTE-240: A Randomized, Double-Blind, Phase III Trial. J. Clin. Oncol. 2020, 38, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Sun, W. Ziv-aflibercept in metastatic colorectal cancer. Biologics 2014, 8, 13–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, P.A.; Moore, M.J. Aflibercept in the treatment of patients with metastatic colorectal cancer: Latest findings and interpretations. Therap. Adv. Gastroenterol. 2013, 6, 459–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sitohy, B.; Nagy, J.A.; Dvorak, H.F. Anti-VEGF/VEGFR therapy for cancer: Reassessing the target. Cancer Res. 2012, 72, 1909–1914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadenhe-Chiweshe, A.; Papa, J.; McCrudden, K.W.; Frischer, J.; Bae, J.O.; Huang, J.; Fisher, J.; Lefkowitch, J.H.; Feirt, N.; Rudge, J.; et al. Sustained VEGF blockade results in microenvironmental sequestration of VEGF by tumors and persistent VEGF receptor-2 activation. Mol. Cancer Res. 2008, 6, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrara, N. Role of myeloid cells in vascular endothelial growth factor-independent tumor angiogenesis. Curr. Opin. Hematol. 2010, 17, 219–224. [Google Scholar] [CrossRef]
- Abdalla, A.M.E.; Xiao, L.; Ullah, M.W.; Yu, M.; Ouyang, C.; Yang, G. Current Challenges of Cancer Anti-angiogenic Therapy and the Promise of Nanotherapeutics. Theranostics 2018, 8, 533–548. [Google Scholar] [CrossRef]
- Ribatti, D. Mast cells and macrophages exert beneficial and detrimental effects on tumor progression and angiogenesis. Immunol. Lett. 2013, 152, 83–88. [Google Scholar] [CrossRef]
- Raffaghello, L.; Vacca, A.; Pistoia, V.; Ribatti, D. Cancer associated fibroblasts in hematological malignancies. Oncotarget 2015, 6, 2589–2603. [Google Scholar] [CrossRef] [Green Version]
- De Palma, M.; Venneri, M.A.; Galli, R.; Sergi Sergi, L.; Politi, L.S.; Sampaolesi, M.; Naldini, L. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell 2005, 8, 211–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shojaei, F.; Wu, X.; Malik, A.K.; Zhong, C.; Baldwin, M.E.; Schanz, S.; Fuh, G.; Gerber, H.P.; Ferrara, N. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat. Biotechnol. 2007, 25, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Azam, F.; Mehta, S.; Harris, A.L. Mechanisms of resistance to antiangiogenesis therapy. Eur. J. Cancer 2010, 46, 1323–1332. [Google Scholar] [CrossRef]
- Casanovas, O.; Hicklin, D.J.; Bergers, G.; Hanahan, D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 2005, 8, 299–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.; Ding, Y.; Zhou, M.; Rini, B.I.; Petillo, D.; Qian, C.N.; Kahnoski, R.; Futreal, P.A.; Furge, K.A.; Teh, B.T. Interleukin-8 mediates resistance to antiangiogenic agent sunitinib in renal cell carcinoma. Cancer Res. 2010, 70, 1063–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, A.S.; Wu, X.; Zhuang, G.; Ngu, H.; Kasman, I.; Zhang, J.; Vernes, J.M.; Jiang, Z.; Meng, Y.G.; Peale, F.V.; et al. An interleukin-17-mediated paracrine network promotes tumor resistance to anti-angiogenic therapy. Nat. Med. 2013, 19, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- Rigamonti, N.; Kadioglu, E.; Keklikoglou, I.; Wyser Rmili, C.; Leow, C.C.; De Palma, M. Role of angiopoietin-2 in adaptive tumor resistance to VEGF signaling blockade. Cell Rep. 2014, 8, 696–706. [Google Scholar] [CrossRef] [Green Version]
- Keunen, O.; Johansson, M.; Oudin, A.; Sanzey, M.; Rahim, S.A.; Fack, F.; Thorsen, F.; Taxt, T.; Bartos, M.; Jirik, R.; et al. Anti-VEGF treatment reduces blood supply and increases tumor cell invasion in glioblastoma. Proc. Natl. Acad. Sci. USA 2011, 108, 3749–3754. [Google Scholar] [CrossRef] [Green Version]
- Leenders, W.P.; Kusters, B.; Verrijp, K.; Maass, C.; Wesseling, P.; Heerschap, A.; Ruiter, D.; Ryan, A.; de Waal, R. Antiangiogenic therapy of cerebral melanoma metastases results in sustained tumor progression via vessel co-option. Clin. Cancer Res. 2004, 10, 6222–6230. [Google Scholar] [CrossRef] [Green Version]
- Rossi, D.; Hermanowicz, M.; Serment, G.; Khouzami, A.; Bretheau, D.; Ducassou, J. Urethral diverticula in women. Apropos of 7 cases. Ann. Urol. 1989, 23, 352–353. [Google Scholar]
- Navis, A.C.; Bourgonje, A.; Wesseling, P.; Wright, A.; Hendriks, W.; Verrijp, K.; van der Laak, J.A.; Heerschap, A.; Leenders, W.P. Effects of dual targeting of tumor cells and stroma in human glioblastoma xenografts with a tyrosine kinase inhibitor against c-MET and VEGFR2. PLoS ONE 2013, 8, 58262. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, P.; Ulbricht, U.; Bohlen, P.; Brockmann, M.A.; Fillbrandt, R.; Stavrou, D.; Westphal, M.; Lamszus, K. Inhibition of glioma angiogenesis and growth in vivo by systemic treatment with a monoclonal antibody against vascular endothelial growth factor receptor-2. Cancer Res. 2001, 61, 6624–6628. [Google Scholar]
- Madar-Balakirski, N.; Tempel-Brami, C.; Kalchenko, V.; Brenner, O.; Varon, D.; Scherz, A.; Salomon, Y. Permanent occlusion of feeding arteries and draining veins in solid mouse tumors by vascular targeted photodynamic therapy (VTP) with Tookad. PLoS ONE 2010, 5, 10282. [Google Scholar] [CrossRef] [PubMed]
- Mohajeri, A.; Sanaei, S.; Kiafar, F.; Fattahi, A.; Khalili, M.; Zarghami, N. The Challenges of Recombinant Endostatin in Clinical Application: Focus on the Different Expression Systems and Molecular Bioengineering. Adv. Pharm. Bull. 2017, 7, 21–34. [Google Scholar] [CrossRef]
- Allen, R.T.; Cluck, M.W.; Agrawal, D.K. Mechanisms controlling cellular suicide: Role of Bcl-2 and caspases. Cell Mol. Life Sci. 1998, 54, 427–445. [Google Scholar] [CrossRef] [PubMed]
- Qiu, B.; Ji, M.; Song, X.; Zhu, Y.; Wang, Z.; Zhang, X.; Wu, S.; Chen, H.; Mei, L.; Zheng, Y. Co-delivery of docetaxel and endostatin by a biodegradable nanoparticle for the synergistic treatment of cervical cancer. Nanoscale Res. Lett. 2012, 7, 666. [Google Scholar] [CrossRef] [Green Version]
- Mohajeri, A.; Pilehvar-Soltanahmadi, Y.; Pourhassan-Moghaddam, M.; Abdolalizadeh, J.; Karimi, P.; Zarghami, N. Cloning and Expression of Recombinant Human Endostatin in Periplasm of Escherichia coli Expression System. Adv. Pharm. Bull. 2016, 6, 187–194. [Google Scholar] [CrossRef] [Green Version]
- Boehm, T.; Folkman, J.; Browder, T.; O’Reilly, M.S. Antiangiogenic therapy of experimental cancer does not induce acquired drug resistance. Nature 1997, 390, 404–407. [Google Scholar] [CrossRef]
- Luo, H.; Xu, M.; Zhu, X.; Zhao, J.; Man, S.; Zhang, H. Lung cancer cellular apoptosis induced by recombinant human endostatin gold nanoshell-mediated near-infrared thermal therapy. Int. J. Clin. Exp. Med. 2015, 8, 8758–8766. [Google Scholar]
- Danafar, H.; Davaran, S.; Rostamizadeh, K.; Valizadeh, H.; Hamidi, M. Biodegradable m-PEG/PCL Core-Shell Micelles: Preparation and Characterization as a Sustained Release Formulation for Curcumin. Adv. Pharm. Bull. 2014, 4, 501–510. [Google Scholar] [CrossRef]
- Jiang, L.P.; Zou, C.; Yuan, X.; Luo, W.; Wen, Y.; Chen, Y. N-terminal modification increases the stability of the recombinant human endostatin in vitro. Biotechnol. Appl. Biochem. 2009, 54, 113–120. [Google Scholar] [CrossRef]
- Chen, W.; Hu, S. Suitable carriers for encapsulation and distribution of endostar: Comparison of endostar-loaded particulate carriers. Int. J. Nanomed. 2011, 6, 1535–1541. [Google Scholar] [CrossRef] [Green Version]
- Hu, S.; Zhang, Y. Endostar-loaded PEG-PLGA nanoparticles: In vitro and in vivo evaluation. Int. J. Nanomed. 2010, 5, 1039–1048. [Google Scholar] [CrossRef] [Green Version]
- Sennino, B.; Ishiguro-Oonuma, T.; Wei, Y.; Naylor, R.M.; Williamson, C.W.; Bhagwandin, V.; Tabruyn, S.P.; You, W.K.; Chapman, H.A.; Christensen, J.G.; et al. Suppression of tumor invasion and metastasis by concurrent inhibition of c-Met and VEGF signaling in pancreatic neuroendocrine tumors. Cancer Discov. 2012, 2, 270–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fredrickson, J.; Serkova, N.J.; Wyatt, S.K.; Carano, R.A.; Pirzkall, A.; Rhee, I.; Rosen, L.S.; Bessudo, A.; Weekes, C.; de Crespigny, A. Clinical translation of ferumoxytol-based vessel size imaging (VSI): Feasibility in a phase I oncology clinical trial population. Magn. Reson. Med. 2017, 77, 814–825. [Google Scholar] [CrossRef] [Green Version]
- Cortes-Santiago, N.; Hossain, M.B.; Gabrusiewicz, K.; Fan, X.; Gumin, J.; Marini, F.C.; Alonso, M.M.; Lang, F.; Yung, W.K.; Fueyo, J.; et al. Soluble Tie2 overrides the heightened invasion induced by anti-angiogenesis therapies in gliomas. Oncotarget 2016, 7, 16146–16157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, Y.J.; Kim, H.Z.; Hwang, S.I.; Lee, J.E.; Oh, N.; Jung, K.; Kim, M.; Kim, K.E.; Kim, H.; Lim, N.K.; et al. Double antiangiogenic protein, DAAP, targeting VEGF-A and angiopoietins in tumor angiogenesis, metastasis, and vascular leakage. Cancer Cell 2010, 18, 171–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reardon, D.A.; Lassman, A.B.; Schiff, D.; Yunus, S.A.; Gerstner, E.R.; Cloughesy, T.F.; Lee, E.Q.; Gaffey, S.C.; Barrs, J.; Bruno, J.; et al. Phase 2 and biomarker study of trebananib, an angiopoietin-blocking peptibody, with and without bevacizumab for patients with recurrent glioblastoma. Cancer 2018, 124, 1438–1448. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AngiomiR | Molecular Function | Reference |
|---|---|---|
| miR-15b, miR-16, miR-20a, miR-20b | Have no known functions. They might contribute in regulation of VEGF. | [10] |
| miR-21, miR-31 | Triggers mobilisation of EC. | [11] |
| miR-17-92 | Dysregulation of miR-17-92 in cancer cells promote growth. | [12] |
| miR-130a | Induces angiogenesis by supressing GAX and HOXA5 | [13] |
| miR-296 | Animal studies showed that by acting on HGS, miR-296 stimulate angiogenesis. | [14] |
| miR-320 | Suppression of miR-320 in diabetic cells trigger angiogenesis by stimulating EC proliferation. | [15] |
| miR-210 | In hypoxic cell culture, miR-210 promote EC proliferation and survival. | [16] |
| miR-378 | Support tumour growth by improving vascularisation via angiogenesis. | [17] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saman, H.; Raza, S.S.; Uddin, S.; Rasul, K. Inducing Angiogenesis, a Key Step in Cancer Vascularization, and Treatment Approaches. Cancers 2020, 12, 1172. https://doi.org/10.3390/cancers12051172
Saman H, Raza SS, Uddin S, Rasul K. Inducing Angiogenesis, a Key Step in Cancer Vascularization, and Treatment Approaches. Cancers. 2020; 12(5):1172. https://doi.org/10.3390/cancers12051172
Chicago/Turabian StyleSaman, Harman, Syed Shadab Raza, Shahab Uddin, and Kakil Rasul. 2020. "Inducing Angiogenesis, a Key Step in Cancer Vascularization, and Treatment Approaches" Cancers 12, no. 5: 1172. https://doi.org/10.3390/cancers12051172
APA StyleSaman, H., Raza, S. S., Uddin, S., & Rasul, K. (2020). Inducing Angiogenesis, a Key Step in Cancer Vascularization, and Treatment Approaches. Cancers, 12(5), 1172. https://doi.org/10.3390/cancers12051172