1. Introduction
Prostate cancer (PCa) is the most commonly diagnosed cancer in men, except for superficial skin cancer, and the second leading cause of death due to cancer among American men [
1,
2]. Prostate cancer has a complex disease spectrum, ranging from clinically indolent to aggressive subtypes with a high degree of molecular and cellular heterogeneity [
3,
4,
5,
6,
7]. In order to provide optimal personalized management of the disease, both the physician and the patient need to consider if the disease is likely or unlikely to progress based on biomarkers and imaging tests and to then select the best course of action, either active surveillance for benign and low risk tumor or treatment for a tumor that is likely to progress. Several relevant aspects of current clinical practice are suboptimal. Prostate cancer screening based on serum prostate-specific antigen (PSA) results in many false positives, biopsy complications, and overdiagnosis that ultimately leads to overtreatment [
8,
9]. In addition, conventional primary and repeat biopsies can be inaccurate and pose unnecessary risks. Application of multi parametric MRI (mpMRI) in the context of diagnosis and active surveillance improved risk stratification and the identification of target lesions [
10], while increasing the overall rate of cancer detection in higher grade groups. Currently, combining mpMRI and conventional biopsies provides the highest detection rate [
11,
12].
Early detection of PCa in isolation is not sufficient to reduce mortality from the disease, as a large proportion of screen-detected lesions are indolent. There is a critical unmet need to distinguish early between indolent and aggressive PCa, so that the proper treatment can be selected while overtreatment of indolent disease can be avoided. Although recent decades have seen the development of several RNA-based panels directed at prognosis for PCa [
5,
13,
14], none of these has yet received endorsement as the early marker of choice for future progression, and the genes/proteins represented in the various assays do not overlap. In addition to mRNA-based biomarker panels, recent efforts have also explored the possibility of using protein-based biomarkers in PCa, most focused on the identification and verification of differentially abundant proteins in case-control studies, including the development of urinary protein signatures for extracapsular PCa [
15]. While promising, none of these assays has yet entered clinical practice or demonstrated a significant improvement in prognostic accuracy over more traditional methods of assessing risk.
In this study, we have focused on the identification of reliable early molecular markers capable of accurately predicting future aggressive disease progression. Our objective was to develop a panel of protein biomarkers, detectable in radical prostatectomy (RP) samples from men with organ-confined PCa, that would improve the ability to stratify patients for risk of progression, defined as either biochemical recurrence (BCR) after prostatectomy or distant metastasis (DM). The feasibility of generating protein expression data for low abundance proteins from formalin-fixed parafilm-embedded (FFPE) specimens from primary prostate tumors was demonstrated using an antibody-independent targeted proteomics analysis (high-pressure, high-resolution separations coupled with intelligent selection and multiplexing-selected reaction monitoring (PRISM-SRM)) developed by our team [
16]. Differential protein abundance was then used to identify proteins associated with PCa aggressiveness. The predictive accuracy of a proteomic classifier in predicting local and distant cancer progression was validated in a cohort of men with long-term follow-up data and detailed clinical annotation. The addition of the proteomic classifier to the traditional, Standard of Care (SOC) variables was examined using training and testing analysis to determine the ability of the classifier to improve performance in the study cohort.
3. Discussion
Improved clinical management of prostate cancer is based not only on early detection of neoplastic lesions in the prostate but also very significantly on the early discrimination of indolent prostate cancer, which can be effectively managed by active surveillance from aggressive forms of prostate cancer which could rapidly progress to castration resistance and/or metastatic disease. This project focused on identifying robust biomarkers to predict patients at risk for future cancer progression, including biochemical recurrence and distant metastasis, to enhance existing biopsy or pathology factors used for risk stratification and to provide additional support for safe delay (i.e., active surveillance) of invasive treatment versus early, aggressive therapy.
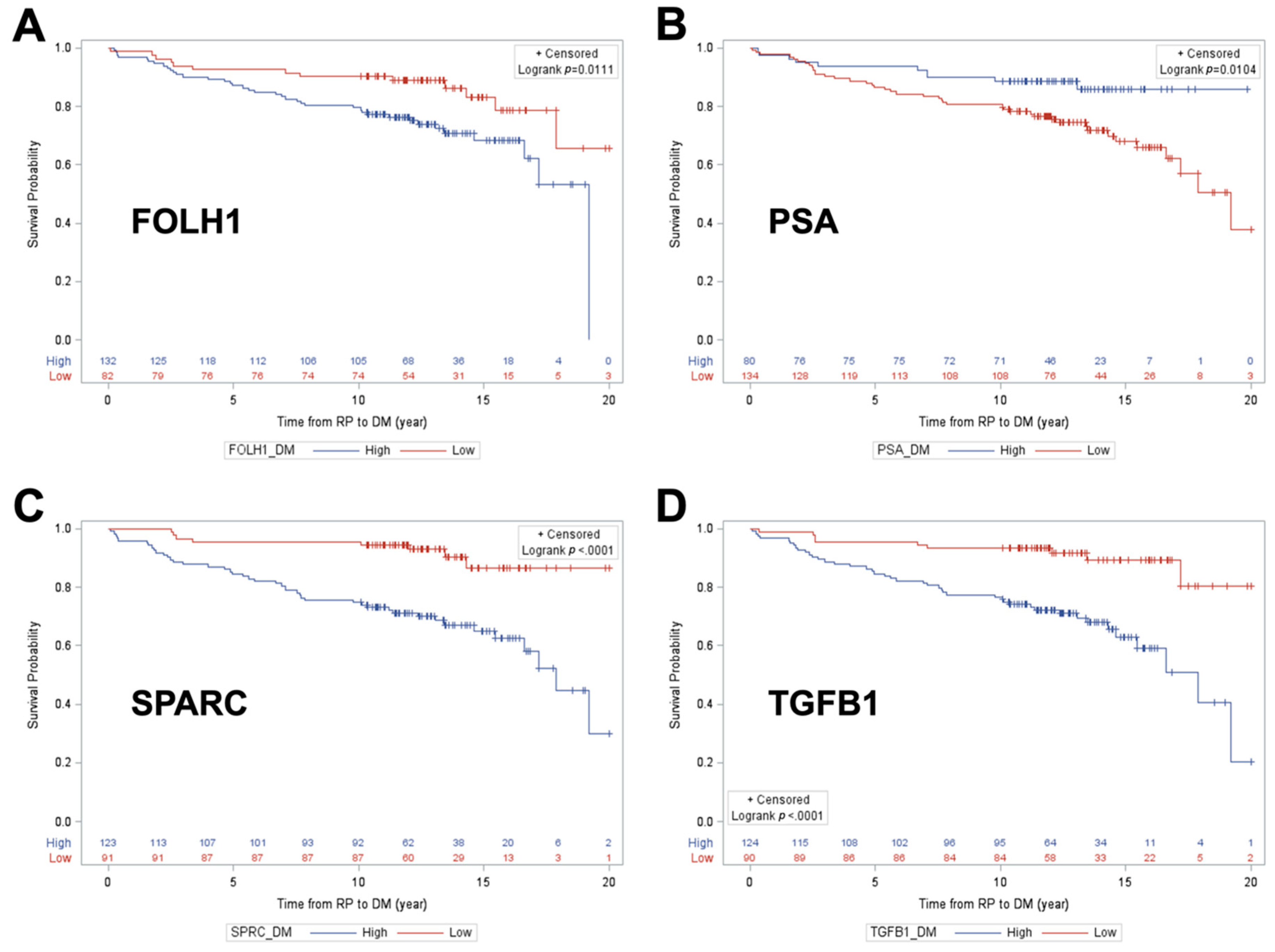
The experimental approach in this study was designed to raise the threshold for biomarker selection in a series of filters, with each providing increased stringency. The starting panel of candidate biomarkers, chosen on the basis of curated knowledge, was initially filtered on the basis of differential expression at the mRNA level using NanoString. Targeted antibody-independent proteomic assays (PRISM-SRM assays) were then developed for the 42 differentially expressed genes and tested for the ability to detect the cognate proteins in FFPE-preserved RP specimens from a small patient cohort with long-term follow-up, documenting DM, BCR, and no progression of events. Of the 42 proteins which could be detected and quantified, 16 were identified as differentially abundant across study outcomes in a combined cohort of 338 patients and finally in a testing-and-training cohort set. Three candidate proteins were robustly associated with BCR, four were associated with DM, and six were associated with high GG; across these proteins, three proteins were common in all endpoints: FOLH1, SPARC, and TGFB1. FOLH1, folate hydrolase 1, is also known as Prostate-Specific Membrane Antigen (PSMA) and has been extensively studied as a potential biomarker of PCa [
25,
26]. FOLH1 is involved in folate uptake, thus indirectly affects DNA synthesis, and is known to be correlated with aggressive PCa. SPARC, also known as osteonectin, has been frequently observed as an upregulated protein in multiple cancers including PCa [
27,
28]. SPARC/osteonectin plays an important role in bone mineralization and may serve to facilitate the metastasis of PCa to bone. TGFB1, transforming growth factor beta1, is a ubiquitous cytokine with well-documented effects on immune function that have been associated with tumor-promoting stromal interactions [
29,
30]. Thus, the consistent observation of these three proteins across models serves to further support their importance in tumor biology.
The final study cohort of 338 patients is one of the largest tested for validation of such proteomic biomarkers, and the final training and testing analysis ensures a thorough examination of the performance of the developed classifier. Moreover, it is important to note that the biomarker measurements in all the case-control studies presented in this report were solely based on antibody-independent MS assays. Such assays are highly sensitive, are precise, and circumvent the necessity to develop a high-quality ELISA for each of the candidate markers [
16,
31,
32,
33,
34]. Albeit, an important limitation that must be noted is the possibility of model overfitting, since a limited number of study endpoints were examined in this single study.
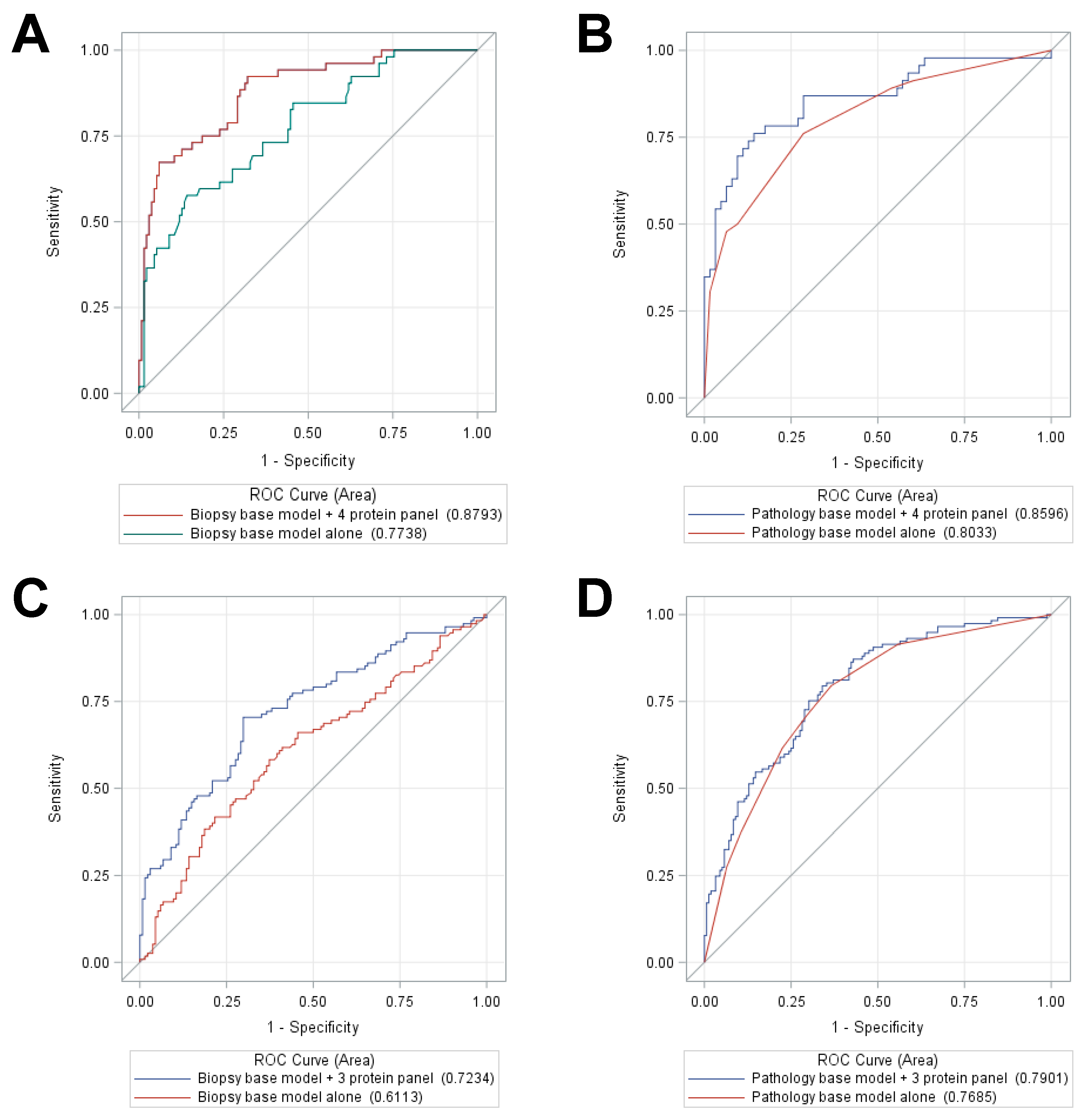
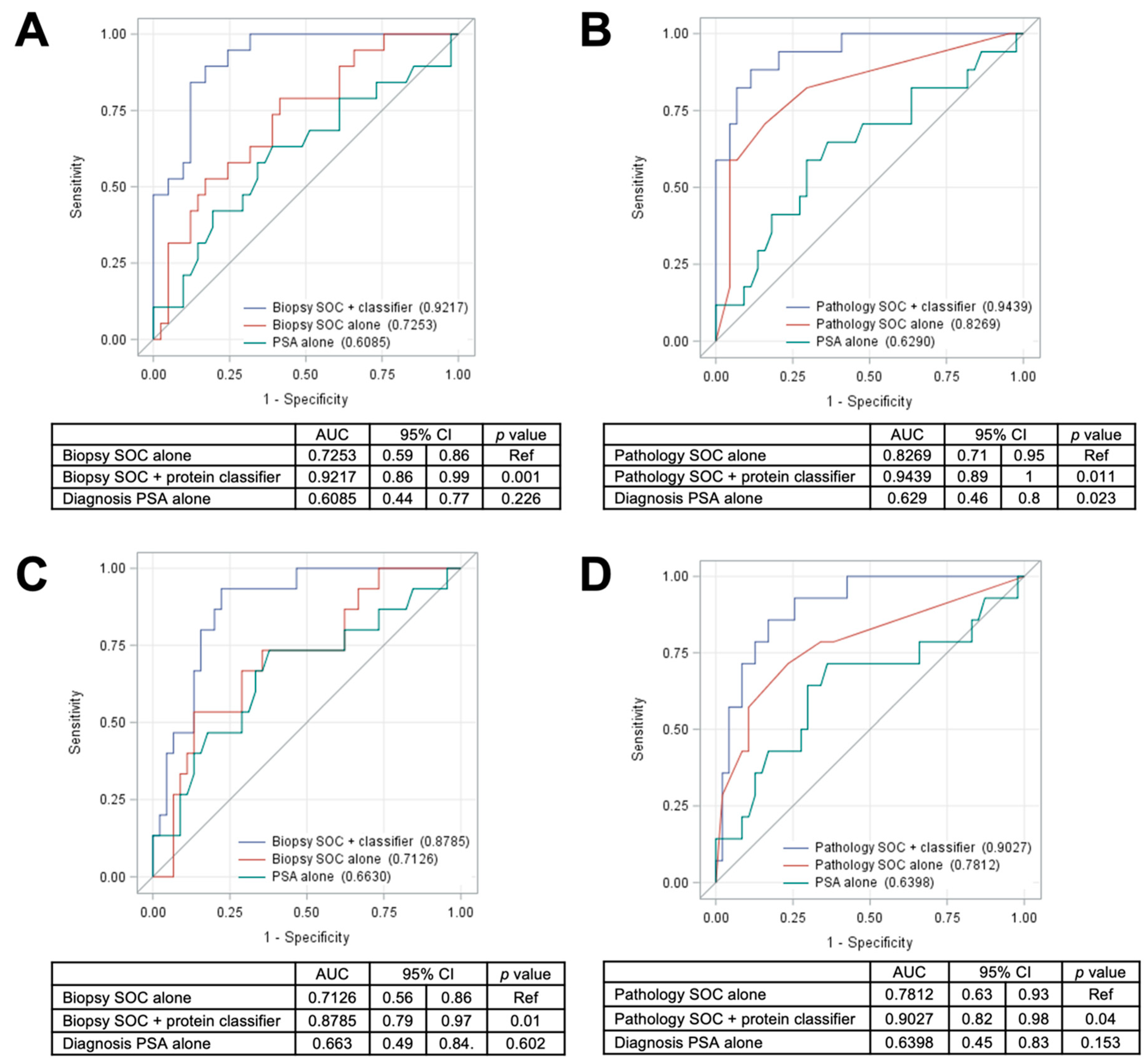
The current standard of care for PCa involves the initial use of biopsies and the biopsy base model to assess the risk of aggressive PCa in patients identified by PSA screening. Those patients exceeding a specific risk threshold are then referred for RP, and the pathological characteristics of the excised tumor are frequently used to guide further treatment. Improvements in the predictive power of the biopsy base model have the potential to significantly reduce the need for RP, and the tissue proteomic classifiers developed in this study significantly improved the performance of the biopsy base model for predicting either BCR or metastasis. Addition of the 5 protein classifier to the biopsy base model provided a significantly improved AUC of 0.92 (95% CI = 0.86–0.99,
p = 0.001; versus 0.73 with the biopsy base model alone or 0.61 with PSA alone) for predicting DM (
Figure 4A) as well as a significantly improved AUC of 0.88 (95% CI = 0.79–0.97,
p = 0.01; versus 0.71 with the biopsy base model alone or 0.66 with PSA alone) for prediction of BCR (
Figure 4C). While the tissue proteomic biomarkers did slightly improve the performance of the pathology base model for predicting both DM (
Figure 4B) and BCR (
Figure 4D), we postulate that the most valuable clinical application is likely to be in the analysis of prostate biopsy samples, prior to the acquisition of pathology metrics from RP. Although it remains to be demonstrated, prostate biopsy samples theoretically contain sufficient material for proteomic analysis of the panels developed in this study. If the predictive power of the proteomic classifier is also observed in biopsy samples, it may be possible to more accurately assess the risk of aggressive PCa from biopsy samples alone, potentially sparing RP in borderline risk scenarios. Although direct comparisons are complicated by differences in aspects such as study time period, cohort size, racial composition, specimen type, and methodologies, an initial comparison with the other existing commercial tests provides evidence of superior performance using the 5-protein classifier developed in this study (
Table S18).
Prostate cancer molecular biomarkers, including prognostic ones, have been extensively reviewed [
14,
35,
36,
37,
38,
39,
40]. The development of these biomarkers has created new opportunities for improving on current clinical practice but, at the same time, also poses challenges for the selection and incorporation of the most appropriate new assays into prostate cancer care. In addition to molecular biomarkers, prostate imaging (mpMRI) has recently emerged as a new tool to guide clinicians in managing prognostic evaluation of patients with localized prostate cancer [
41,
42,
43]. An individualized risk-based decision making process needs to be established in the clinical practice, with contribution from both biomarkers and imaging [
35,
44,
45]. Future research directions in support of this objective would include prospective studies of targeted protein marker expression in diagnostic biopsy specimens at the time when the option of active surveillance is still available [
44,
45,
46].
4. Materials and Methods
4.1. Study Cohort
A retrospective cohort study was conducted using the Walter Reed National Military Medical Center (WRNMMC) prostate cancer Biospecimen Repository linked to the Center for Prostate Disease Research (CPDR) Multi-center National Database. In brief, specimens in the WRNMMC Biospecimen Repository were collected from PCa patients who underwent RP at WRNMMC and who provided informed consent to donate prostatectomy specimens to the repository and enrollment in the CPDR Multi-center National Database clinical data repository. The Multi-center National Database contains detailed demographic, clinical, treatment, pathologic, and outcomes information. Further details about these databases have been reported previously [
47]. Both repositories and multi-center national database have Institutional Review Board (IRB) approval at the WRNMMC and the Uniformed Services University of the Health Sciences (USUHS), respectively. The IRB code for this study is Ref #908925 with an approval date of 18 June 2019 (at the Center for Prostate Disease Research).
4.2. Demographic, Clinical, and Treatment Variables
Variables were age at PCa diagnosis (years), self-reported race (African American, Caucasian American, and “Other”), PSA at PCa diagnosis (ng/mL), clinical T stage (T1-T2a, T2b-T2c, and T3a-T4), biopsy Gleason sum (≤6, 7, and 8–10), NCCN-defined risk strata (low, intermediate risk, and high risk), time from diagnosis to RP (months), and post-RP follow-up time (months).
4.3. RP Specimen Processing and Pathologic Variable Measurement
All RP specimens were processed by whole mount and sectioned at 2.2 mm intervals, as previously described [
48,
49]. Pathologic parameters were measured based on evaluation by central pathology review (by I.S.), including pathologic T stage (pT2 and pT3-pT4), grade group (GG1-5), and surgical margin status (negative and positive).
4.4. Dependent Study Outcomes
To ascertain whether targeted protein marker expression in FFPE tissues could be used to predict PCa progression, the study outcomes included BCR and DM after RP. A BCR event was defined in the following manner: a post-RP PSA level ≥0.2 ng/mL followed by a successive, confirmatory PSA level ≥ 0.2 or the initiation of salvage radiation or hormonal therapy after a rising PSA and excluding PSA values drawn within eight weeks of the RP. Presence of DM was ascertained by physician’s review of each patient’s complete imaging history, including bone scan, CT scan, MRI, and/or bone biopsy results. Subjects who had no evidence of BCR or metastasis at the end of study period with at least 10 years of post-RP follow-up were defined as “nonevents”.
4.5. Protein Digestion of FFPE Tissue Samples
The FFPE human prostate tissue samples were first deparaffinized by adding 500 µL of xylene (Sigma Aldrich, St. Louis, MO, USA) and by incubating for 5 min at room temperature with 300 rpm shaker speed. The solution was removed, and the xylene addition and incubation were repeated. After removing the solution for a second time, 500 µL of 190 proof ethanol (Decon Laboratories, King of Prussia, PA, USA) was added and incubated for 5 min at room temperature with 300 rpm shaker speed. The solution was removed. Finally, 500 µL of 80% ethanol was added and incubated for 5 min at room temperature with 300 rpm shaker speed. The solution was removed, and the samples were dried for 15 min in Speed-Vac (Thermo Fisher Scientific, Waltham, MA, USA).
Once dried, 50 µL of 2,2,2-Trifluoroethanol (TFE) (Sigma Aldrich) was added to the samples. Then, 50 µL of 600 mM Tris-HCl was added to the samples to give a final concentration of 50% TFE. The samples were homogenized with a Kontes® Pellet Pestle® (VWR, Radnor, PA, USA) for 30 s, keeping the samples cool on an ice block during homogenization and afterwards for 3 min. The samples were transferred to a 1.5-mL screw top tube before incubating with a Thermomixer (Eppendorf, Hamburg, Germany) at 99 °C for 90 min with 1000 rpm shaker speed. The samples were allowed to cool to room temperature. The protein concentrations of the samples were determined using bicinchoninic acid (BCA) assay (Thermo Fisher Scientific).
Proteins were reduced with 5 mM Dithiothreitol at 37 °C for 1 h and alkylated using 10 mM iodoacetamide at room temperature for 1 h in the dark. The samples were diluted with water and digested with sequencing grade modified trypsin (Promega Corporation, Madison, WI, USA) at a 1:50 trypsin:protein ratio. The samples were incubated at 37 °C for 4 h; then, a second 1:50 trypsin addition was made, and the samples were incubated overnight at 37 °C. The digestion was stopped by addition of 10% formic acid to have reach final concentration of 1% formic acid.
The samples were centrifuged at 14,000 rpm at 4 °C prior to the final solid-phase extraction (SPE) based desalting step using 50 mg, 1 mL C-18 SPE cartridges (Strata, Phenomenex, Torrance, CA, USA) and a manual vacuum manifold (Supelco, Sigma Aldrich). The cartridges were preconditioned using 3 mL of 100% methanol followed by 2 mL of 0.1% trifluoroacetic acid (TFA). The sample was loaded and slowly passed through the cartridge at a rate no faster than 1 mL per minute. The cartridge was then washed with 4 mL of 5% acetonitrile (ACN), 0.1% TFA, and 1 mL of 1% formic acid to remove any residual TFA. The desalted peptide sample was eluted into a 2.0-mL microcentrifuge tube using 1.5 mL of 80% can and 0.1% formic acid. The eluted sample was placed in the Speed-Vac and concentrated. The peptide concentration was determined using the BCA assay, and the final concentration was adjusted to 0.3 µg/µL. The sample was then frozen in liquid nitrogen and stored at −70 °C until needed for peptide spiking and SRM analysis.
4.6. PRISM-SRM Assay Configuration and Measurements
A total of 110 tryptic peptides for the 52 protein candidates were selected based on well-accepted criteria for targeted proteomics analysis [
50]. Pure stable isotope-labeled heavy peptides (purity >97%) with C-terminal (U-13C6, 15N2) lysine or (U-13C6, 15N4) arginine were synthesized (AQUA QuantPro, ThermoFisher Scientific, Waltham, MA, USA) for PRISM-SRM assay development and measurements. The peptide list is provided in
Table S2. The peptides were received at a concentration of 5 pmol/µL in 5% ACN. Equal volume of these 110 peptides were mixed together to create a heavy peptide mixture stock, and the final peptide concentration in the stock is 45 fmol/µL.
The transitions and collision energy of individual peptides were first optimized by direct infusion experiments on a TSQ Vantage triple quadrupole mass spectrometer (Thermo Fisher Scientific) and furthered evaluated by LC-SRM using a nanoACQUITY UPLC
® system (Waters Corporation, Milford, MA, USA) and a TSQ Vantage triple quadrupole mass spectrometer. Three best transitions with minimal interference and highest sensitivity were retained for each peptide in the final SRM assays. The list of best transitions, optimized collision energy, and LC elution times of each peptide are provided in
Table S3.
Heavy peptides were spiked in digested and cleaned FFPE samples, and they were separated following the PRISM workflow using high pH reversed-phase capillary LC on a nanoACQUITY UPLC
® system as described previously [
16]. Briefly, separations were performed using a capillary column packed in-house (3 μm Jupiter C18 bonded particles, 200 μm i.d. × 50 cm long) at a flow rate of 2.2 μL/min on binary pump systems, using 10 mM ammonium formate (pH 9) as mobile phase A and 10 mM ammonium formate in 90% ACN (pH 9) as mobile phase B. Forty-five µL of each sample (35 µg) were loaded onto the column and separated using a binary gradient of 5%–15% B in 15 min, 15%–25% B in 25 min, 25%–45% B in 25 min, and 45%–90% B in 38 min. The samples were separated into 96 fractions (1-min elution time per fraction), and the fractions were collected using a LEAP’s Collect PAL fraction collector (LEAP Technologies, Carrboro, NC, USA). Prior to peptide fraction collection, ~20 μL of water was added to each well in the plate to avoid peptide loss and to dilute the peptide fraction for LC-SRM analysis.
Configuration 1:110 peptides (52 proteins) in the first 105 samples. To facilitate the high-throughput PRISM-SRM analysis of 110 peptides in the first batch of 105 samples, the 96 fractions were concatenated into 24 fractions. These 24 fractions were analyzed individually on the second dimension LC-SRM using a nanoACQUITY UPLC® system coupled to TSQ Vantage triple quadrupole mass spectrometer. Briefly, separations were performed using a ACQUITY UPLC M-Class Peptide BEH C18 Column, 300 Å, 1.7 µm, 100 µm × 100 mm (Waters Cooperation) at a flow rate of 0.4 μL/min and a temperature of 42 °C on binary pump systems, using 0.1% formic acid in water as mobile phase A and 0.1% formic acid in ACN as mobile phase B. Four µL of each sample was loaded onto the column at a flow rate of 0.5 µL/min for 10 min and separated using a binary gradient of 0.5–5% in 0.5 min, 5–20% B in 26.5 min, 20–25% B in 10 min, 25–38.5% B in 8 min, and 38.5–95% B in 1 min. The TSQ Vantage was operated with ion spray voltages of 2400 V and a capillary inlet temperature of 370 °C. Tube lens voltages were obtained from automatic tuning and calibration without further optimization. Both Q1 and Q3 were set at unit resolution of 0.7 FWHM, and Q2 gas pressure was 1.5 mTorr. A scan width of 0.002 m/z was used. Because of the large number of transitions to be scanned, we used a scheduled SRM method with RT window set to be 4 min and cycle time of 1 s.
Configuration 2: 16 peptides (16 proteins) in the remaining 233 samples. For the remaining 233 samples in the cohort, we reduced the number of protein candidates from 52 to 16. In order to achieve similar or even higher sensitivity with higher throughput, we selected only the target-containing fractions (roughly 16 fractions) during PRISM via online SRM monitoring of the heavy peptides, instead of concatenating into 24 fractions, for the second dimension LC-SRM analysis; we also used a faster LC gradient for the LC-SRM analysis of the PRISM fractions. Briefly, separations were performed at a flow rate of 0.4 μL/min and a temperature of 42 °C on binary pump systems, using 0.1% formic acid in water as mobile phase A and 0.1% formic acid in ACN as mobile phase B. Four µL of each sample was loaded onto the column at a flow rate of 0.5 µL/min for 10 min and separated using a binary gradient of 0.5–10% B in 0.5 min, 10–15% B in 1 min, 15–25% B in 6 min, 25–35% B in 3 min, and 35–95% B in 2 min. A nonscheduled SRM method with dwell time of 10 ms for each transition was used for analysis of the much smaller set of 16 peptides, while other MS conditions remained the same.
In order to evaluate the consistency between the first and second PRISM-SRM configurations, we used 3 individual samples from the first set of 105 and analyze them to quantify the final 16 peptides (of 16 proteins) using both configurations. The average measurement variations of 3 samples between two configurations for all the 16 peptides are between 2% and 29% with a median of 5%, which demonstrated the consistency in peptide quantification between two PRISM-SRM configurations.
4.7. Response Curves for the PRISM-SRM Assays
The sensitivity and linearity of the PRISM-SRM assay were determined by measuring heavy isotope-labeled peptide standards spiked into a sample pooled from all the remaining study samples to final concentrations of 0, 0.6, 3, 12, 60, 300, 1500, 3000, 6000, 12,000, 24,000 and 48,000 amol/µg. Each of the above samples were subjected to the same PRISM-SRM workflow as mentioned in configuration 2, with 3 injection replicates. The response curves of each peptide were generated using the heavy-over-light peak area ratios and the heavy peptides concentration as mentioned above. Signal-to-noise ratio (S/N) was calculated by the peak apex intensity over the highest background noise in a retention time region of ±15 s for the target peptides. The background noise levels were conservatively estimated by visually inspecting chromatographic peak regions. The lower limit of detection (LOD) and quantification (LOQ) were defined as the lowest concentration point of target proteins at which the S/N of surrogate peptides was at least 3 and 10, respectively. Additionally, LOQs also require a coefficient of variation (CV) of less than 20%. The final LOD and LOQ values of each assay were provided in
Table S4. Note that, given the significant interference for the heavy peptide transitions of TGFB1 peptide GGEIEGFR (shown in
Figure S2), the LOD and LOQ of GGEIEGFR cannot be accurately determined; however, we made sure that the S/N ratios of the endogenous (light) peptides are acceptable through manual inspection.
4.8. SRM Data Analysis
The entire cohort of FFPE samples was randomized and analyzed by PRISM-SRM at the Pacific Northwest National Laboratory (PNNL) in a blinded fashion (patient outcome data were restricted at CPDR during the entire analysis from the experimental design through statistical analysis). The raw data acquired on the TSQ Vantage triple quadrupole MS were imported into Skyline software (Version 20.1) [
51] for visualization and quantification. The total peak area ratio between endogenous peptides and their heavy isotope labeled peptide standards was used for quantification. Peak detection and integration were based on two criteria: (1) the same retention time and (2) approximately the same relative peak intensity ratios across multiple transitions between endogenous peptide and heavy isotope labeled peptide standards. All data were manually inspected to ensure correct peak detection and accurate integration.
4.9. Endogenous Concentration Calculation
The final endogenous peptide concentrations (amol/µg) for all the samples were calculated using the response curves. The steps we took to calculate the final concentration of peptides in the study samples are provided below.
Step 1. Fit the calibration curve using linear regression (Y =
Slope × X +
Intercept), where X is the heavy peptide concentration in amol/µg and Y is the heavy over light peak area ratio (H/L). The final
slope and
intercept values are provided in
Table S4.
Step 2. Calculate light peptide concentration of each peptide in the matrix (Clight in response curve) using the response curve obtained above and data at three heavy peptide spike-in levels (300, 1500, and 3000 amol/µg) and obtain the average of calculated light peptide concentrations (amol/µg).
Step 3. Calculate the final endogenous peptide concentration in the study samples (
Cendogenous in sample) using the
slope,
intercept, and
Clight in response curve of the response curves. Equation (1) is as follows:
where
L/H Ratioin sample is the light over heavy peptide peak area ratio (
Supplementary Materials Table S5) obtained directly from PRISM-SRM measurements and
Cheavy in sample is the heavy peptide concentration spiked in the study samples (amol/µg) (
Supplementary Materials Table S6). The final endogenous concentrations of the peptides in all the study samples are provided in
Table S7.
4.10. Initial Evaluation of Performance of Protein Biomarker Panels
All statistical analysis was performed using SAS version 9.4 (Cary, NC, USA), and statistical significance was set at p < 0.05 (except for univariable analysis of individual protein markers, described below, that underwent Bonferroni adjustment).
Analysis of variance (ANOVA) and Kruskal–Wallis testing was used to examine differences in the distribution of continuous variables (i.e., age at diagnosis, time from diagnosis to RP, and total follow-up time) across event groups, while Chi-square or Fisher exact tests were used to evaluate the associations of categorical variables (e.g., self-reported patient race, PSA at diagnosis, clinical T stage, biopsy grade, NCCN risk stratum, pathological T stage, Grade Group (GG), and surgical margin status) across event groups.
AUC values were computed for each of the 16 proteins markers for the ability of the markers to discriminate event group comparisons, including distant metastasis vs. nonevents, BCR versus nonevents, and high versus low GG (3–5 vs. 1–2). Only markers that achieved an AUC value ≥0.60 were retained in subsequent models, per event group comparison. For predicting distant metastasis, among those markers with an AUC ≥ 0.60, an optimal cutpoint was derived for maximizing sensitivity while satisfying the following critical thresholds: Negative Predictive Value (NPV) > 80% and Specificity > 40%. The 95% confidence intervals for each AUC value were constructed using nonparametric, bootstrapping with 1000 replicates. Using the Bonferroni method, the type I error threshold for determining statistical significance was adjusted for evaluating 16 protein markers as follows: p = 0.05/16 = 0.003125.
Unadjusted Kaplan–Meier survival curve analysis with log-rank testing was used to examine distant metastasis-free survival as a function of each individual protein classifier cut point that achieved the NPV/Spec requirements, including FOLH1, PSA, SPARC, and TGFB1 for DM. In KM analysis for BCR-free survival, the same approach was used for two protein classifier cut points: SPARC and TGFB1.
ROC curves were then examined to predict distant metastasis and BCR, using SOC variables, alone and then in combination with protein panels constructed by using principal components analysis to identify panel candidates, defined as those markers that achieved a principal components Eigenvalue > 1 per study endpoint (BCR and DM). For DM, four proteins (FOLH1, PSA, SPARC, and TGFB1) were selected from univariable analysis to form a protein panel; for BCR, three proteins (FOLH1, SPARC, and TGFB1) were selected. Two types of SOC models were examined: (i) a base model with biopsy features (i.e., age at PCa diagnosis, patient race, and NCCN risk stratum) and (ii) a base model with pathology variables (i.e., age at PCa diagnosis, patient race, pathological T stage, GG, and surgical margin status). The comparison between the AUCs statistic for the base models versus base models in combination with protein panels, for predicting of each study endpoint (DM and BCR) was performed using maximum likelihood ratio tests.
Finally, for multivariable logistic regression analysis, ROC curves were generated to evaluate prediction accuracy based on AUC statistics of each set of protein marker “panels” for each outcome: metastasis, BCR, and high GG. Four multivariable Cox proportional hazards (PH), models were constructed to examine distant metastasis-free survival and BCR-free survival, each as a function of the protein markers specific to the study endpoint, adjusting first for the biopsy “base” SOC variables, followed by adjustment for the pathologic “base” SOC variables.
4.11. Protein Classifier Development and Evaluation
Among the 338 patients, 53 developed DM after RP, 124 developed BCR but did not metastasize, and 161 are nonevents (did not develop BCR or metastasis after at least 10 years of follow-up post-RP). Two hundred and fourteen patients (53 DM and 161 nonevents) were used to develop a biomarker panel classifier to predict DM. This 214-patient cohort was randomly split into training and testing data sets (70% vs. 30%): a training cohort of 149 patients (33 DM and 116 nonevents) and a testing cohort of 65 patients (20 DM and 45 nonevents).
Biomarker selection, classifier construction, and optimal cutoff development in the training cohort were performed as followed: (1) Biomarker selection: Univariable logistic regression analysis was used to select those biomarkers which are significantly predicting DM status (p < 0.05 and AUC > 0.65). They are CAMKK2, FOLH1, PSA, SPRC, and TGFB1; (2) protein classifier construction: Fitted multivariable logistic regression model including the 5-protein panel to get parameter estimates for 5-protein panel classifier construction in predicting DM, scaled from 0 to 100; (3) classifier threshold: bootstrapped multivariable logistic regression (with 1000 replicates) was used to search for optimal cutoff of protein classifier in predicting DM, and the optimal threshold was defined as a cut point which maximizes sensitivity, with at least 90% NPV and at least 35% specificity.
Testing of the protein classifier and its threshold in the testing cohort was performed as followed: (1) Protein classifier testing: those parameter estimates which were generated in training cohort were applied to the testing cohort to construct a protein classifier. First, we tested 5-protein panel classifier’s prediction accuracy in predicting DM using logistic regression and ROC curve. Second, we evaluated the effects of adding the 5-protein classifier to biopsy SOC model (age, race, and NCCN-risk strata) and pathology SOC models (pathological T stage, GG, and surgical margin) on prediction accuracy based on PSA, using multivariable logistic regression analysis, ROC curve, and Mantel–Haenszel Chi-square tests. (2) Classifier threshold testing: The threshold for the 5-protein panel classifier in predicting DM-free survival was tested using univariable Kaplan–Meier curve and log-rank test. The classifier was tested alone, and when added to both the biopsy SOC model and the pathology SOC models, using multivariable-Cox proportional hazard analysis, the proportional hazard assumption of each covariate was checked and met.
The same method was used to evaluate the performance of the classifier for predicting BCR in the training and testing cohorts.

 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}