2. Methods
2.1. Cell Culture
Raji cells, a human Burkitt lymphoma cell line, were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA) and maintained in RPMI supplemented with 10% fetal bovine serum (FBS) and antibiotics (Sigma, St. louis, MO, USA). The cells were maintained at 37 °C in a humidified 5% CO2 incubator. MINO, U2932, and Jeko-1 (mantle cell lymphoma) cells were also maintained under the same condition.
2.2. Cell Cycle Analysis
After 1 day of irradiation, the cells were harvested for analysis. The cells were pelleted by centrifugation at. 1200 rpm for 3 min, fixed by the dropwise addition of 5 mL ice-cold 70% ethanol with vortexing, and then incubated at 4 °C for at least 1 h. The fixed cells were pelleted by centrifugation, rinsed with PBS, and pelleted again. The cells were then incubated with propidium iodide ReadyProbesTM reagent (2 drops/1 × 106 cells, Thermo Fisher Scientific, Eugene, OR, USA) containing 100 mg RNase (1 mg/mL in PBS; Qiagen) at 37 °C for 30 min. Finally, 10,000 cells of each sample were analyzed by fluorescence-activated cell sorter (BD Biosciences, San Jose, CA, USA) and CellQuest software (BD Biosciences, San Jose, CA, USA) to determine their cell cycle stage.
2.3. Lymphoma Xenograft Subcutaneous Model
Five-week-old female NOD/SCID mice were obtained from Animal Resource Center (WA, Australia). The mice were maintained in temperature-controlled clean racks with a 12-h light/dark cycle and allowed to acclimatize for 1 week before the start of the experiment. Approximately 1 × 107 Raji cells were injected subcutaneously into the right flank. When the tumor size reached approximately 200 mm3, the mice were randomized into each group (n = 4–5/group). The tumor size was measured at the indicated times by using a digital caliper, and the tumor volume was calculated using the formula width2 × length × 0.4. To monitor potential toxicity, body weight was measured. The mice were euthanized when the tumor size exceeded the volume of 1,500 mm3 or the body weight loss was > 20% of the original weight.
2.4. Conjugation of Alexa Fluor 488 to Rituximab
A solution of Alexa Fluor 488 (Invitrogen, Carlsbad, CA, USA) in dimethyl sulfoxide with 1% acetic acid was prepared. This solution was immediately added to 500 μL (10 mg/mL) dissolved in 1 M of sodium bicarbonate solution, pH 8.4. The solution was mixed thoroughly and left to stand for 1 h at room temperature. This reaction solution was purified by using a size exclusion PD-10 column (GE Healthcare, IL, USA) with phosphate-buffered saline (PBS) as the elution buffer. The protein concentration of the purified solution was quantified by using a Nano-drop spectrophotometer.
2.5. In Vivo Antibody Penetration Studies
When the tumor size reached ~200 mm3, Alexa488-RTX was intravenously injected as a single dose (150 μg), and ATV (12 μg/day in PBS) (approximately equivalent to 40 mg/day in human treatment) was administered via oral gavage for 5 days. After 5 days, the mice were exsanguinated by cardiac puncture and dissected. The tumors were isolated from the mice and immediately fixed with 4% paraformaldehyde overnight at 4 °C. Then, 8-μm tumor sections from three different regions were cut using a Leica CM 1850 cryostat (Leica microsystems, Wetzlar, Germany) to obtain representative sections throughout the tumor. After three washes with 200 μL PBS, TUNEL-positive cells were stained with Click-iT® TUNEL Alexa Fluor® 647 Imaging Assay kit (Invitrogen, Carlsbad, CA, USA). For antibody penetration, we imaged and calculated Alexa488-RTX in whole tumor images. And apoptotic cells was calculated by TUNEL assay.
2.6. 131I-Radiolabeling with Rituximab
Pierce pre-coated iodination tubes (Thermo Scientific, Eugene, OR, USA) were used for 131I radiolabeling of RTX. 131I (100 μL; 59.2 MBq) was added in a pre-coated iodination tube and incubated for 10 min with shaking at 18–21 °C (room temperature). Subsequently, 200 μg of RTX was added to the tube and reacted for 10 min at room temperature. After labeling, an instant thin layer chromatography (solvent: 100% C3H6O) test showed that the radiochemical purity of 131I-RTX was > 95%. The immunoreactivity of 131I-RTX was determined as 87.7% by using a cell-binding assay and the specific activity was 86.2 ± 11.8 MBq/mg.
2.7. Radioimmunotherapy and Tumor Growth Delay
When the tumor volume in Raji-bearing mice reached ~200 mm3, the mice were randomly divided into five groups (n = 5–6 per group). Each group was treated with a single dose of PBS, ATV (12 μg/day in PBS), 131I-RTX (150 μg, 12.95 MBq), and 131I-RTX (150 μg, 12.95 MBq) plus ATV (12 μg/day in PBS).
2.8. SPECT/CT Image of 131I-Rituximab
All SPECT scans in this study were performed by using a Mediso nanoSPECT/CT scanner (Mediso, Budapest, Hungary). When the tumor size reached ~ 200 mm3, ATV (12 μg/day in PBS) was orally administered daily for a total of 10 days; PBS was administered to the control group. 131I-RTX (150 μg, 12.1–14.6 MBq/200 μL) was intravenously injected after 5 days of administration of ATV or PBS. SPECT data were obtained at 2, 24, 48, and 72 h after the injection of 131I-RTX.
2.9. Autoradiography
Immediately after SPECT/CT scanning, the tumor tissues were isolated and frozen in an optimal cutting temperature (OCT) compound. After decaying for 48 h, the frozen tumors were sectioned at 20 μm thickness by using a cryostat microtome and exposed on an imaging plate for 24 h. The plates were scanned with BAS-5000 (Fujifilm Life science Co., Japan). The intensities of tumor uptake were quantified as units of photostimulated luminescence per square millimeter (PSL/μm2) computed by using Multi Gauge software (version 3.0; Fujifilm, Los Altos, CA, USA).
2.10. In Vivo Optical Imaging of Orthotopic Models
Luciferase-expressing Raji cells (1 × 106) were injected into the tail vein of NOD/SCID mice for the establishment of orthotopic model. Seven days after cell injection, 131I-RTX (150 μg, 12.1–14.6 MBq/ 200 μL) was intravenously injected and ATV (12 μg/day in PBS) was orally administered daily for a total of 5 days; PBS was administered to the control group. For in vivo optical imaging, scans using IVIS Spectrum (Perkinelmer, Waltham, MA, USA) were performed 14 days after the cell injection. Bioluminescence images were acquired 6 min post-injection. Depending on bioluminescence intensity, the images were collected under various conditions with exposure times ranging from 1 to 60 s, binning from 4 to 16, and a 13.5-field of view. The total number of photons emitted per second was quantified using Living Image analysis software (v 4.5.1, Perkinelmer, Waltham, MA, USA).
2.11. In Vivo Tumor Model and Administration of ATV and miR-346 Mimic
Therapy was started when the tumor volume reached ~200 mm3. The tumor-bearing mice were randomly assigned to four groups (n = 5 each): (1) control mice receiving intraperitoneal (i.p.) injection of 100 μL PBS and intratumoral injection of 5 μg miRNA negative mimic; (2) mice receiving i.p. intratumoral injection of 5 μg has-miR-346 mimic; (3) mice receiving orally administered ATV (12 μg/day in PBS) and intratumoral injection of 5 μg has-miR-346 mimic; (4) mice receiving ATV (12 μg/day in PBS), intratumoral injection of 5 μg has-miR-346 mimic, and intratumoral injection of 5 μg has-miR-346 inhibitor. For in vivo administration of miRNA mimic, the negative control or miR-346 mimic was complexed with in vivo-JetPEI at an N/P ratio of 6 in 5% glucose solution (a total of 20 μL) for intratumoral injection. The mice were treated twice a week.
2.12. Immunocytochemistry of γH2AX
The cells were cultured with 5 μM ATV in 6-well plates for 2 days and then irradiated. At 3 h after 8 Gy of irradiation, the cells were fixed in 3.7% formaldehyde, blocked with 1% BSA/0.2% Triton-X-100 diluted in PBS, and stained with anti-γH2AX antibody (Abcam, Cambridge, UK) followed by a secondary antibody (anti-rabbit IgG-TRITC, Sigma, St. louis, MO, USA). The cells were then washed, loaded into a pre-assembled cytospin cuvette, and centrifuged at. 1000 rpm for 5 min in Shadon Cytospin 3 (Thermo Scientific, Eugene, OR, USA) to attach Raji cells. The cells were cover-slipped with mounting medium containing a 40, 6-diamidino-2-phenylindole dihydrochloride (DAPI, Invitrogen, Carlsbad, CA, USA) solution (Vector Laboratories, Inc., Burlingame, CA, USA). All groups were then assessed for γH2AX by using confocal microscopy (LSM 710, Carl Zeiss, Germany) with filters specific for DAPI (excitation/emission: 359/461 nm) and TRITC (excitation/emission: 555/580 nm).
2.13. Whole Code Set Heat Map and Cluster Analysis
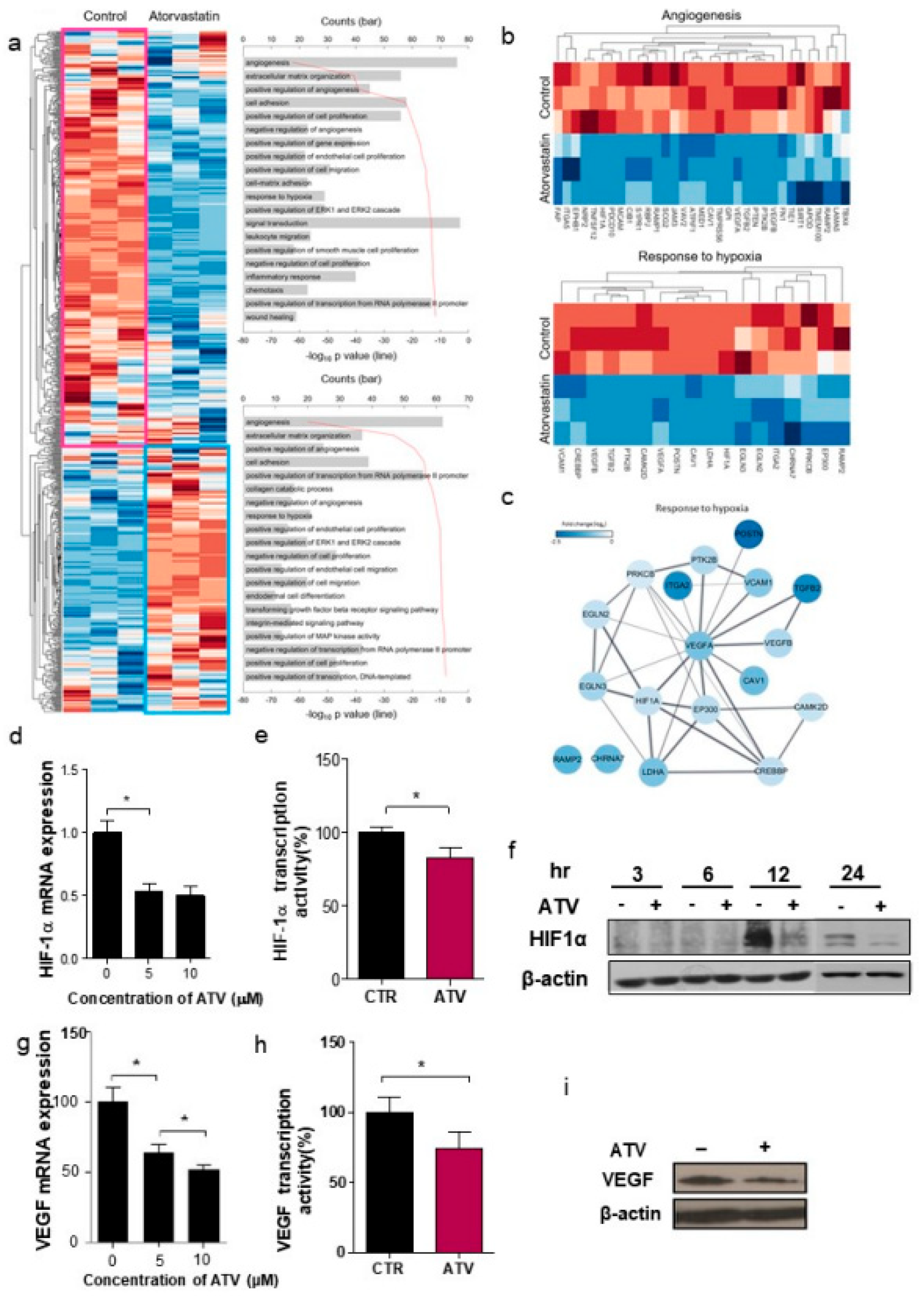
Unsupervised hierarchical analysis of the genes from control (n = 3) and ATV-treated mice (n = 3) tumor tissues was performed using NanoString nCounter gene expression assay. Z score corrected expression data were presented. Lower signal of expression was indicated in blue and higher signal in red. Two large clusters, cluster 1 (439 genes) and cluster 2 (279 genes), were identified (shown as pink and sky-blue). The top 10 gene ontology (GO) terms associated with cluster 1 (pink box) and cluster 2 (sky-blue boxes) from the unsupervised hierarchical cluster analysis were presented.
2.14. NanoString nCounter Preparation, Operation, and Analysis
Seven minutes prior to euthanization and tumor removal, the total RNA was isolated from the tumor tissue by using QIAGEN Allprep kit (QIAGEN, Hilden, Germany). NanoString nCounter assay was performed using 300 ng of purified RNA from control and ATV-treated mice samples. The hybridization reaction was performed according to the manufacturer’s instruction, and the sample was incubated at 65 ℃ for at least 12 h before direct processing. After the experimental process and NanoString nCounter digital reading, the counts for all RNAs were extracted, normalized, and analyzed using a NanoString nSolver software and in-house MATLAB (Mathworks, MA, USA) code. The genes with a p-value < 0.05 were selected. For normalization, we used nCounter-nSolver analysis software (version 4.0, nanoString, Seattle, WA, USA), which included normalization process using housekeeping gene/positive control/negative control.
2.15. Bioinformatics Analyses
The cluster genes were analyzed using the DAVID web toolkit (
http://david.avcc.ncifcrf.goc) to assign GO categories and plotted by homemade MATLAB code, following the DAVID manual. To interrogate the interactions between our interesting cluster gene set and pathways, we used STRING according to the manual (Search Tool for Recurring Instances of Neighbouring Genes).
2.16. Cell Viability Assay and Cell Lysate Analysis
To examine the cytotoxicity induced by ATV and RTX in various lymphoma cells, we treated Raji, Jeko-1, Mino (mantle cell lymphoma), and U2932 cells with ATV for 2 days and RTX for 1 h in. To determine the concentration of ATV for viability of Raji cells, we treated the cells with various concentrations (0, 0.5, 1, 2, 5, 10, 20, and 50 μM) of ATV for 48 h. Cell viability was measured at an emission wavelength of 590 nm using a microplate reader. Next, to examine the cytotoxicity induced by ATV and RTX in various lymphoma cells, we treated the cells with ATV for 2 days and RTX for 1 h. Among variable doses, 10 μM showed effective cytotoxicity, corresponding to an inhibitory concentration of 25% at 48 h exposure.
Next, cytotoxicity induced by poly (ADP-ribose) polymerase-1, which is a unique characteristic of apoptotic cell death, was proven by immunoblotting of cell lysates using the indicated antibodies.
2.17. Western Blotting
The cells were subsequently lysed with RIPA buffer (Thermo Scientific, Eugene, OR, USA), and proteins were separated by sodium-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. The membranes were blocked with 1% (v/v) non-fat dried milk in Tris-buffered saline with 0.05% Tween 20 and incubated with the required antibodies. Primary antibodies were used at a 1:1000 dilution (5% bovine serum albumin) and secondary antibodies at a 1:5000 dilution (5% skim milk). Immunoreactive protein bands were visualized via enhanced chemiluminescence (Amersham Biosciences, Freiburg, Germany) and scanned.
2.18. Reagents and Antibodies
The miR-346 mimics, control mimics, miR-346 inhibitors, and control inhibitors were all purchased from Bioneer (Daejeon, Korea). In vivo-jetPEI®in vivo DNA and siRNA delivery reagent was purchased from Polyplus-transfection® SA (Polyplus, New York, NY) and lipofectamine (Thermo Fishers Scientific, Eugene, OR, USA). The primary antibodies used for western blot analyses were obtained as follows: Anti-HIF1alpha (BD Transduction Laboratories, San Jose, CA, USA); anti-VEGF, anti-cyclin B, anti-β-actin (Santa Cruz Biotechnology, Dallas, TX, USA); anti-cleaved PARP, anti-caspase-3, anti-p-cdc2 (Cell Signaling Technology Danvers, MA, USA); and anti-γ- H2AX (Millipore, Billerica, MA, USA). ATV was purchased from Sigma-Aldrich (St. Louis, MO, USA).
2.19. Matrigel In Vitro Endothelial tube Formation Assay
Endothelial cell tube formation assays were performed on Matrigel-coated chamber slides and the results were photographed (Nikon Eclipse Ti microscope with DS-Fi1 camera) at ×40 magnification. Tube formation was quantified by counting the number of connected cells in randomly selected fields at ×400 magnification and dividing that number by the total number of cells in the same field.
2.20. Quantitative RT-PCR Analysis
To verify the array results, quantitative RT-PCR (qRT-PCR) analysis was performed by using total RNA obtained from Raji cells treated with different concentrations of ATV. The total RNA was isolated by using RNeasy isolation Kit (QIAGEN, Hilden, Germany) and copy DNA (cDNA) was synthesized from 1 mg of RNA by using the miScript II RT kit (QIAGEN, Hilden, Germany) in accordance with the manufacturer’s instructions. qRT-PCR was performed by using miScript Primer Assays (QIAGEN, Hilden, Germany) and SYBR Green qPCR Master Mix (QIAGEN, Hilden, Germany) at 95 °C for 3 min and 40 cycles of 95 °C for 15 s and 62 °C for 30 s on. 7500 Real time PCR System (Applied Biosystems, Waltham, Massachusetts, USA). Each miRNA expression was represented as a fold-change relative to the expression of small RNA 5S rRNA, which was used as an internal control of qRT-PCR. The difference in miRNA expression was determined as a fold change using the 2−ΔΔCt method.
2.21. Measurement of HIF-1α Subunit and VEGF Production by Enzyme-Linked Immunosorbent Assay
Intracellular HIF-1α and VEGF activity were assayed using the SimpleStep enzyme-linked immunosorbent assay (ELISA) kit (Abcam, Cambridge, UK) according to manufacturer’s instructions with plates assayed on a 96-well plate reader at 450 nm.
2.22. Microarray and Data Analysis
Using 250 ng of total RNA, the labeling process was initiated with poly-A tailing of each RNA strand with poly-A polymerase, followed by the ligation of biotin-labeled 3DNA dendrimer. Biotinylated RNA strands were hybridized at 48 °C for 18 h on an Affymetrix GeneChip miRNA 4.0 Array (Affymetrix, Hatfield, PA, USA). The GeneChip miRNA 4.0 Array was washed and stained in Affymetrix Fluidics Station 450. The fluorescence signals amplified by the branched structure of the 3DNA dendrimer were scanned by using an Affymetrix GeneChip Scanner. 3000 7G. The arrays were analyzed by using an Agilent scanner and the associated software. miRNA expression levels were calculated by using Expression Console 1.4 (Affymetrix, Hatfield, PA, USA). The relative signal intensities for each miRNA were generated by using the Robust Multi-Array Average algorithm. The target predictions were computed and analyzed by using TargetScan and microRNA.org databases.
2.23. miRNA and Transient Transfection
The cells were transiently transfected with 60 nM control or miR-346 mimics, or with 120 nM control or miR-346 inhibitors, using X-treme GENE siRNA Transfection Reagent (Roche, Indianapolis, IN, USA). We used miRNA scramble as a control. The sequence of miRNA-346 mimic was 3′ UCUCCGUCCGUACGCCCGUCUGU 5′. For irradiation analysis, at 2 days after transfection of the miRNA inhibitor and treatment with ATV, the cells were exposed to various doses of ionizing radiation.
2.24. Luciferase Assay
The cells were seeded into a 24-well plate and HIF1 3′UTR reporter constructs were co-transfected with miR-346 and pRL-CMV-Renilla internal control plasmid (Promega, Madison, WI, USA) using Lipofectamine. 2000. The luciferase activity was determined by the Dual-GloTM Luciferase assay system (Promega, Madison, WI, USA).
2.25. Irradiation
Raji cells were administered with/without 5 μM ATV and incubated at 37 ℃ for 2 days. Then, Raji cells were exposed to various doses of ionizing radiation from a 137Cs γ-ray source in Exposure Instrument Cammacell-40 (Atomic Energy of Canada, Ltd., Canada) at a dose rate of 3.81 Gy/min.
2.26. Three-dimensional (3D) Culture System
The cells were seeded in 96-well plates at a density of 1 × 104 cells/well. In the 3D culture model, 96-well plates were pre-coated with Matrigel as a basement membrane by adding 40 µL of Matrigel to each well followed by incubation at 37 °C for 30 min. The cells were plated onto the gel in appropriate medium, and the wells were photographed after 10 days.
2.27. Neutral Comet Assay
To detect double-strand breaks (DSBs), a neutral comet (single-cell gel electrophoresis) assay was performed according to the manufacturer’s instructions (Trevigen, Gaithersburg, MD, USA). The cells were plated in 100 mm tissue culture dishes at 1 × 106 cells/dish and incubated overnight. After ATV or miR-346 inhibitor exposure for 24 h, the cells were irradiated and incubated for 24 h. The cells were then immediately lysed at 4 °C for 1 h in lysis buffer (2.5 M NaCl, 100 mM ethylenediamine tetraacetic acid, 10 mM Tris-HCl, 1% N-lauroylsarcosine, 1% Triton X-100, 10% DMSO, pH 10.0) and subjected to neutral electrophoresis buffer at 4 °C. To detect DNA, the slides were stained with ethidium bromide and examined for fluorescence emission using a 515–560 nm excitation filter and 590 nm barrier filter. DNA damage was quantified through computer-assisted image analysis (Komet analysis software, ver. 3.1; Kinetic Imaging, Liverpool, UK) to integrate fluorescence intensity.
2.28. Invasion Assay
The invasive ability was measured in vitro using transwell chambers, according to the manufacturer’s protocol. Briefly, cells were seeded onto the membrane of the upper chamber of the transwell at a concentration of 4 × 105 cells/mL in 150 μL of medium and left untreated or treated with ATV, radiation, miR-346 inhibitor, or a combination of both for 24 h. The medium in the upper chamber was serum-free, while the medium in the lower chamber contained 10% FBS as a source of chemo-attractants. The cells that passed through the Matrigel-coated membrane were stained with Cell Stain Solution containing crystal violet supplied in the transwell invasion assay (Chemicon, Millipore, Billerica, GA, USA) and photographed after 24 h of incubation.
2.29. Dosimetry
The radiation dose per unit of administered activity (mSv/MBq), effective dose in organ, and absorbed dose for the tumor region on the 131I-RTX SPECT in mice were calculated by using OLINDA/EXM software (OLINDA; Vanderbilt University, Nashville, TN, USA). To calculate the absorbed dose in the tumor, the sphere model of OLINDA was used. The tumor volume was calculated from 131I-RTX SPECT data with multiple slices of ROIs. The ROIs were delineated in the heart, stomach, intestine, liver, spleen, kidney, bladder, lung, bone, muscle, and tumor regions. After the delineation of ROIs on X-ray CT, ROIs were copied to the 131I-RTX SPECT data. Time activity curves (TACs) were obtained for each organ.
2.30. Ethics Approval and Consent to Participate
All experiments were performed under a protocol approved by IACUC (KIRAMS 2013-0099, KIRAMS 2015-0026, KIRAMS 2016-0019) of the Korea Institute of Radiological and Medical Sciences (KIRAMS) and by IACUC (2017-106) of Osong Medical Innovation Foundation.
2.31. Statistical Analysis
The data were presented as the mean ± SD and calculated using Student’s t test. Statistical significance was accepted for p values of < 0.05 and 0.005.
4. Discussion
In the present study, we reported for the first time that ATV improved the therapeutic effect of RIT using 131I-RTX. In addition, we proposed three hypotheses to elucidate the cause of the enhanced effect by ATV.
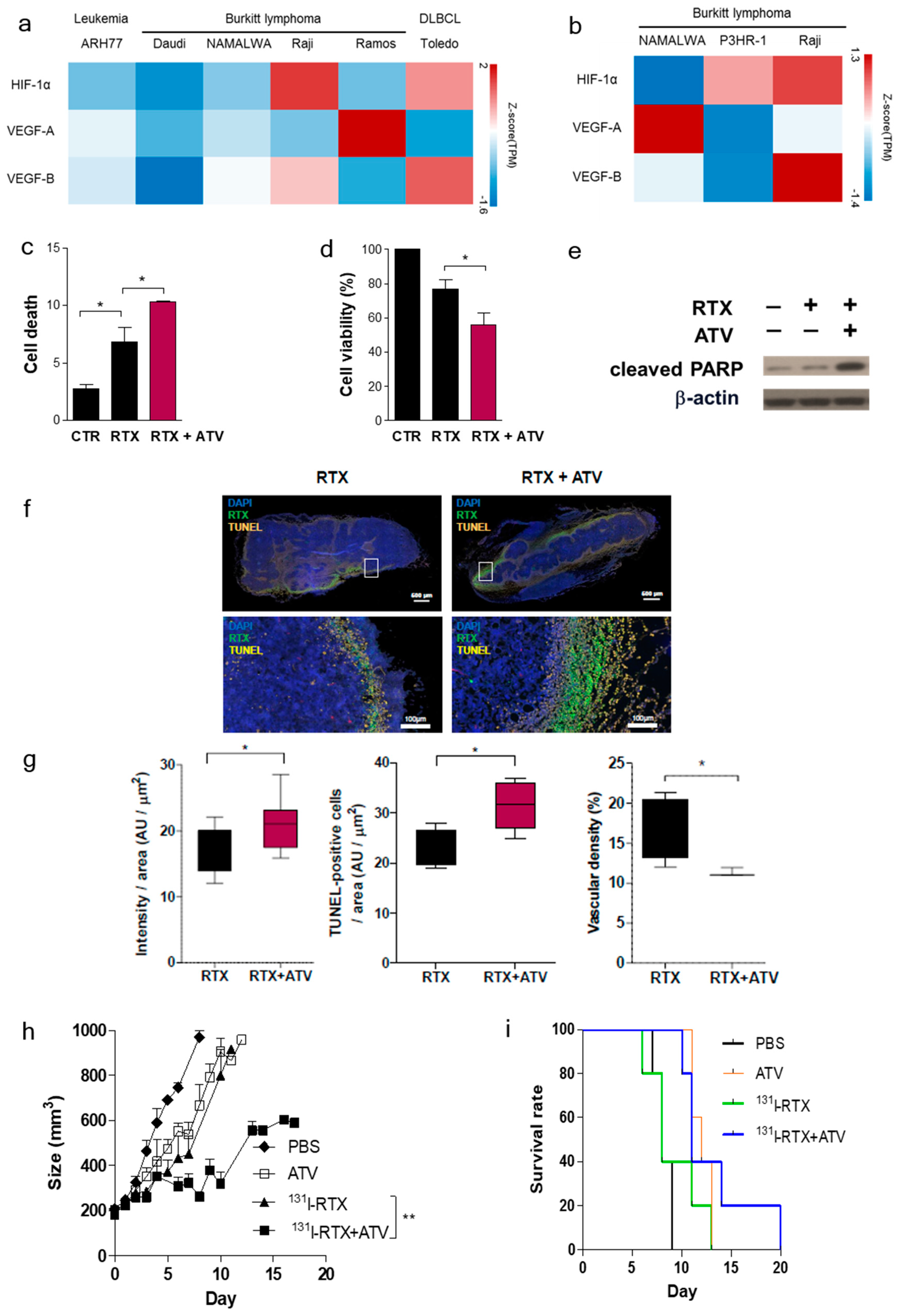
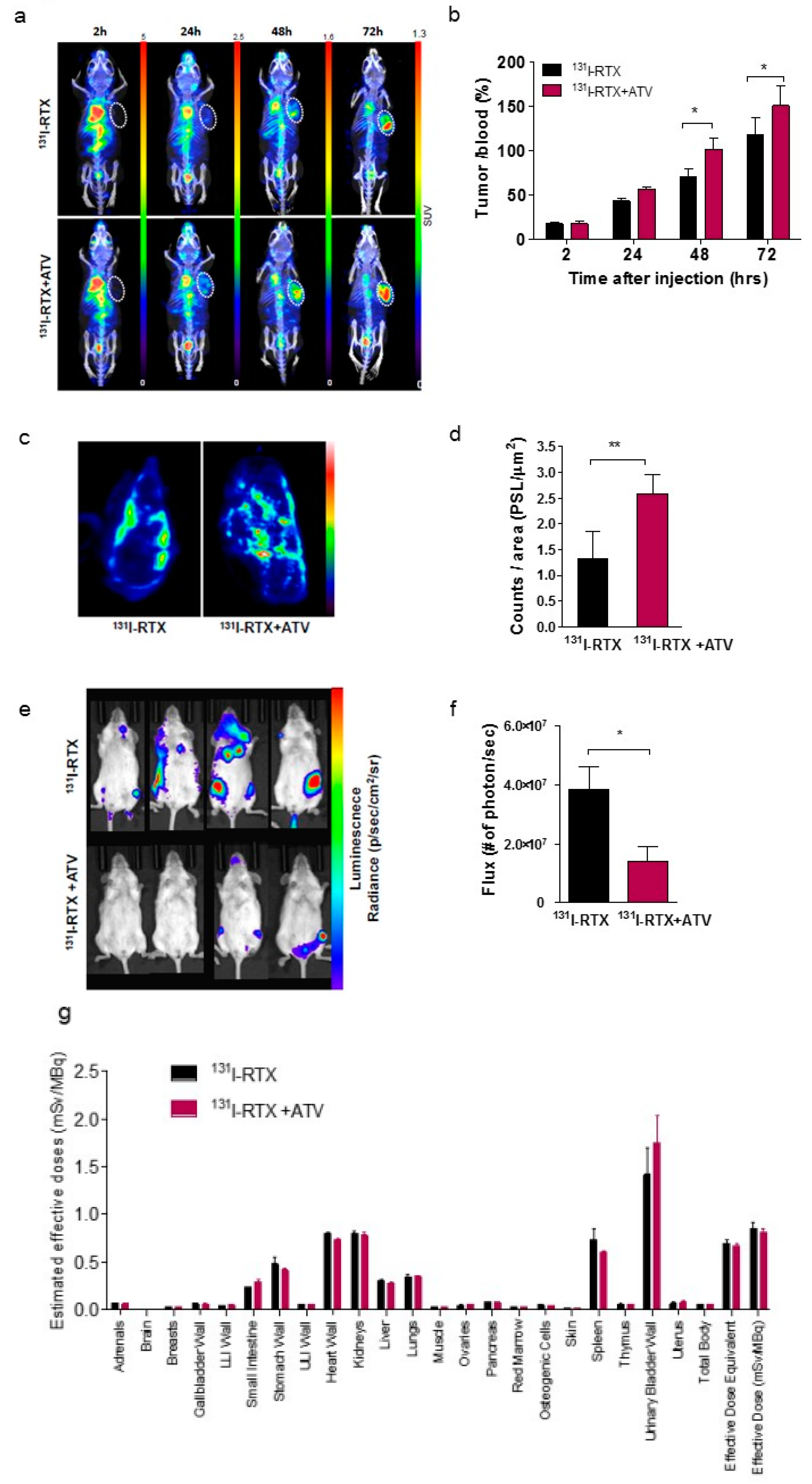
First, we investigated whether ATV improved the penetration of antibody into tumor by analyzing the fluorescence images of Alexa488-RTX and SPECT images of
131I-RTX (
Figure 1f and
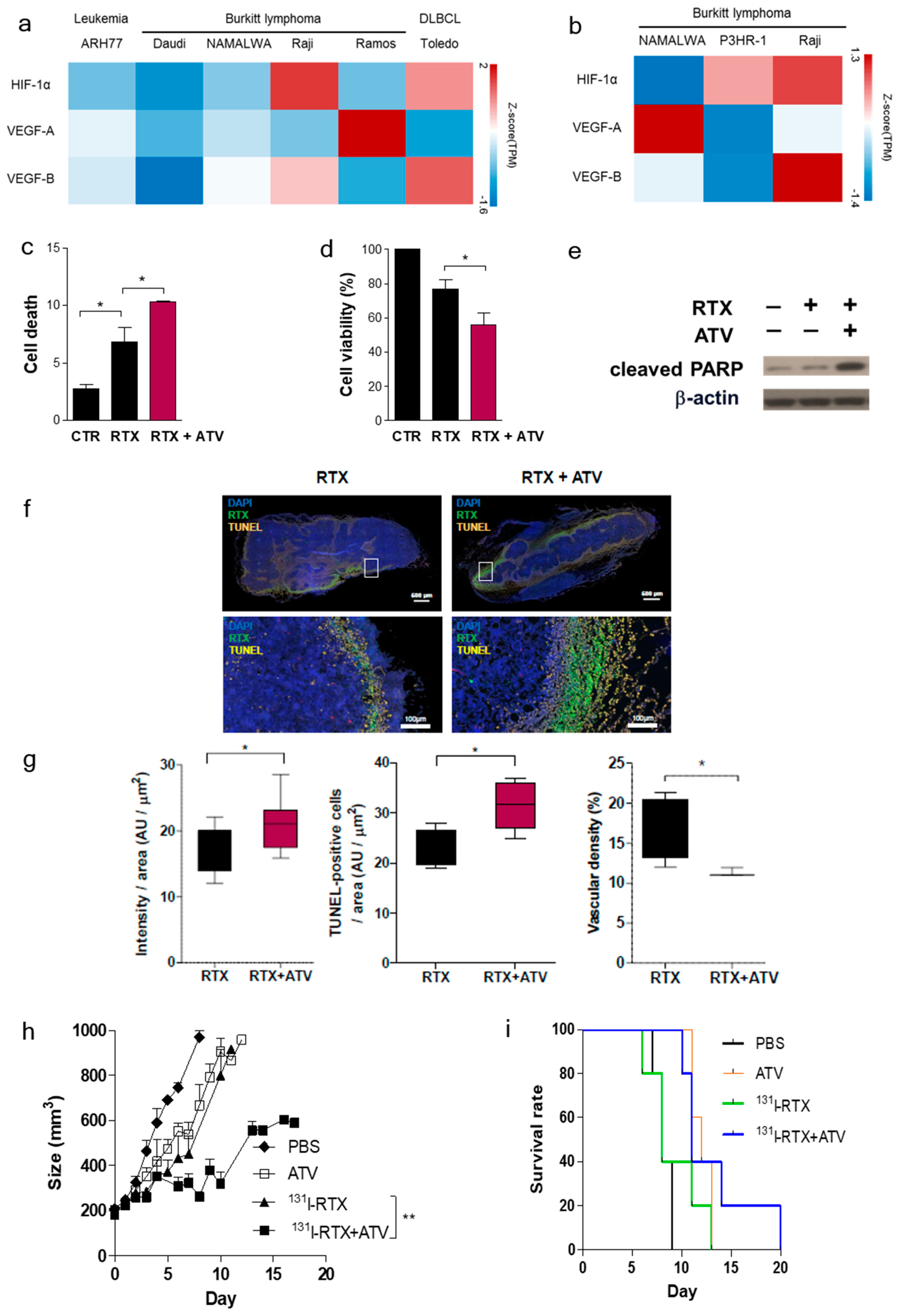
Figure 2a). Statins have been demonstrated to induce apoptosis by regulating Akt, Erk, and p38 signaling through the suppression of mevalonate pathway in lymphoma cells [
20]. We confirmed the increased apoptosis of ATV-treated lymphoma cells by using cell viability assay (
Figure S2a) and upregulated expression of miR-1182 and miR-608, which was related to inhibition of tumor growth and induction of cell death [
21,
22], according to ATV concentration by microarray and qRT-PCR (
Figure 5a and
Figure S6c,d). The induction of cancer cell death enhanced intratumoral penetration via formation of void space in tumor tissues [
23]. An increase in penetration of
131I-RTX resulted in increased absorption of the tumor dose, as presented by SPECT images, leading to DNA damage. DNA damage was proven by comet assay, western blotting of γH2AX, and colony assay (
Figure 6f–h). Tumor absorbed dose was calculated using OLIDNA program [
24]. The sphere model in OLINDA program provides the absorbed dose for the sphere mass in the range of 0.01–6000 g [
24]. Based on the absorbed dose, tumors in the PBS group received 175.5 mGy irradiation and those in the ATV-treated group received 210.7 mGy irradiation. We presumed that ATV may induce 20% more DSBs because the number of lesions induced in the DNA of a cell by a dose of 1–2 Gy was approximately 40 DSBs in cell killing [
25]. We speculated that the apoptosis increase by ATV resulted in increased penetration of
131I-RTX, which led to enhanced therapeutic effect of RIT in Raji cells.
Second, we found that vascular formation decreased in vitro and in vivo in ATV-treated Raji cells (
Figure 1g and
Figure S3b). Additionally, we found that the gene expression related to angiogenesis, for example, VEGF, was reduced in ATV-treated Raji cells. VEGF signaling promotes the formation and branching of neovascularization in tumors, leads to rapid tumor growth, and facilitates metastatic potential [
26]. Thus, we suggested that ATV induced anti-angiogenesis effects.
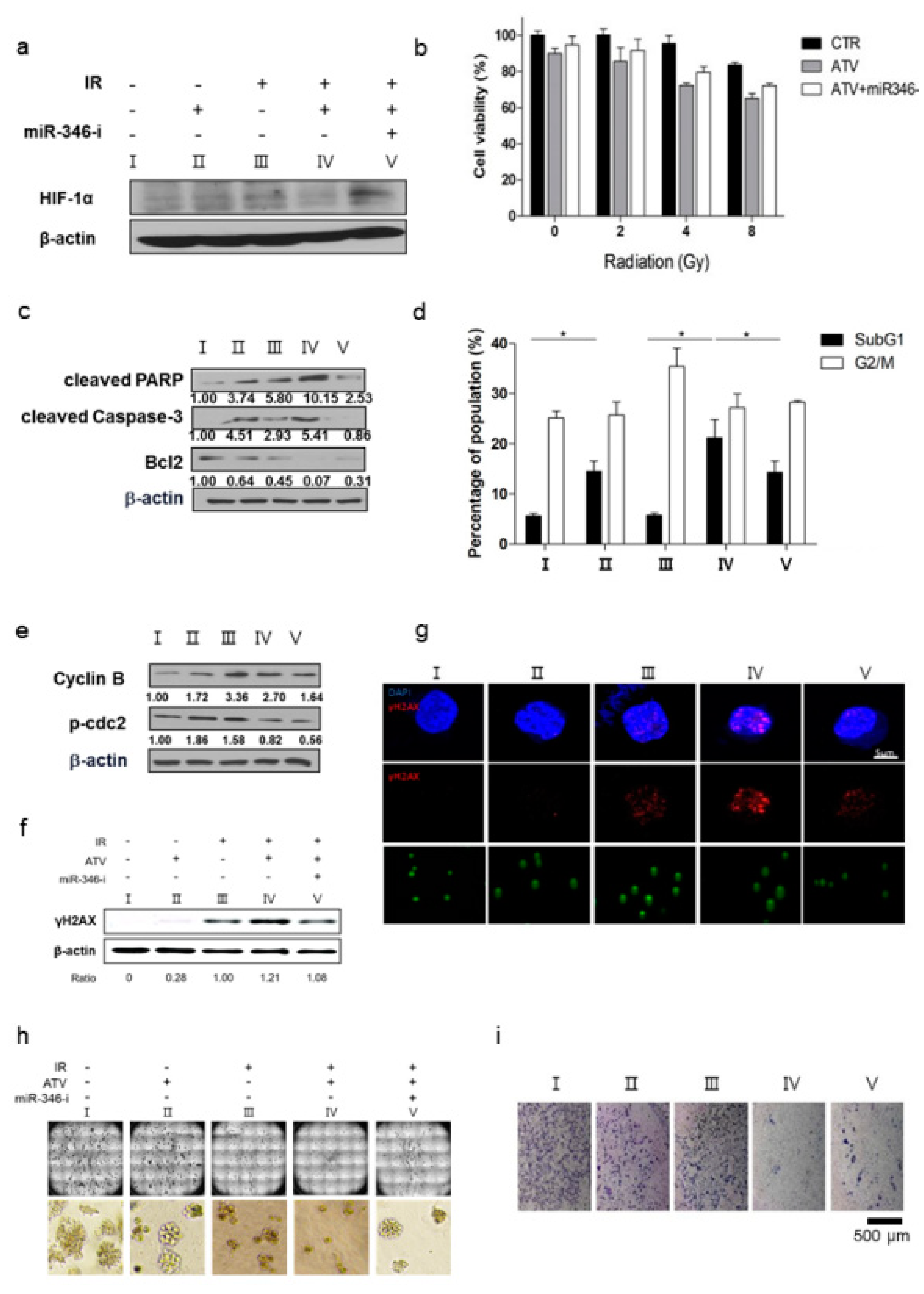
Third, we confirmed the improvement of radiosensitivity by ATV by using cell viability assays, colony assays, cell cycle analysis, and γH2AX formation (
Figure 6). When the same dose of radiation was irradiated, radiosensitive cells were subjected to more DNA damage than radioresistant cells because irradiation-induced DSBs led to apoptotic or mitotic death in cancer cells [
27]. Therefore, the enhancement of radiosensitivity by ATV improved the therapeutic effect of RIT in Raji cells. However, ATV treatment as an option for combination therapy with
131I-RTX should be tailored according to the cell subtype. In the present study, we found that ATV was the most effective in Raji lymphoma cells based on the results of cell viability assay: ATV exhibited the highest cytotoxicity against Raji cells when compared with other cells such as MINO, U2932 (DLBCL), and Jeko-1 in (
Figure S2a). This finding indicated that selection of ATV for combination therapy with
131I-RTX should be personalized according to the cell subtype.
The enhanced uptake of
131I-RTX can be explained by the results of Tunel assay (
Figure 1g). TUNEL assay showed that the proportion of apoptotic cells increased after treatment with ATV. The total uptake of
131I-RTX could be increased after reducing the interstitial pressure [
28] due to apoptotic effect of ATV treatment. Similarly, enhanced uptake of mAb can be found after combinatorial treatment with chemotherapy drugs. After treatment of paclitaxel and HIFU, tumor accumulation and penetration of
90Y-B3 were enhanced [
29,
30]. Reduced interstitial pressure has been reported to be another reason for this enhanced uptake of mAb [
29,
30].
Combinatorial treatment approach for the enhancement of mAb could be applied during RIT in solid tumor. The barriers of tumor microenvironment, such as extracellular matrix, cell to cell junction, and high interstitial pressure, should be overcome during RIT in solid tumor [
31,
32]
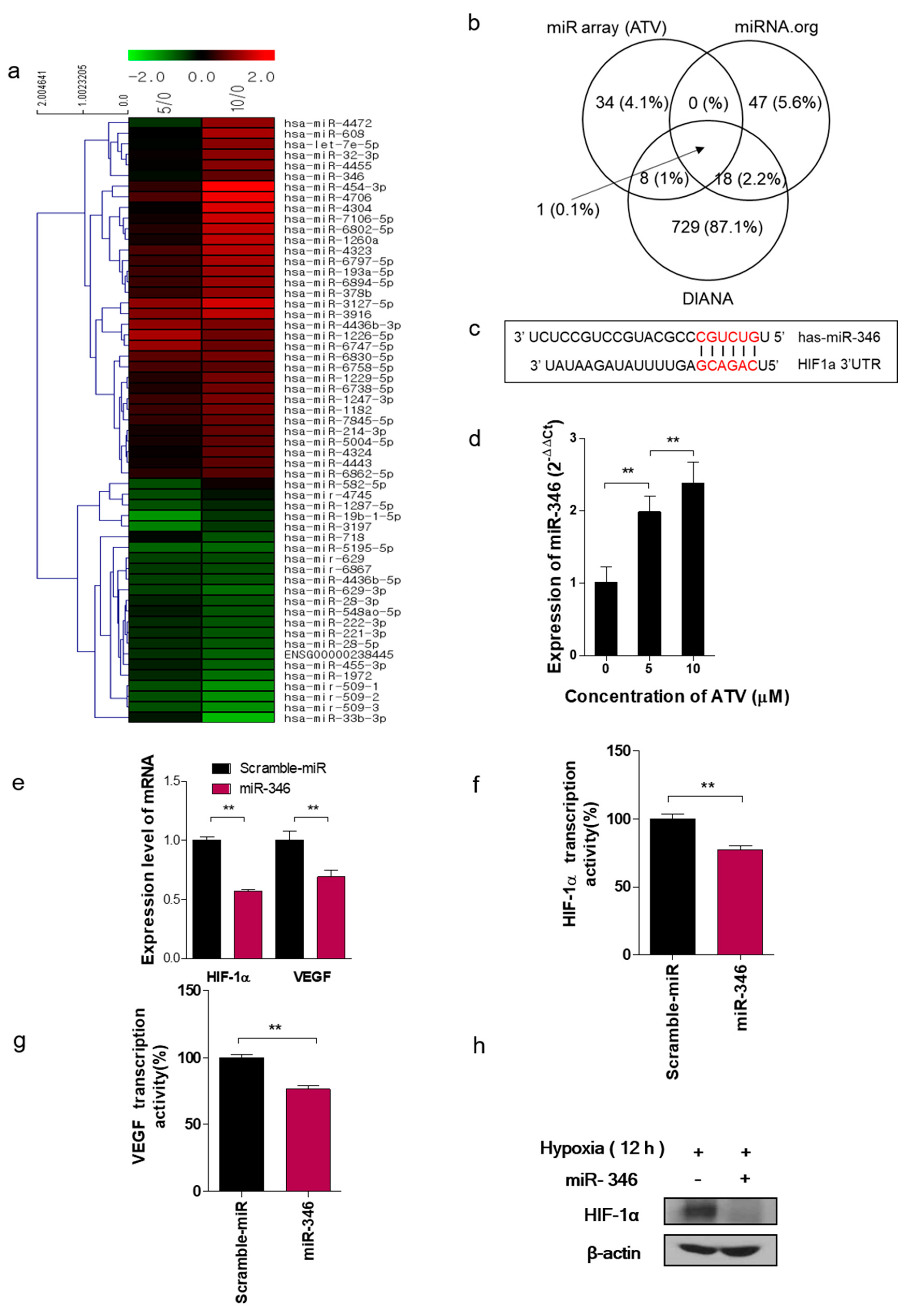
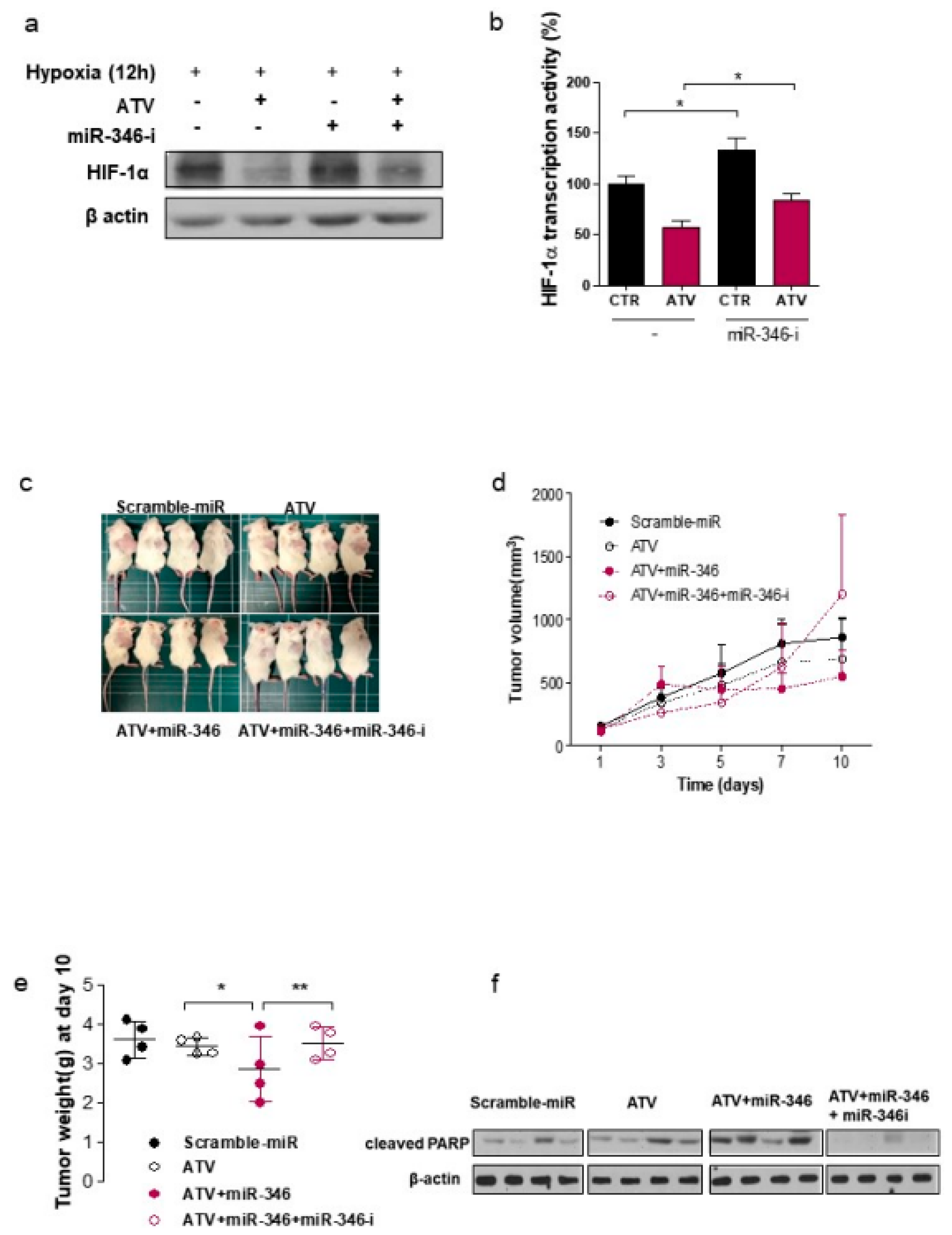
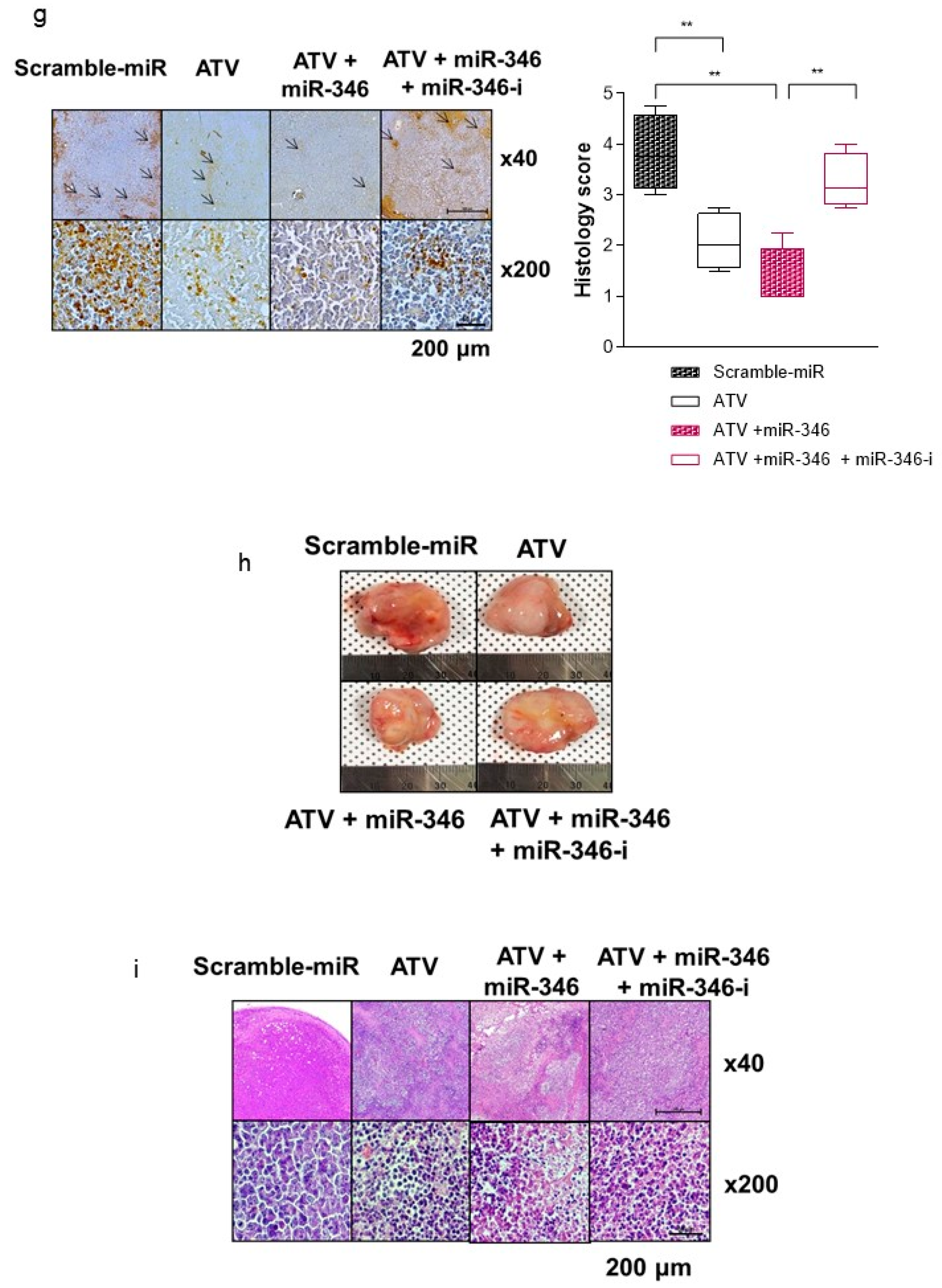
In this study, we noted the changes in miRNA after ATV treatment to elucidate the mechanism of enhanced radiosensitivity in Raji cells. No report on the relationship between ATV and miRNAs has been published to date. In the present study, overexpression mimics of miR-346 in Raji cells abrogated the mRNA levels of the angiogenesis-related proteins HIF-1α and VEGF under hypoxia and the relative increase of HIF-1α protein levels in cells transfected with an miR-346 inhibitor [
33], demonstrating that miR-346 targets the HIF-1α/VEGF axis (
Figure 4e). Consistently, ATV attenuated VEGF expression at the protein level, reduced VEGF secretion, and reduced HIF-1α expression in Raji cells transfected with miR-346 mimics compared with the untreated control, indicating the anti-angiogenic property of miR-346. Moreover, we confirmed that miR-346 was upregulated by ATV in Raji cells as determined by miRNA microarray and qRT-PCR analysis. Furthermore, the overexpression of miR-346 synergistically reduced VEGF production by lymphoma cells, implying an important role of miR-346 in ATV-induced anti-angiogenic activity. In the mouse models, increased expression of miR-346 induced by ATV treatment promoted significant tumor growth delay compared with that in the control. These findings suggest that increasing the expression of miR-346 could be a potential therapeutic strategy. Furthermore, we identified that miR-346 inhibition negated the enhanced radiosensitivity induced by ATV via stimulation of IR-induced DNA damage, apoptosis, G2/M cell cycle arrest, and angiogenesis (
Figure 6d).

 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}