Harnessing Gene Expression Profiles for the Identification of Ex Vivo Drug Response Genes in Pediatric Acute Myeloid Leukemia

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
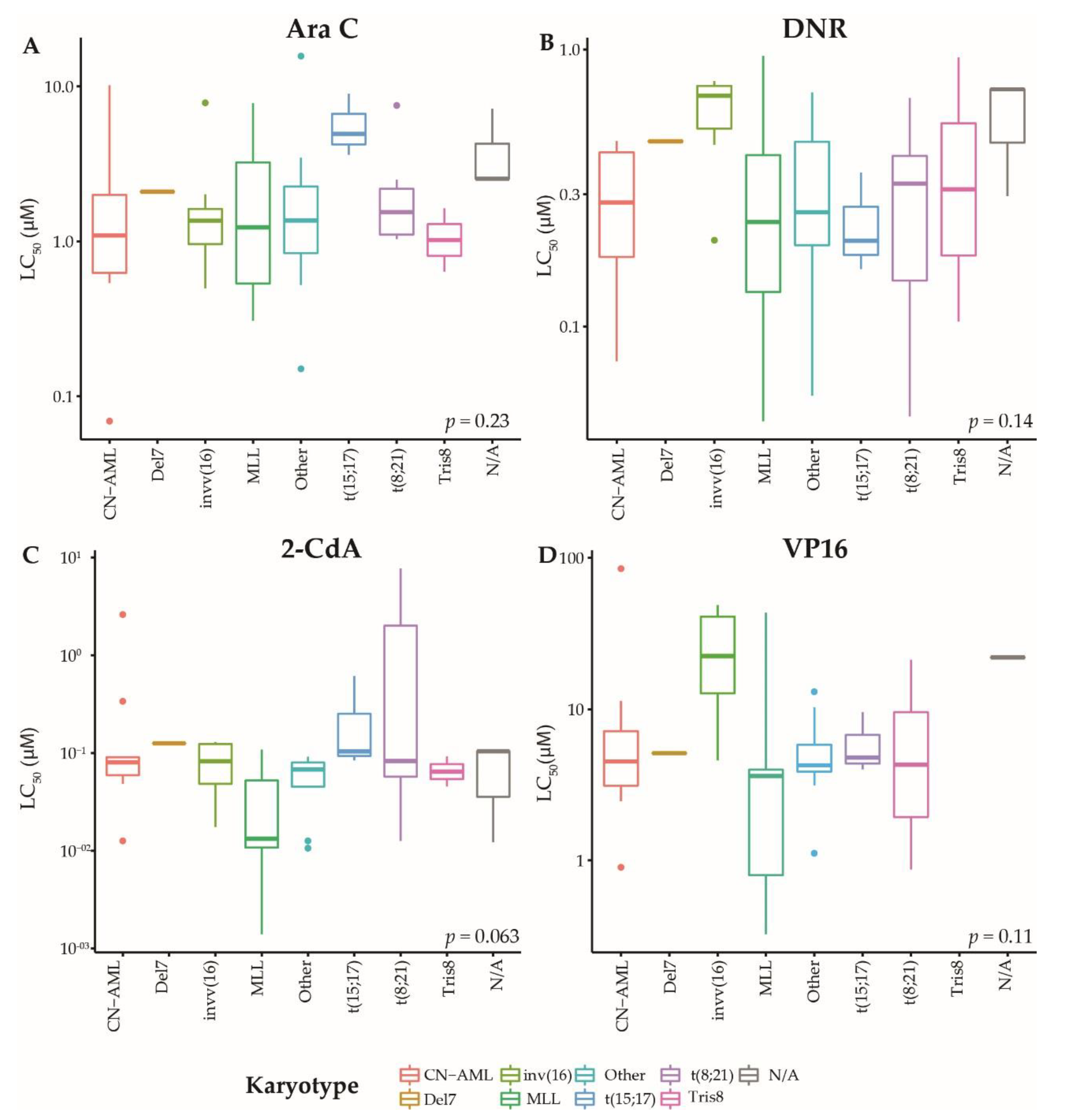
2.1. Patients and Ex Vivo Drug Response of Primary AML Blasts
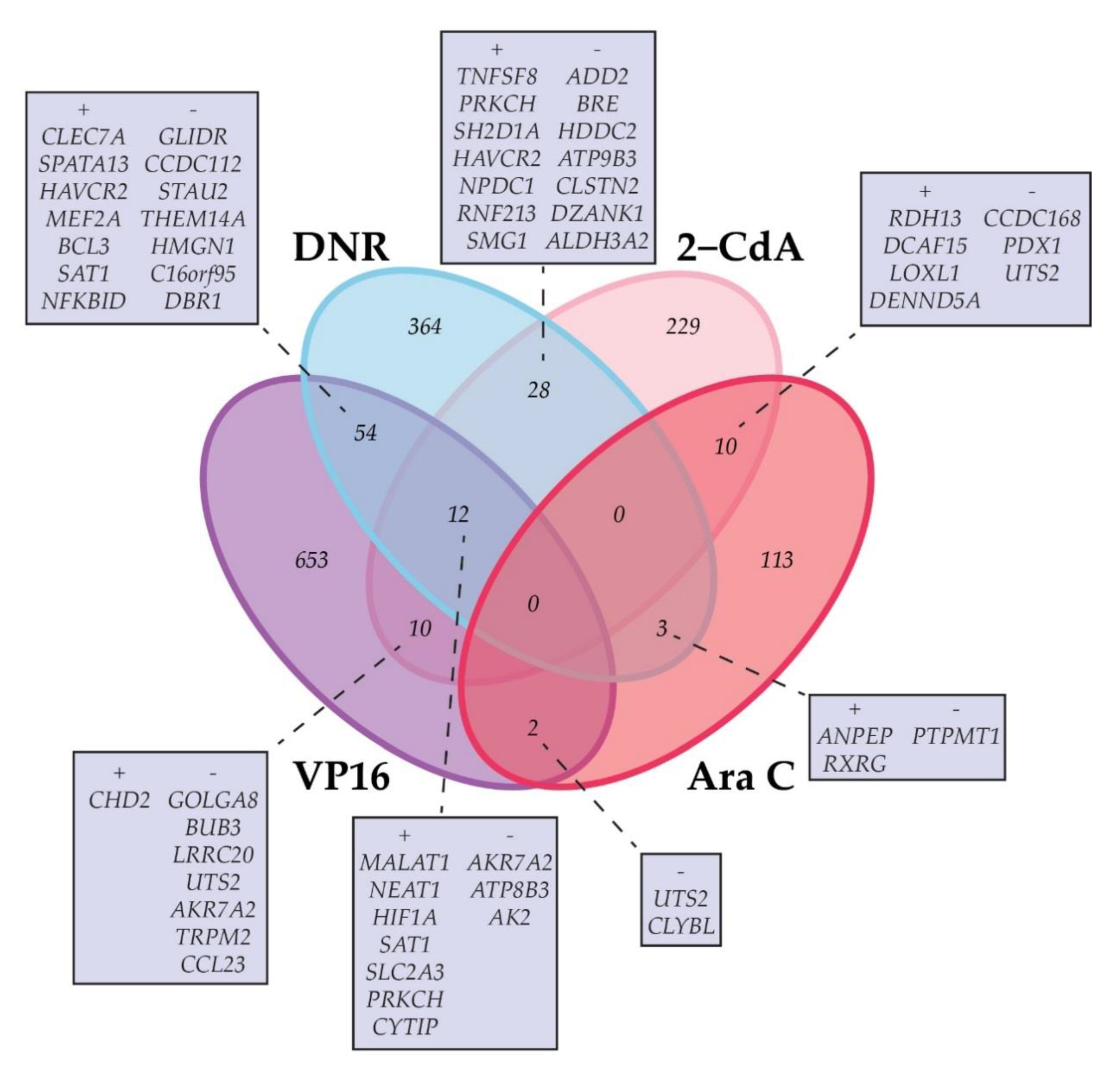
2.2. Correlation of Gene Expression with Ex Vivo Drug Response
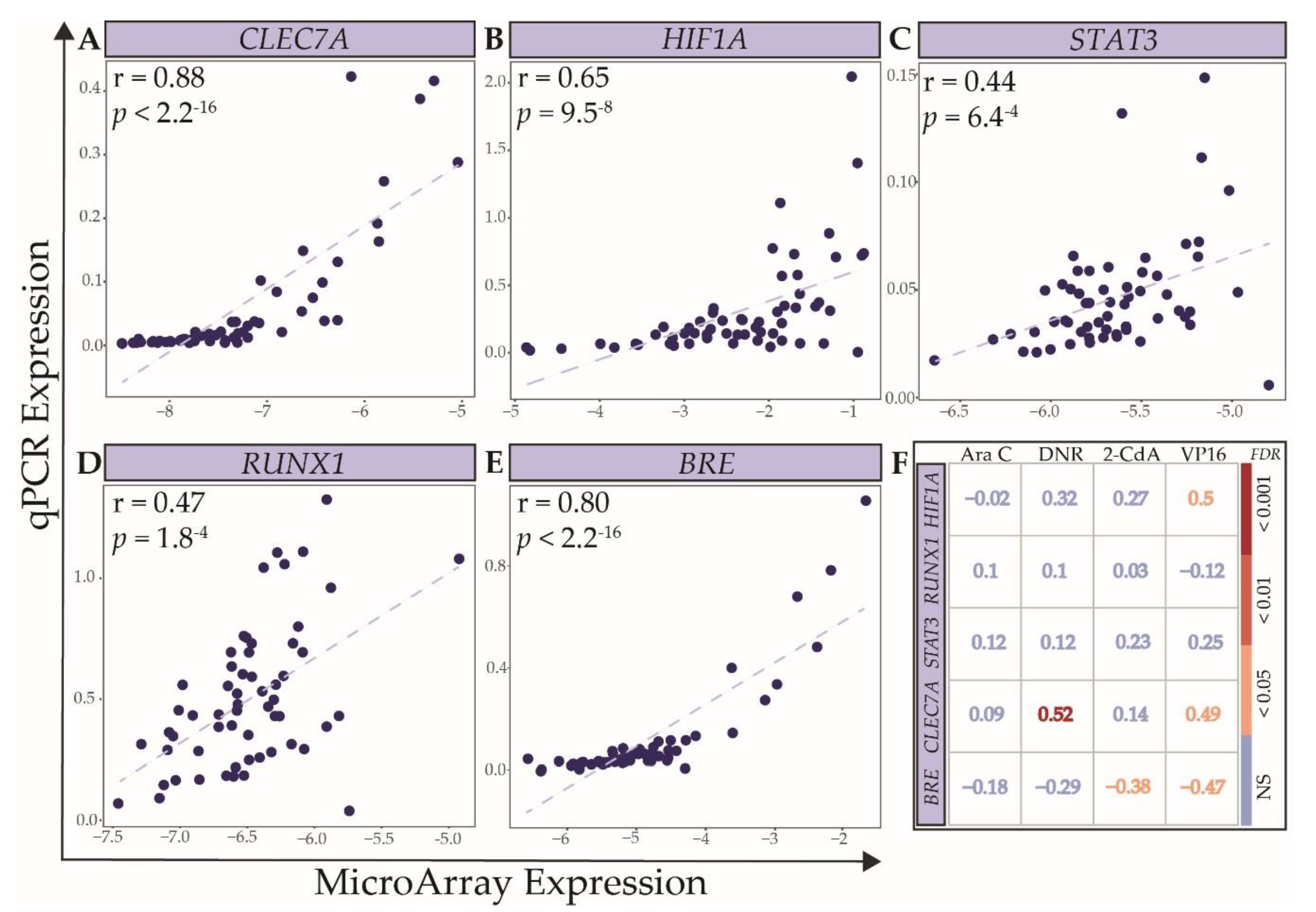
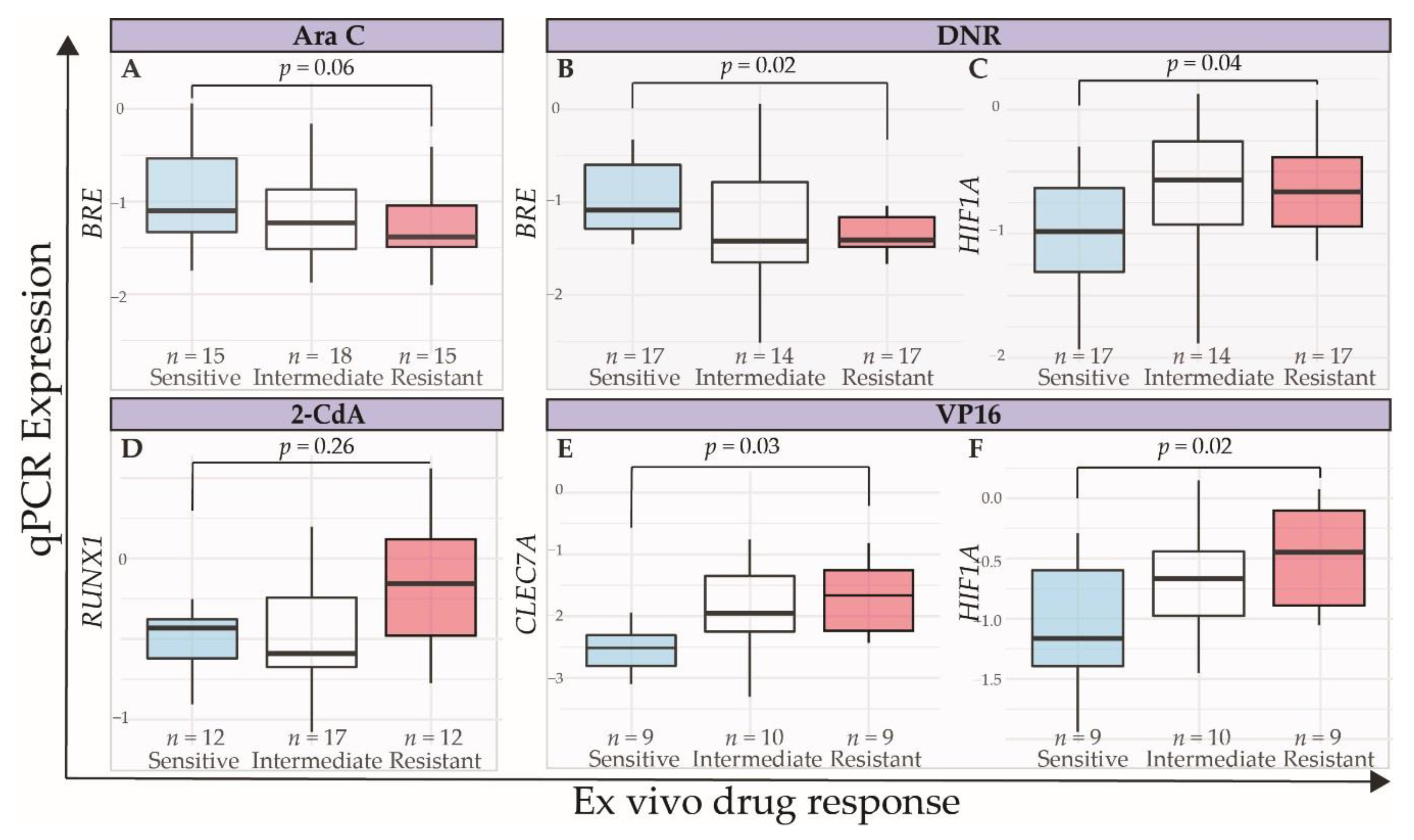
2.3. Technical and Independent Validation of the Association between Expression Levels of Selected Genes and Ex Vivo Drug Response
3. Discussion
4. Materials and Methods
4.1. Patient Samples
4.2. Ex Vivo Drug Response
4.3. qPCR
4.4. Data Preprocessing and Statistical Analyses
4.5. Software
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Molecular Aberration | Discovery Set n = 73 | Validation Set n = 48 | ||
|---|---|---|---|---|---|
| median (range) | median (range) | ||||
| Age | 8.90 | (0.0–15.8) | 7.33 | (0.0–16.9) | |
| % leukemia blasts | 87 | (33–98) | 78.00 | (33–98) | |
| Time to relapse (months) | 9.8 | (0.6–125.5) | 20.1 | (2.1–107.0) | |
| Overall survival (months) | 26.1 | (2.6–148.5) | 37.5 | (0.1–195.8) | |
| N (%) | N (%) | ||||
| Males, n (%) | 45 | 63.4 | 30 | 62.5 | |
| Cytogenetics | Normal | 16 | 38.1 | 10 | 21.7 |
| t(8;21) | 7 | 21.9 | 2 | 4.3 | |
| MLL rearranged | 19 | 43.2 | 10 | 21.7 | |
| Abnormal 16 | 8 | 25.0 | 1 | 2.2 | |
| -7/7q- | 1 | 4.2 | 1 | 2.2 | |
| -5/5q- | 0 | 0.0 | 0 | 0.0 | |
| 8 | 2 | 8.0 | 3 | 6.5 | |
| Complex | 0 | 0.0 | 3 | 6.5 | |
| Other | 14 | 38.9 | 14 | 30.4 | |
| No data | 2 | 8.3 | 2 | 4.3 | |
| Gene mutations | FLT3/ITD | 15 | 40.5 | 7 | 24.1 |
| NPM1 | 6 | 22.2 | 2 | 6.9 | |
| CEBPA | 5 | 20.0 | 2 | 6.9 | |
| WT1 | 5 | 20.8 | 6 | 20.7 | |
| NRAS/KRAS | 15 | 44.1 | 8 | 27.6 | |
| CKIT | 9 | 32.1 | 3 | 10.3 | |
| PTPN11 | 0 | 0.0 | 1 | 3.4 | |
| Category | Molecular Aberration | n | Ara C | DNR | 2-CdA | VP16 |
|---|---|---|---|---|---|---|
| Total | 38–73 | 1.48 | 0.33 | 0.07 | 4.55 | |
| Cytogenetics | ||||||
| Normal | 16 | 1.09 | 0.30 | 0.08 | 4.51 | |
| t(8;21) | 7 | 1.54 | 0.33 | 0.08 | 11.05 | |
| MLL rearranged | 19 | 1.23 | 0.24 | 0.01 | 3.62 | |
| Abnormal 16 | 8 | 1.37 | 0.68 | 0.08 | 22.48 | |
| -7/7q- | 1 | 2.09 | 0.47 | 0.13 | 5.12 | |
| Tris8 | 2 | 1.14 | 0.52 | 0.08 | 0.00 | |
| Other | 14 | 1.37 | 0.26 | 0.06 | 4.25 | |
| Gene mutations | ||||||
| FLT3/ITD | 15 | 1.07 | 0.34 | 0.08 | 4.51 | |
| NPM1 | 6 | 1.07 | 0.27 | 0.21 | 4.51 | |
| CEBPA | 5 | 0.79 | 0.41 | 0.01 | 3.99 | |
| WT1 | 5 | 0.79 | 0.42 | 0.03 | 4.37 | |
| NRAS/KRAS | 15 | 1.29 | 0.43 | 0.09 | 13.08 | |
| CKIT | 9 | 1.54 | 0.36 | 0.08 | 15.85 |
| Rank | Ara C | DNR | 2-CdA | VP16 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Rspearman | p | Gene | Rspearman | p | Gene | Rspearman | p | Gene | Rspearman | p | Gene | |
| Positive correlation | ||||||||||||
| 1 | 0.54 | 1.51 × 10−6 | RDH13 | 0.58 | 1.42 × 10−7 | CLEC7A | 0.54 | 1.00 × 10−5 | TAB2 | 0.67 | 3.70 × 10−6 | MALAT1 |
| 2 | 0.50 | 1.25 × 10−5 | LOC339988 | 0.53 | 2.22 × 10−6 | TNFSF8 | 0.54 | 1.10 × 10−5 | CYTIP | 0.66 | 8.08 × 10−6 | CLEC7A |
| 3 | 0.50 | 1.27 × 10−5 | PML | 0.52 | 5.90 × 10−6 | PRKCH | 0.53 | 1.28 × 10−5 | BNIP3L | 0.65 | 1.23 × 10−5 | C1orf115 |
| 4 | 0.49 | 1.91 × 10−5 | TMEM259 | 0.51 | 7.11 × 10−6 | TNFSF8 | 0.52 | 2.13 × 10−5 | PIK3IP1 | 0.63 | 2.15 × 10−5 | SYNJ2 |
| 5 | 0.48 | 2.59 × 10−5 | KMT2D | 0.51 | 7.30 × 10−6 | SH2D1A | 0.52 | 2.14 × 10−5 | TCF7L2 | 0.63 | 2.29 × 10−5 | SPATA13 |
| 6 | 0.47 | 3.88 × 10−5 | DCAF15 | 0.51 | 8.31 × 10−6 | HAVCR2 | 0.52 | 2.52 × 10−5 | LOXL1 | 0.62 | 2.94 × 10−5 | ATP1B3 |
| 7 | 0.47 | 4.59 × 10−5 | ATG9A | 0.51 | 8.33 × 10−6 | NPDC1 | 0.52 | 2.70 × 10−5 | MALAT1 | 0.61 | 5.10 × 10−5 | NFKBID |
| 8 | 0.46 | 5.59 × 10−5 | ARFGAP1 | 0.50 | 1.23 × 10−5 | TNFAIP3 | 0.51 | 4.09 × 10−5 | GLS | 0.61 | 5.32 × 10−5 | ZBTB18 |
| 9 | 0.46 | 7.32 × 10−5 | KMT2B | 0.49 | 2.21 × 10−5 | TNFSF8 | 0.51 | 4.17 × 10−5 | FOXO3 | 0.60 | 6.73 × 10−5 | PPP1R16B |
| 10 | 0.45 | 8.24 × 10−5 | BAZ2A | 0.49 | 2.28 × 10−5 | RNF213 | 0.50 | 4.66 × 10−5 | OSER1 | 0.60 | 8.08 × 10−5 | USP53 |
| Negative correlation | ||||||||||||
| 10 | −0.44 | 1.38 × 10−4 | TMEM38B | −0.53 | 3.22 × 10−6 | CBX3 | −0.49 | 8.74 × 10−5 | GOLGA8CP | −0.65 | 1.17 × 10−5 | FAR1 |
| 9 | −0.44 | 1.18 × 10−4 | FAM162A | −0.53 | 2.81 × 10−6 | UNC13B | −0.49 | 7.51 × 10−5 | C5orf63 | −0.65 | 9.87 × 10−6 | NOP16 |
| 8 | −0.44 | 1.14 × 10−4 | PPA2 | −0.53 | 2.73 × 10−6 | C6orf99 | −0.50 | 6.10 × 10−5 | GOLGA8N | −0.65 | 9.69 × 10−6 | RCAN3 |
| 7 | −0.45 | 8.11 × 10−5 | STRN | −0.53 | 2.64 × 10−6 | CCDC125 | −0.50 | 5.07 × 10−5 | HOXA9 | −0.66 | 7.36 × 10−6 | IKBKAP |
| 6 | −0.46 | 7.21 × 10−5 | NXPE3 | −0.53 | 2.28 × 10−6 | FAM127A | −0.50 | 4.89 × 10−5 | LOC101927851 | −0.66 | 5.61 × 10−6 | NMNAT3 |
| 5 | −0.46 | 5.60 × 10−5 | UTS2 | −0.54 | 2.06 × 10−6 | TMEM237 | −0.51 | 4.03 × 10−5 | CCDC168 | −0.66 | 5.56 × 10−6 | IMP3 |
| 4 | −0.48 | 2.63 × 10−5 | GPR78 | −0.54 | 1.87 × 10−6 | KCTD15 | −0.51 | 3.82 × 10−5 | ZBTB24 | −0.67 | 3.62 × 10−6 | ENTPD1-AS1 |
| 3 | −0.49 | 1.93 × 10−5 | VWA8 | −0.54 | 1.35 × 10−6 | PARL | −0.51 | 3.11 × 10−5 | H6PD | −0.68 | 2.36 × 10−6 | LOC100506730 |
| 2 | −0.49 | 1.74 × 10−5 | PTCHD1 | −0.56 | 6.70 × 10−7 | PON2 | −0.52 | 2.60 × 10−5 | CCL23 | −0.69 | 1.61 × 10−6 | FEN1 |
| 1 | −0.51 | 5.19 × 10−6 | CLCC1 | −0.60 | 6.76 × 10−8 | PON2 | −0.54 | 1.12 × 10−5 | CCL23 | −0.75 | 6.74 × 10−8 | HNRNPAB |
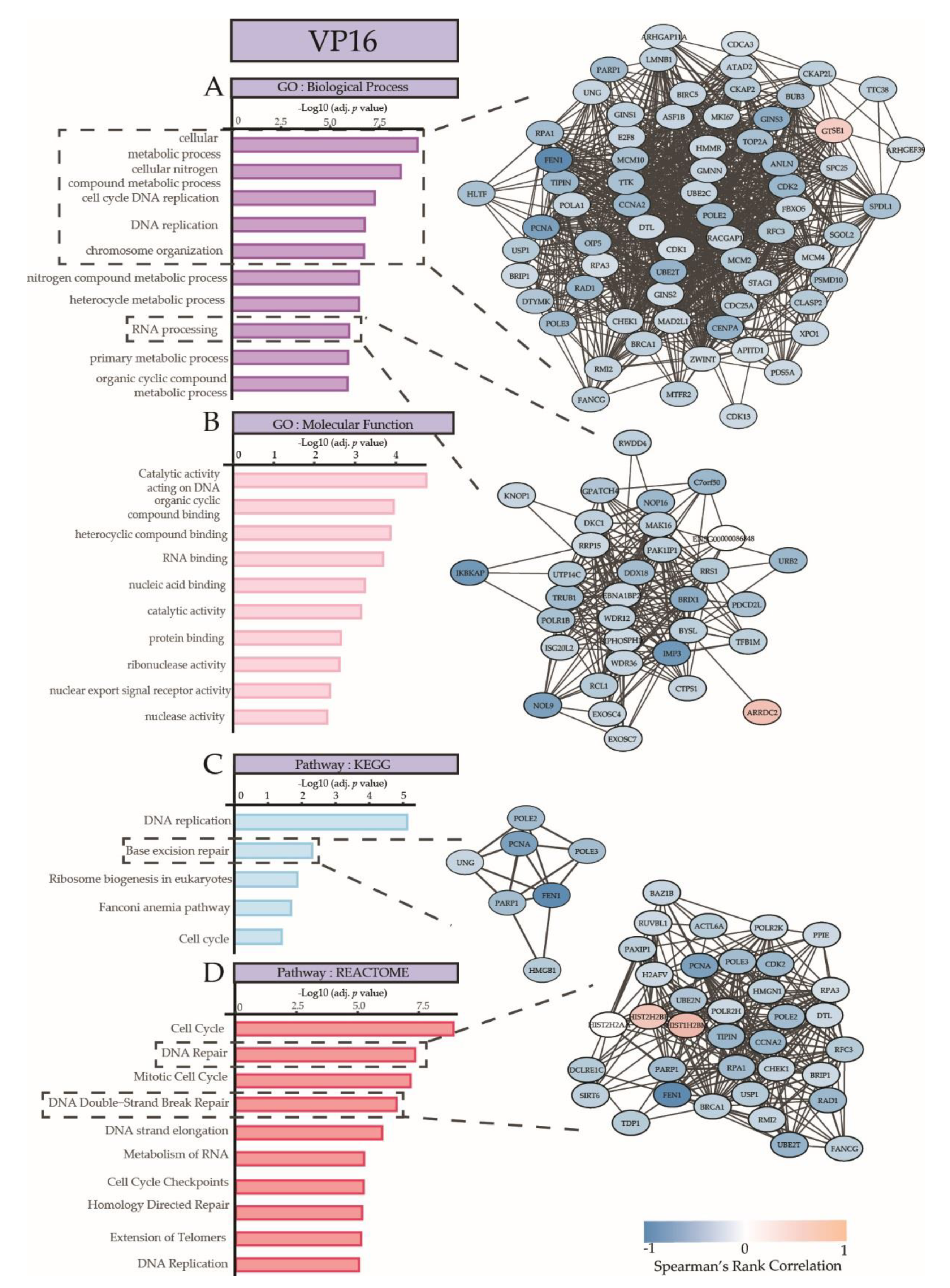
| Drug | Top 3 GO Biological Processes | Top 3 GO Molecular Functions | Top 3 KEGG Pathways | Top 3 REACTOME Pathways |
|---|---|---|---|---|
| Ara C | None | histone methyltransferase activity (H3-K4 specific) | None | RUNX1 regulates genes involved in megakaryocyte differentiation and platelet function |
| DNR | snRNA 3’-end processing | electron transfer activity | DNA replication | Growth hormone receptor signaling |
| snRNA processing | Alzheimer disease | |||
| snRNA metabolic process | Non-alcoholic fatty liver disease (NAFLD) | |||
| + 2 GO terms | + 2 KEGG pathways | |||
| 2-CdA | regulation of transcription by RNA polymerase II | DNA-binding transcription factor activity, RNA polymerase II-specific | Immune System | |
| system development | RNA polymerase II proximal promoter sequence-specific DNA binding | Transcriptional misregulation in cancer | ||
| transcription by RNA polymerase II | kinase binding | |||
| … + 5 GO terms | ||||
| VP16 | cellular metabolic process | catalytic activity, acting on DNA | DNA replication | Cell Cycle |
| cellular nitrogen compound metabolic process | organic cyclic compound binding | Base excision repair | DNA Repair | |
| cell cycle DNA replication | heterocyclic compound binding | Ribosome biogenesis in eukaryotes | Cell Cycle, Mitotic | |
| + 56 GO terms | + 10 GO terms | + 1 KEGG Pathways | \+ 42 REACTOME Pathways |
| Technique | Drug | Discovery Cohort | Validation Cohort | ||
|---|---|---|---|---|---|
| n of Patients (%) | n of Patients (%) | ||||
| MicroArray-based gene expression data | 73 | 100.0 | - | - | |
| MTT | Ara C | 70 | 95.9 | 48 | 100.0 |
| DNR | 69 | 94.5 | 45 | 93.8 | |
| 2-CdA | 59 | 80.8 | 41 | 85.4 | |
| VP16 | 38 | 52.1 | 26 | 54.2 | |
| qPCR | 58 | 79.5 | 48 | 100.0 | |
References
- Kaspers, G.J. Pediatric Acute Myeloid Leukemia. Expert. Rev. Anticancer Ther. 2013, 12, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Rubnitz, J.E.; Razzouk, B.I.; Lensing, S.; Pounds, S.; Pui, C.H.; Ribeiro, R.C. Prognostic Factors and Outcome of Recurrence in Childhood Acute Myeloid Leukemia. Cancer 2007, 109, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Rasche, M.; Zimmermann, M.; Borschel, L.; Bourquin, J.P.; Dworzak, M.; Klingebiel, T.; Lehrnbecher, T.; Creutzig, U.; Klusmann, J.H.; Reinhardt, D. Successes and Challenges in the Treatment of Pediatric Acute Myeloid Leukemia: A Retrospective Analysis of the Aml-Bfm Trials from 1987 to 2012. Leukemia 2018, 32, 2167–2177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaspers, G.J.; Zimmermann, M.; Reinhardt, D.; Gibson, B.E.; Tamminga, R.Y.; Aleinikova, O.; Armendariz, H.; Dworzak, M.; Ha, S.Y.; Hasle, H.; et al. Improved Outcome in Pediatric Relapsed Acute Myeloid Leukemia: Results of a Randomized Trial on Liposomal Daunorubicin by the International Bfm Study Group. J. Clin. Oncol. 2013, 31, 599–607. [Google Scholar] [CrossRef]
- Bachas, C.; Schuurhuis, G.J.; Assaraf, Y.G.; Kwidama, Z.J.; Kelder, A.; Wouters, F.; Snel, A.N.; Kaspers, G.J.; Cloos, J. The Role of Minor Subpopulations within the Leukemic Blast Compartment of Aml Patients at Initial Diagnosis in the Development of Relapse. Leukemia 2012, 26, 1313–1320. [Google Scholar] [CrossRef]
- Daver, N.; Schlenk, R.F.; Russell, N.H.; Levis, M.J. Targeting Flt3 Mutations in Aml: Review of Current Knowledge and Evidence. Leukemia 2019, 33, 299–312. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Gong, Y. Isocitrate Dehydrogenase Inhibitors in Acute Myeloid Leukemia. Biomark Res 2019, 7, 22. [Google Scholar]
- Rubnitz, J.E.; Inaba, H. Childhood Acute Myeloid Leukaemia. Br. J. Haematol. 2012, 159, 259–276. [Google Scholar] [CrossRef]
- Van den Heuvel-Eibrink, M.M.; Van der Holt, B.; Burnett, A.K.; Knauf, W.U.; Fey, M.F.; Verhoef, G.E.; Vellenga, E.; Ossenkoppele, G.J.; Lowenberg, B.; Sonneveld, P. Cd34-Related Coexpression of Mdr1 and Bcrp Indicates a Clinically Resistant Phenotype in Patients with Acute Myeloid Leukemia (Aml) of Older Age. Ann. Hematol. 2007, 86, 329–337. [Google Scholar] [CrossRef] [Green Version]
- Macanas-Pirard, P.; Broekhuizen, R.; Gonzalez, A.; Oyanadel, C.; Ernst, D.; Garcia, P.; Montecinos, V.P.; Court, F.; Ocqueteau, M.; Ramirez, P.; et al. Resistance of Leukemia Cells to Cytarabine Chemotherapy Is Mediated by Bone Marrow Stroma, Involves Cell-Surface Equilibrative Nucleoside Transporter-1 Removal and Correlates with Patient Outcome. Oncotarget 2017, 8, 23073–23086. [Google Scholar] [CrossRef] [Green Version]
- Dow, L.W.; Dahl, G.V.; Kalwinsky, D.K.; Mirro, J.; Nash, M.B.; Roberson, P.K. Correlation of Drug Sensitivity in Vitro with Clinical Responses in Childhood Acute Myeloid Leukemia. Blood 1986, 68, 400–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norgaard, J.M.; Langkjer, S.T.; Palshof, T.; Pedersen, B.; Hokland, P. Pretreatment Leukaemia Cell Drug Resistance Is Correlated to Clinical Outcome in Acute Myeloid Leukaemia. Eur. J. Haematol. 2001, 66, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Zwaan, C.M.; Kaspers, G.J.; Pieters, R.; Hahlen, K.; Huismans, D.R.; Zimmermann, M.; Harbott, J.; Slater, R.M.; Creutzig, U.; Veerman, A.J. Cellular Drug Resistance in Childhood Acute Myeloid Leukemia Is Related to Chromosomal Abnormalities. Blood 2002, 100, 3352–3360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Styczynski, J.; Wysocki, M. Ex Vivo Drug Resistance in Childhood Acute Myeloid Leukemia on Relapse Is Not Higher than at First Diagnosis. Pediatr Blood Cancer 2004, 42, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Hongo, T.; Okada, S.; Watanabe, C.; Fujii, Y.; Ohzeki, T. Clinical Relevance of in Vitro Chemoresistance in Childhood Acute Myeloid Leukemia. Leukemia 2001, 15, 1892–1897. [Google Scholar] [CrossRef] [Green Version]
- Niewerth, D.; Jansen, G.; Riethoff, L.F.; van Meerloo, J.; Kale, A.J.; Moore, B.S.; Assaraf, Y.G.; Anderl, J.L.; Zweegman, S.; Kaspers, G.J.; et al. Antileukemic Activity and Mechanism of Drug Resistance to the Marine Salinispora Tropica Proteasome Inhibitor Salinosporamide a (Marizomib). Mol. Pharmacol. 2014, 86, 12–19. [Google Scholar] [CrossRef] [Green Version]
- Wojtuszkiewicz, A.; Raz, S.; Stark, M.; Assaraf, Y.G.; Jansen, G.; Peters, G.J.; Sonneveld, E.; Kaspers, G.J.; Cloos, J. Folylpolyglutamate Synthetase Splicing Alterations in Acute Lymphoblastic Leukemia Are Provoked by Methotrexate and Other Chemotherapeutics and Mediate Chemoresistance. Int. J. Cancer 2016, 138, 1645–1656. [Google Scholar] [CrossRef] [Green Version]
- Hubeek, I.; Peters, G.J.; Broekhuizen, R.; Zwaan, C.M.; Kaaijk, P.; van Wering, E.S.; Gibson, B.E.; Creutzig, U.; Janka-Schaub, G.E.; den Boer, M.L.; et al. In Vitro Sensitivity and Cross-Resistance to Deoxynucleoside Analogs in Childhood Acute Leukemia. Haematologica 2006, 91, 17–23. [Google Scholar]
- Fiegl, M.; Zimmermann, I.; Lorenz, I.; Hiddemann, W.; Braess, J. In Vitro Cross-Resistance to Nucleoside Analogues and Inhibitors of Topoisomerase 1 and 2 in Acute Myeloid Leukemia. Ann. Hematol. 2008, 87, 27–33. [Google Scholar] [CrossRef]
- Kaspers, G.J.; Veerman, A.J.; Pieters, R.; Broekema, G.J.; Huismans, D.R.; Kazemier, K.M.; Loonen, A.H.; Rottier, M.A.; van Zantwijk, C.H.; Hahlen, K.; et al. Mononuclear Cells Contaminating Acute Lymphoblastic Leukaemic Samples Tested for Cellular Drug Resistance Using the Methyl-Thiazol-Tetrazolium Assay. Br. J. Cancer 1994, 70, 1047–1052. [Google Scholar] [CrossRef]
- Galmarini, C.M.; Mackey, J.R.; Dumontet, C. Nucleoside Analogues: Mechanisms of Drug Resistance and Reversal Strategies. Leukemia 2001, 15, 875–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubeek, I.; Stam, R.W.; Peters, G.J.; Broekhuizen, R.; Meijerink, J.P.; van Wering, E.R.; Gibson, B.E.; Creutzig, U.; Zwaan, C.M.; Cloos, J.; et al. The Human Equilibrative Nucleoside Transporter 1 Mediates in Vitro Cytarabine Sensitivity in Childhood Acute Myeloid Leukaemia. Br. J. Cancer 2005, 93, 1388–1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galmarini, C.M.; Thomas, X.; Calvo, F.; Rousselot, P.; Rabilloud, M.; el Jaffari, A.; Cros, E.; Dumontet, C. In Vivo Mechanisms of Resistance to Cytarabine in Acute Myeloid Leukaemia. Br. J. Haematol. 2002, 117, 860–868. [Google Scholar] [CrossRef] [PubMed]
- Farge, T.; Saland, E.; de Toni, F.; Aroua, N.; Hosseini, M.; Perry, R.; Bosc, C.; Sugita, M.; Stuani, L.; Fraisse, M.; et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer Discov. 2017, 7, 716–735. [Google Scholar] [CrossRef] [Green Version]
- Lamba, J.K. Genetic Factors Influencing Cytarabine Therapy. Pharmacogenomics 2009, 10, 1657–1674. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.; Oellerich, T.; Baldauf, H.M.; Schwarz, S.M.; Thomas, D.; Flick, R.; Bohnenberger, H.; Kaderali, L.; Stegmann, L.; Cremer, A.; et al. Samhd1 Is a Biomarker for Cytarabine Response and a Therapeutic Target in Acute Myeloid Leukemia. Nat. Med. 2017, 23, 250–255. [Google Scholar] [CrossRef]
- Levin, M.; Stark, M.; Berman, B.; Assaraf, Y.G. Surmounting Cytarabine-Resistance in Acute Myeloblastic Leukemia Cells and Specimens with a Synergistic Combination of Hydroxyurea and Azidothymidine. Cell Death Dis. 2019, 10, 390. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, Y.; Schuetz, J.D. Abc Transporters and Their Role in Nucleoside and Nucleotide Drug Resistance. Biochem. Pharmacol. 2012, 83, 1073–1083. [Google Scholar] [CrossRef] [Green Version]
- Schuurhuis, G.J.; Broxterman, H.J.; Ossenkoppele, G.J.; Baak, J.P.; Eekman, C.A.; Kuiper, C.M.; Feller, N.; van Heijningen, T.H.; Klumper, E.; Pieters, R.; et al. Functional Multidrug Resistance Phenotype Associated with Combined Overexpression of Pgp/Mdr1 and Mrp Together with 1-Beta-D-Arabinofuranosylcytosine Sensitivity May Predict Clinical Response in Acute Myeloid Leukemia. Clin. Cancer Res. 1995, 1, 81–93. [Google Scholar]
- Kaspers, G.J.; Veerman, A.J. Clinical Significance of Cellular Drug Resistance in Childhood Leukemia. Recent. Results Cancer Res. 2003, 161, 196–220. [Google Scholar]
- Zhang, J.; Gu, Y.; Chen, B. Mechanisms of Drug Resistance in Acute Myeloid Leukemia. Oncol. Targets Ther. 2019, 12, 1937–1945. [Google Scholar] [CrossRef] [Green Version]
- McLornan, D.P.; McMullin, M.F.; Johnston, P.; Longley, D.B. Molecular Mechanisms of Drug Resistance in Acute Myeloid Leukaemia. Expert. Opin. Drug Metab. Toxicol. 2007, 3, 363–377. [Google Scholar] [CrossRef] [PubMed]
- Lamba, J.K.; Crews, K.R.; Pounds, S.B.; Cao, X.; Gandhi, V.; Plunkett, W.; Razzouk, B.I.; Lamba, V.; Baker, S.D.; Raimondi, S.C.; et al. Identification of Predictive Markers of Cytarabine Response in Aml by Integrative Analysis of Gene-Expression Profiles with Multiple Phenotypes. Pharmacogenomics 2011, 12, 327–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNeer, N.A.; Philip, J.; Geiger, H.; Ries, R.E.; Lavallee, V.P.; Walsh, M.; Shah, M.; Arora, K.; Emde, A.K.; Robine, N.; et al. Genetic Mechanisms of Primary Chemotherapy Resistance in Pediatric Acute Myeloid Leukemia. Leukemia 2019, 33, 1934–1943. [Google Scholar] [CrossRef] [PubMed]
- Molica, M.; Breccia, M.; Capria, S.; Trisolini, S.; Foa, R.; Jabbour, E.; Kadia, T.M. The Role of Cladribine in Acute Myeloid Leukemia: An Old Drug up to New Tricks. Leuk. Lymphoma 2019, 61, 1–10. [Google Scholar] [CrossRef]
- Holowiecki, J.; Grosicki, S.; Giebel, S.; Robak, T.; Kyrcz-Krzemien, S.; Kuliczkowski, K.; Skotnicki, A.B.; Hellmann, A.; Sulek, K.; Dmoszynska, A.; et al. Cladribine, but Not Fludarabine, Added to Daunorubicin and Cytarabine during Induction Prolongs Survival of Patients with Acute Myeloid Leukemia: A Multicenter, Randomized Phase Iii Study. J. Clin. Oncol. 2012, 30, 2441–2448. [Google Scholar] [CrossRef]
- Martin, M.G.; Welch, J.S.; Augustin, K.; Hladnik, L.; DiPersio, J.F.; Abboud, C.N. Cladribine in the Treatment of Acute Myeloid Leukemia: A Single-Institution Experience. Clin. Lymphoma Myeloma 2009, 9, 298–301. [Google Scholar] [CrossRef]
- Putzer, B.M.; Engelmann, D. E2f1 Apoptosis Counterattacked: Evil Strikes Back. Trends Mol. Med. 2013, 19, 89–98. [Google Scholar] [CrossRef]
- Weijts, B.G.; Van Impel, A.; Schulte-Merker, S.; De Bruin, A. Atypical E2fs Control Lymphangiogenesis through Transcriptional Regulation of Ccbe1 and Flt4. PLoS ONE 2013, 8, e73693. [Google Scholar] [CrossRef]
- Chu, M.; Yin, K.; Dong, Y.; Wang, P.; Xue, Y.; Zhou, P.; Wang, Y.; Wang, Y. Tfdp3 Confers Chemoresistance in Minimal Residual Disease within Childhood T-Cell Acute Lymphoblastic Leukemia. Oncotarget 2017, 8, 1405–1415. [Google Scholar] [CrossRef] [Green Version]
- Kalyuga, M.; Gallego-Ortega, D.; Lee, H.J.; Roden, D.L.; Cowley, M.J.; Caldon, C.E.; Stone, A.; Allerdice, S.L.; Valdes-Mora, F.; Launchbury, R.; et al. Elf5 Suppresses Estrogen Sensitivity and Underpins the Acquisition of Antiestrogen Resistance in Luminal Breast Cancer. PLoS. Biol. 2012, 10, e1001461. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Shan, Y.; Bai, R.; Xue, L.; Eide, C.A.; Ou, J.; Zhu, L.J.; Hutchinson, L.; Cerny, J.; Khoury, H.J.; et al. A Therapeutically Targetable Mechanism of Bcr-Abl-Independent Imatinib Resistance in Chronic Myeloid Leukemia. Sci. Transl. Med. 2014, 6, 252ra121. [Google Scholar] [CrossRef] [Green Version]
- Brown, F.C.; Still, E.; Koche, R.P.; Yim, C.Y.; Takao, S.; Cifani, P.; Reed, C.; Gunasekera, S.; Ficarro, S.B.; Romanienko, P.; et al. Mef2c Phosphorylation Is Required for Chemotherapy Resistance in Acute Myeloid Leukemia. Cancer Discov. 2018, 8, 478–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, F.H.; Putoczki, T.L.; Stylli, S.S.; Luwor, R.B. The Role of Stat3 Signaling in Mediating Tumor Resistance to Cancer Therapy. Curr. Drug Targets 2014, 15, 1341–1353. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Liu, L.; Bai, Q.; Wang, J.; Xi, W.; Qu, Y.; Xiong, Y.; Long, Q.; Xu, J.; Guo, J. Dectin-1 Predicts Adverse Postoperative Prognosis of Patients with Clear Cell Renal Cell Carcinoma. Sci. Rep. 2016, 6, 32657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deynoux, M.; Sunter, N.; Herault, O.; Mazurier, F. Hypoxia and Hypoxia-Inducible Factors in Leukemias. Front. Oncol. 2016, 6, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mimeault, M.; Batra, S.K. Hypoxia-Inducing Factors as Master Regulators of Stemness Properties and Altered Metabolism of Cancer- and Metastasis-Initiating Cells. J. Cell Mol. Med. 2013, 17, 30–54. [Google Scholar] [CrossRef]
- Simon, L.; Lavallee, V.P.; Bordeleau, M.E.; Krosl, J.; Baccelli, I.; Boucher, G.; Lehnertz, B.; Chagraoui, J.; MacRae, T.; Ruel, R.; et al. Chemogenomic Landscape of Runx1-Mutated Aml Reveals Importance of Runx1 Allele Dosage in Genetics and Glucocorticoid Sensitivity. Clin. Cancer Res. 2017, 23, 6969–6981. [Google Scholar] [CrossRef] [Green Version]
- Balgobind, B.V.; Zwaan, C.M.; Reinhardt, D.; Arentsen-Peters, T.J.; Hollink, I.H.; De Haas, V.; Kaspers, G.J.; De Bont, E.S.; Baruchel, A.; Stary, J.; et al. High Bre Expression in Pediatric Mll-Rearranged Aml Is Associated with Favorable Outcome. Leukemia 2010, 24, 2048–2055. [Google Scholar] [CrossRef] [Green Version]
- Noordermeer, S.M.; Sanders, M.A.; Gilissen, C.; Tonnissen, E.; Van der Heijden, A.; Dohner, K.; Bullinger, L.; Jansen, J.H.; Valk, P.J.; Van der Reijden, B.A. High Bre Expression Predicts Favorable Outcome in Adult Acute Myeloid Leukemia, in Particular among Mll-Af9-Positive Patients. Blood 2011, 118, 5613–5621. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Herold, T.; He, C.; Valk, P.J.; Chen, P.; Jurinovic, V.; Mansmann, U.; Radmacher, M.D.; Maharry, K.S.; Sun, M.; et al. Identification of a 24-Gene Prognostic Signature That Improves the European Leukemianet Risk Classification of Acute Myeloid Leukemia: An International Collaborative Study. J. Clin. Oncol. 2013, 31, 1172–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network. Genomic and Epigenomic Landscapes of Adult de Novo Acute Myeloid Leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balgobind, B.V.; Zwaan, C.M.; Pieters, R.; van den Heuvel-Eibrink, M.M. The Heterogeneity of Pediatric Mll-Rearranged Acute Myeloid Leukemia. Leukemia 2011, 25, 1239–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyagi, S.; Herr, W. E2f1 Mediates DNA Damage and Apoptosis through Hcf-1 and the Mll Family of Histone Methyltransferases. EMBO J. 2009, 28, 3185–3195. [Google Scholar] [CrossRef] [Green Version]
- Rosenfeldt, M.T.; Bell, L.A.; Long, J.S.; O’Prey, J.; Nixon, C.; Roberts, F.; Dufes, C.; Ryan, K.M. E2f1 Drives Chemotherapeutic Drug Resistance via Abcg2. Oncogene 2014, 33, 4164–4172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, L.H.; Wei, W.Y.; Cao, W.L.; Zhang, X.S.; Xie, Y.B.; Xiao, Q. Overexpression of E2f1 in Human Gastric Carcinoma Is Involved in Anti-Cancer Drug Resistance. BMC Cancer 2014, 14, 904. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Yeow, W.S.; Ertel, A.; Coleman, I.; Clegg, N.; Thangavel, C.; Morrissey, C.; Zhang, X.; Comstock, C.E.; Witkiewicz, A.K.; et al. The Retinoblastoma Tumor Suppressor Controls Androgen Signaling and Human Prostate Cancer Progression. J. Clin. Investig. 2010, 120, 4478–4492. [Google Scholar] [CrossRef] [Green Version]
- Imai, M.A.; Oda, Y.; Oda, M.; Nakanishi, I.; Kawahara, E. Overexpression of E2f1 Associated with Loh at Rb Locus and Hyperphosphorylation of Rb in Non-Small Cell Lung Carcinoma. J. Cancer Res. Clin. Oncol. 2004, 130, 320–326. [Google Scholar] [CrossRef] [Green Version]
- Reimer, D.; Sadr, S.; Wiedemair, A.; Goebel, G.; Concin, N.; Hofstetter, G.; Marth, C.; Zeimet, A.G. Expression of the E2f Family of Transcription Factors and Its Clinical Relevance in Ovarian Cancer. Ann. N. Y. Acad. Sci. 2006, 1091, 270–281. [Google Scholar] [CrossRef]
- Park, S.A.; Platt, J.; Lee, J.W.; Lopez-Giraldez, F.; Herbst, R.S.; Koo, J.S. E2f8 as a Novel Therapeutic Target for Lung Cancer. J. Natl. Cancer Inst. 2015, 107, djv151. [Google Scholar] [CrossRef] [Green Version]
- Ye, L.; Guo, L.; He, Z.; Wang, X.; Lin, C.; Zhang, X.; Wu, S.; Bao, Y.; Yang, Q.; Song, L.; et al. Upregulation of E2f8 Promotes Cell Proliferation and Tumorigenicity in Breast Cancer by Modulating G1/S Phase Transition. Oncotarget 2016, 7, 23757–23771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Park, Y.R.; Kim, S.H.; Park, E.J.; Kang, M.J.; So, I.; Chun, J.N.; Jeon, J.H. Geraniol Suppresses Prostate Cancer Growth through Down-Regulation of E2f8. Cancer Med. 2016, 5, 2899–2908. [Google Scholar] [CrossRef] [PubMed]
- Thurlings, I.; Martinez-Lopez, L.M.; Westendorp, B.; Zijp, M.; Kuiper, R.; Tooten, P.; Kent, L.N.; Leone, G.; Vos, H.J.; Burgering, B.; et al. Synergistic Functions of E2f7 and E2f8 Are Critical to Suppress Stress-Induced Skin Cancer. Oncogene 2017, 36, 829–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kent, L.N.; Rakijas, J.B.; Pandit, S.K.; Westendorp, B.; Chen, H.Z.; Huntington, J.T.; Tang, X.; Bae, S.; Srivastava, A.; Senapati, S.; et al. E2f8 Mediates Tumor Suppression in Postnatal Liver Development. J. Clin. Investig. 2016, 126, 2955–2969. [Google Scholar] [CrossRef] [PubMed]
- Nitiss, J.L. Targeting DNA Topoisomerase Ii in Cancer Chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.; Fang, L.; Gibbs, S.; Huang, Y.; Dai, Z.; Wen, P.; Zheng, X.; Sadee, W.; Sun, D. Glucose Uptake Inhibitor Sensitizes Cancer Cells to Daunorubicin and Overcomes Drug Resistance in Hypoxia. Cancer Chemother. Pharmacol. 2007, 59, 495–505. [Google Scholar] [CrossRef]
- Ma, H.; Padmanabhan, I.S.; Parmar, S.; Gong, Y. Targeting Cll-1 for Acute Myeloid Leukemia Therapy. J. Hematol. Oncol. 2019, 12, 41. [Google Scholar] [CrossRef]
- Balgobind, B.V.; Van den Heuvel-Eibrink, M.M.; De Menezes, R.X.; Reinhardt, D.; Hollink, I.H.; Arentsen-Peters, S.T.; Van Wering, E.R.; Kaspers, G.J.; Cloos, J.; De Bont, E.S.; et al. Evaluation of Gene Expression Signatures Predictive of Cytogenetic and Molecular Subtypes of Pediatric Acute Myeloid Leukemia. Haematologica 2011, 96, 221–230. [Google Scholar] [CrossRef] [Green Version]
- Van Meerloo, J.; Kaspers, G.J.; Cloos, J. Cell Sensitivity Assays: The Mtt Assay. Methods Mol. Biol. 2011, 731, 237–245. [Google Scholar]
- Bachas, C.; Schuurhuis, G.J.; Zwaan, C.M.; Van den Heuvel-Eibrink, M.M.; Den Boer, M.L.; De Bont, E.S.; Kwidama, Z.J.; Reinhardt, D.; Creutzig, U.; De Haas, V.; et al. Gene Expression Profiles Associated with Pediatric Relapsed Aml. PLoS ONE 2015, 10, e0121730. [Google Scholar] [CrossRef] [Green Version]
- Huber, W.; Von Heydebreck, A.; Sultmann, H.; Poustka, A.; Vingron, M. Variance Stabilization Applied to Microarray Data Calibration and to the Quantification of Differential Expression. Bioinformatics 2002, 18, S96–S104. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex Heatmaps Reveal Patterns and Correlations in Multidimensional Genomic Data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, T.; Simko, V. R Package Corrplot: Visualization of a Correlation Matrix (Version 0.84). 2017. Available online: Https://Github.Com/Taiyun/Corrplot (accessed on 2 February 2020).
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. G:Profiler: A Web Server for Functional Enrichment Analysis and Conversions of Gene Lists (2019 Update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. String V10: Protein-Protein Interaction Networks, Integrated over the Tree of Life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Verfaillie, A.; Imrichova, H.; Janky, R.; Aerts, S. Iregulon and I-Cistarget: Reconstructing Regulatory Networks Using Motif and Track Enrichment. Curr. Protoc. Bioinform. 2015, 52, 1–39. [Google Scholar] [CrossRef]
- Fojo, T. Multiple Paths to a Drug Resistance Phenotype: Mutations, Translocations, Deletions and Amplification of Coding Genes or Promoter Regions, Epigenetic Changes and Micrornas. Drug Resist. Updat. 2007, 10, 59–67. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cucchi, D.G.J.; Bachas, C.; van den Heuvel-Eibrink, M.M.; Arentsen-Peters, S.T.C.J.M.; Kwidama, Z.J.; Schuurhuis, G.J.; Assaraf, Y.G.; de Haas, V.; Kaspers, G.J.L.; Cloos, J. Harnessing Gene Expression Profiles for the Identification of Ex Vivo Drug Response Genes in Pediatric Acute Myeloid Leukemia. Cancers 2020, 12, 1247. https://doi.org/10.3390/cancers12051247
Cucchi DGJ, Bachas C, van den Heuvel-Eibrink MM, Arentsen-Peters STCJM, Kwidama ZJ, Schuurhuis GJ, Assaraf YG, de Haas V, Kaspers GJL, Cloos J. Harnessing Gene Expression Profiles for the Identification of Ex Vivo Drug Response Genes in Pediatric Acute Myeloid Leukemia. Cancers. 2020; 12(5):1247. https://doi.org/10.3390/cancers12051247
Chicago/Turabian StyleCucchi, David G.J., Costa Bachas, Marry M. van den Heuvel-Eibrink, Susan T.C.J.M. Arentsen-Peters, Zinia J. Kwidama, Gerrit J. Schuurhuis, Yehuda G. Assaraf, Valérie de Haas, Gertjan J.L. Kaspers, and Jacqueline Cloos. 2020. "Harnessing Gene Expression Profiles for the Identification of Ex Vivo Drug Response Genes in Pediatric Acute Myeloid Leukemia" Cancers 12, no. 5: 1247. https://doi.org/10.3390/cancers12051247