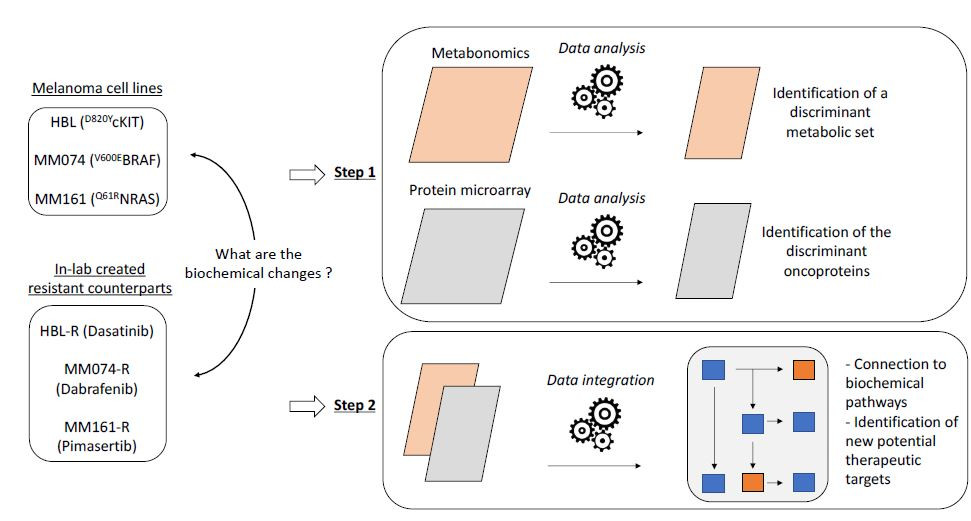
Metabolic Reprogramming in Metastatic Melanoma with Acquired Resistance to Targeted Therapies: Integrative Metabolomic and Proteomic Analysis

 , , ,
, , ,  ,
, 

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
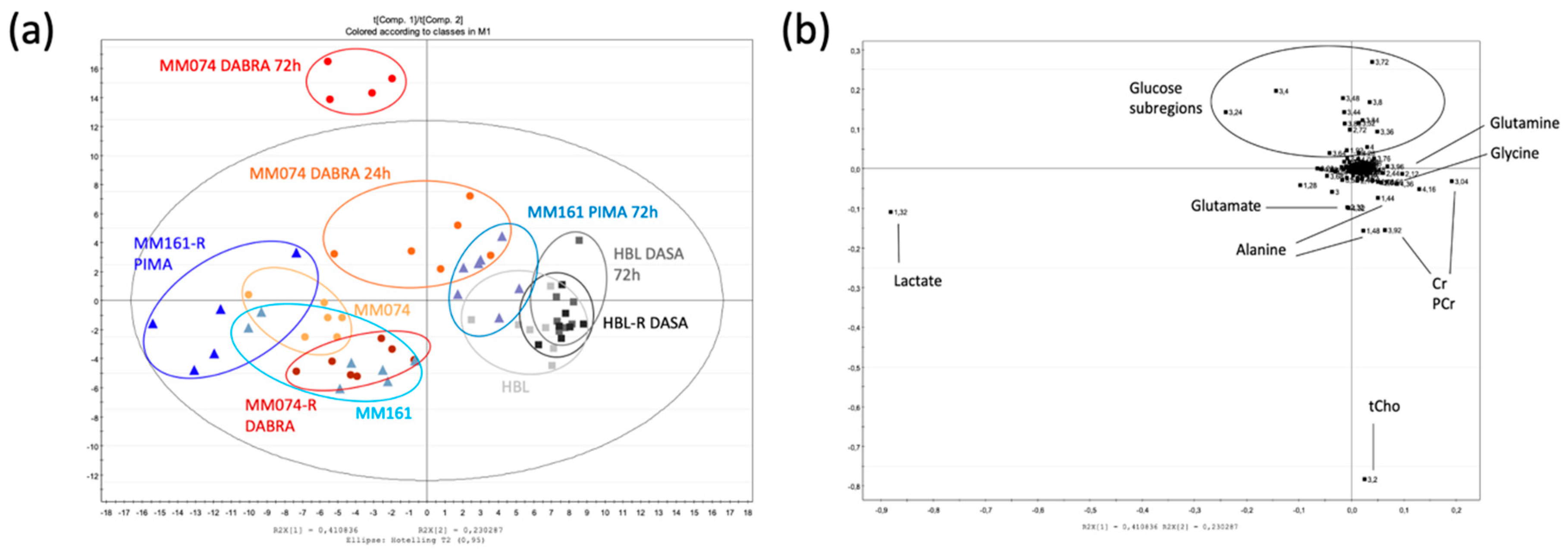
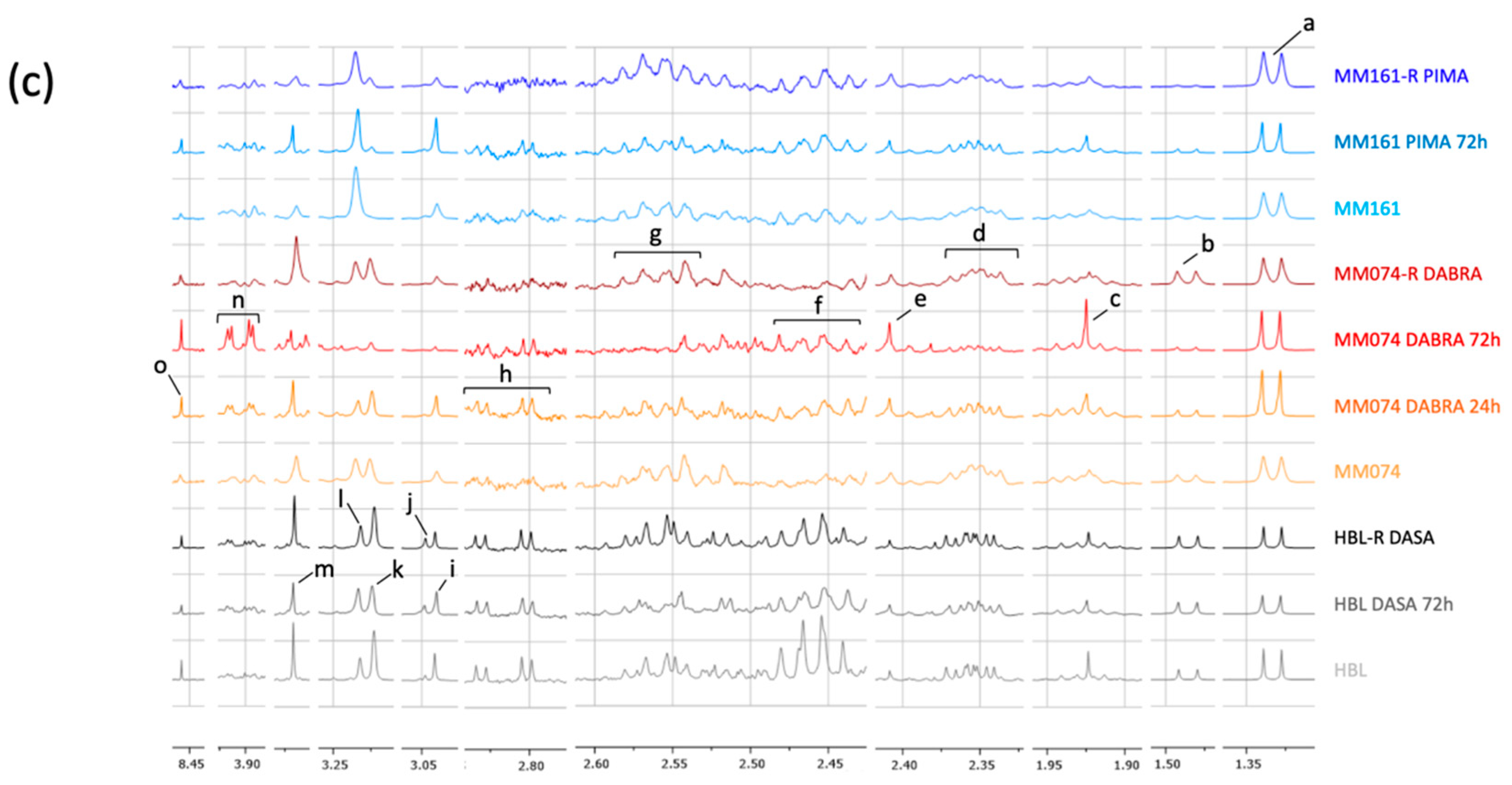
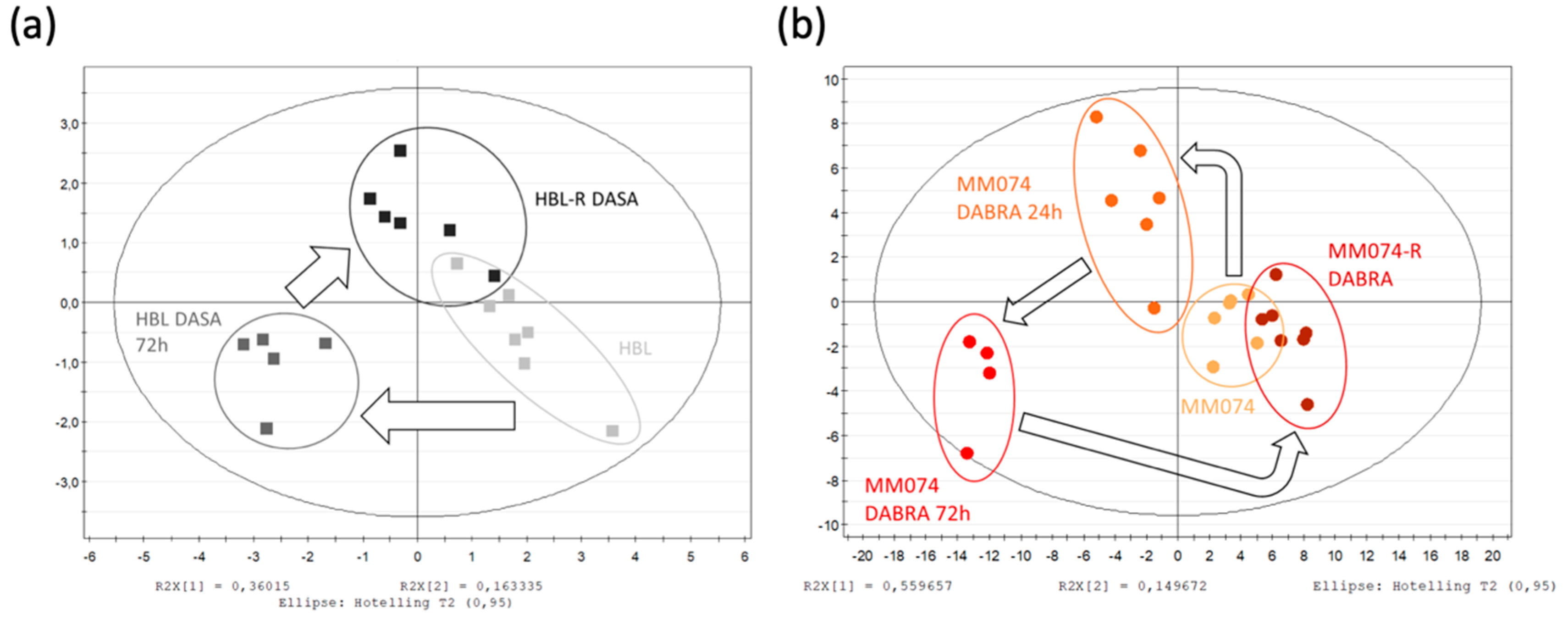
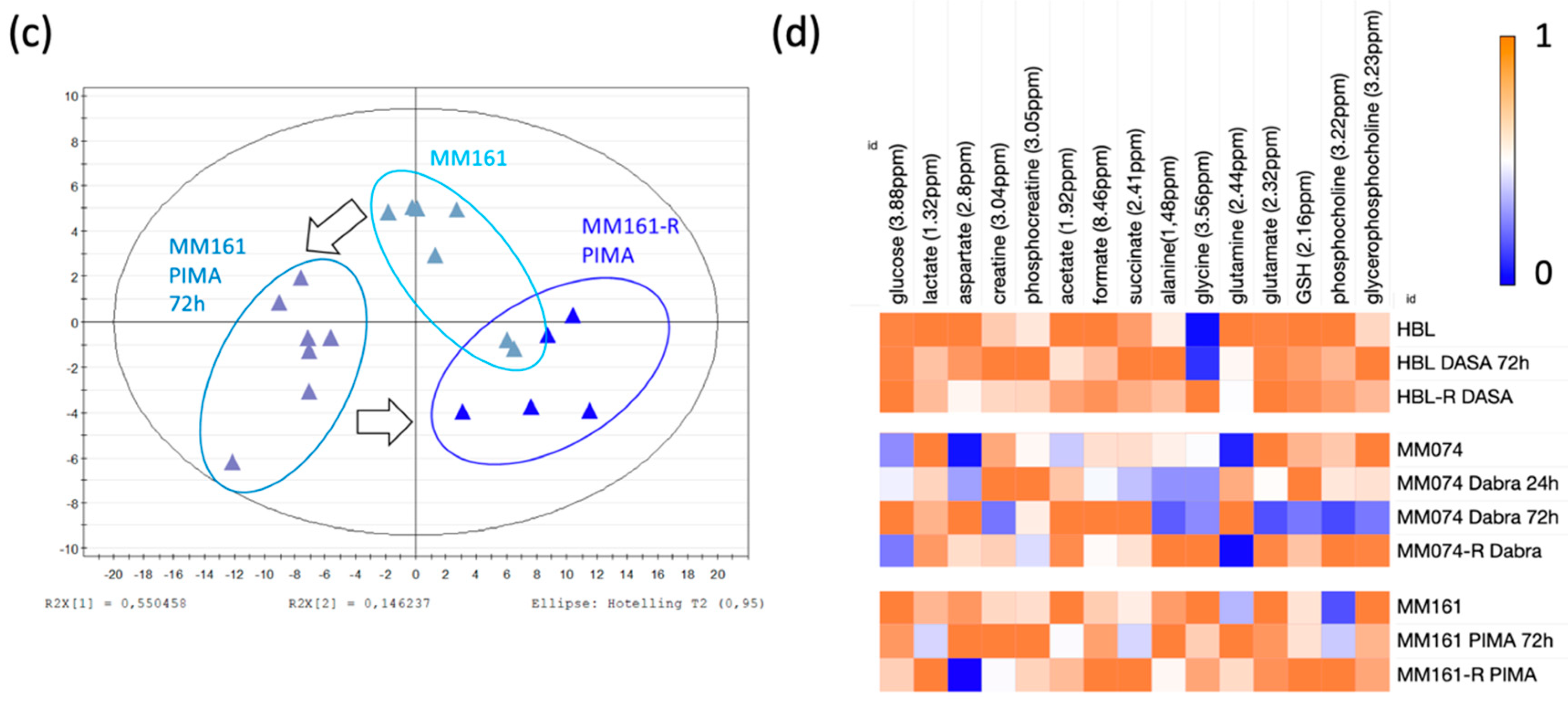
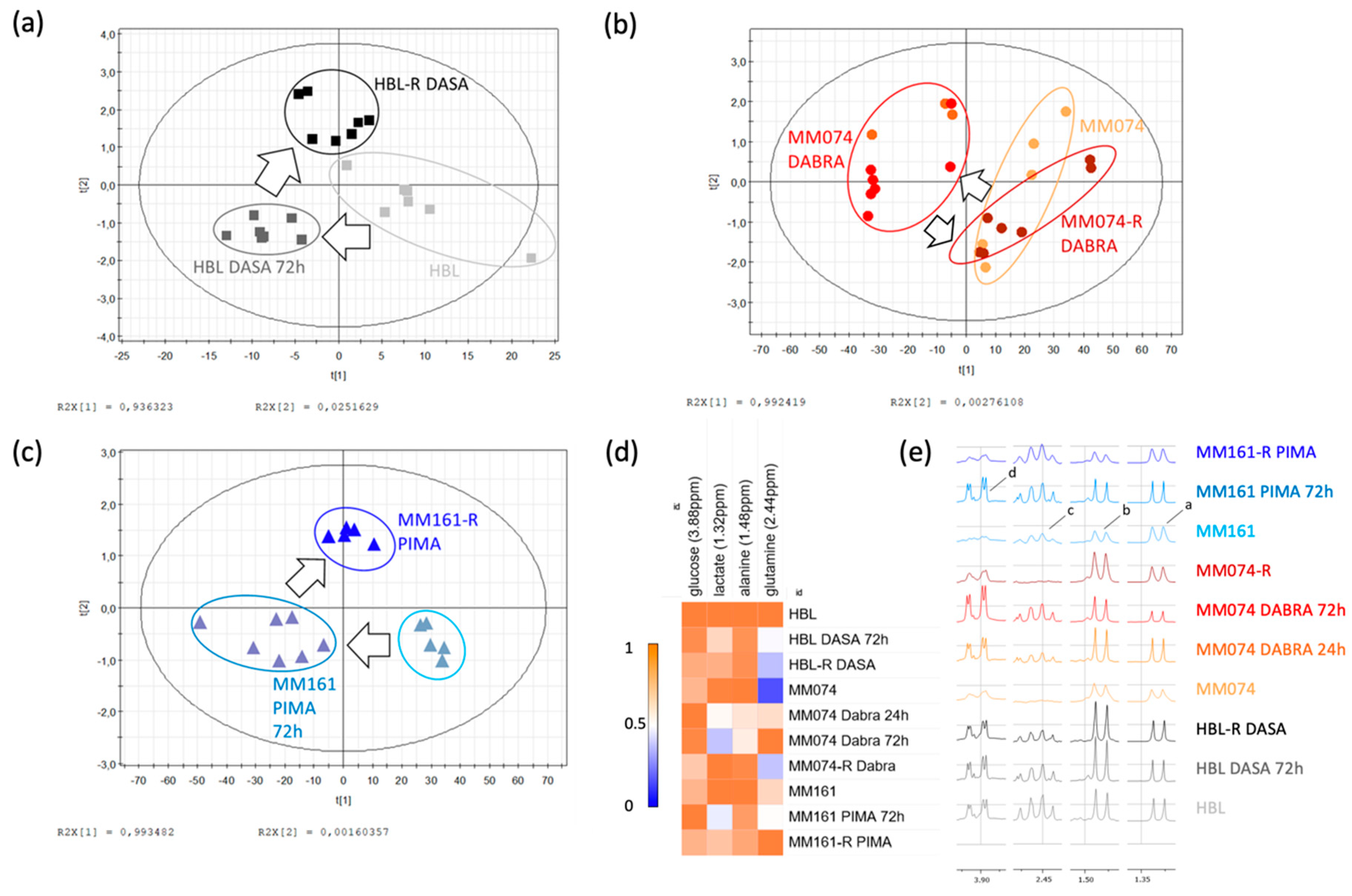
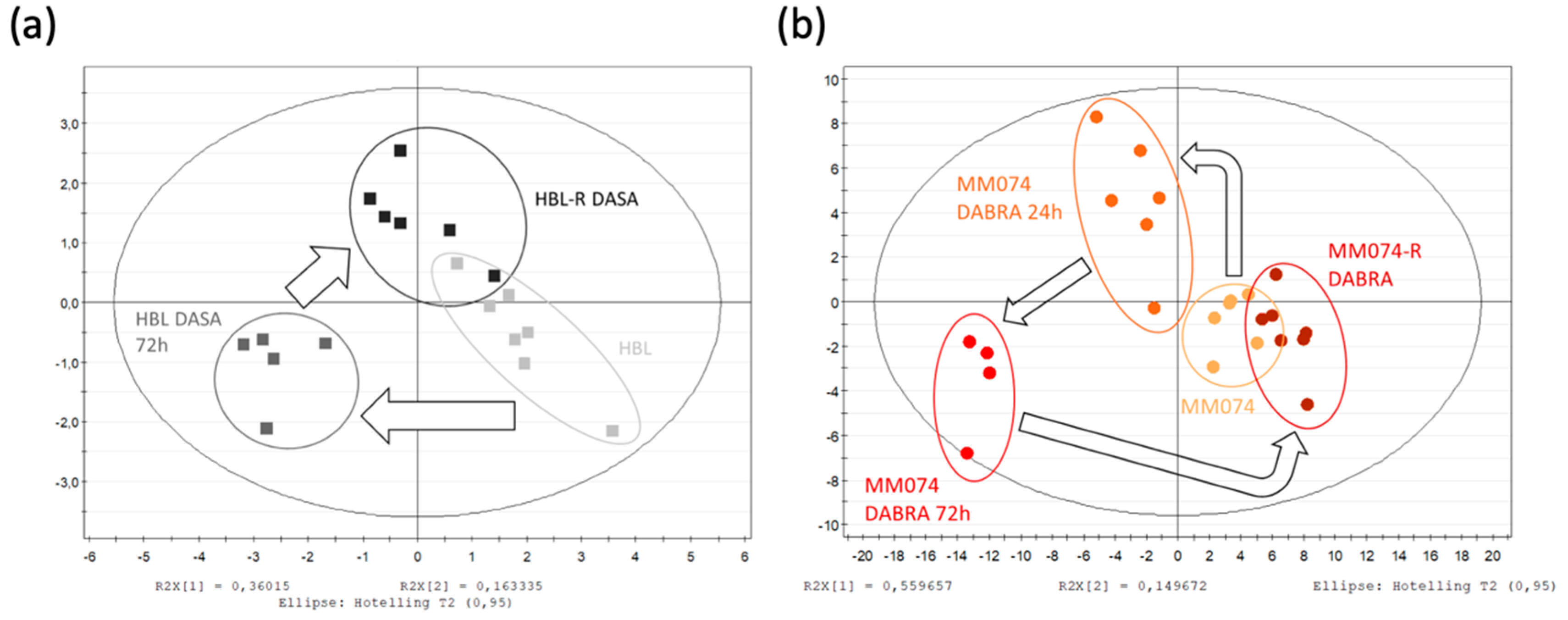
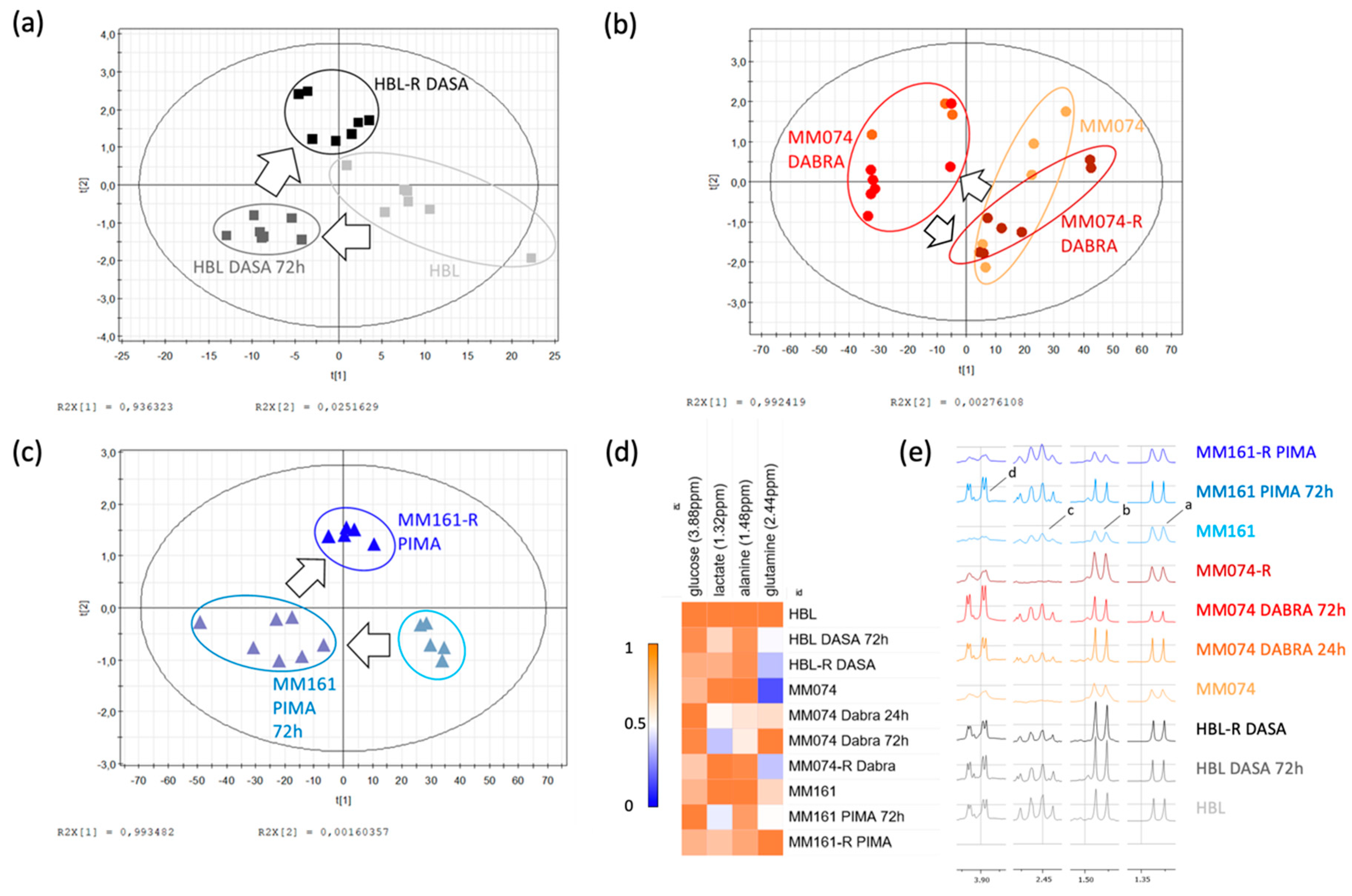
2.1. 1H-NMR Cellular Extract Signature of the Three Types of Cell Lines with Acquired Resistance
2.1.1. Identification of the Discriminant Metabolites Using Multivariate Data Analysis
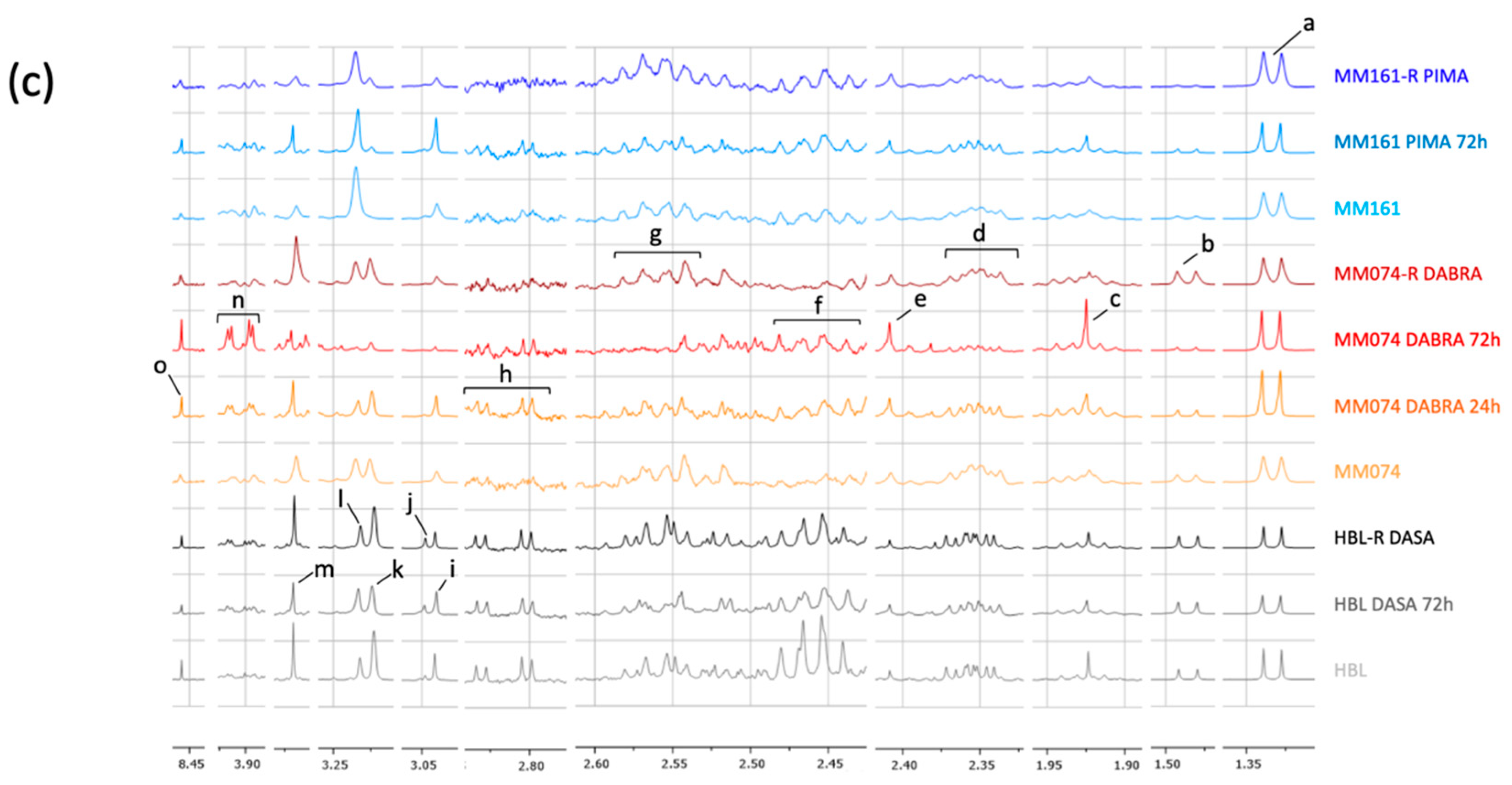
2.1.2. Additional Spectral Investigations
2.1.3. Independent Evaluation of the NRAS, cKIT, and BRAF Resistance Signatures
2.2. 1H-NMR Extracellular Signature Investigations
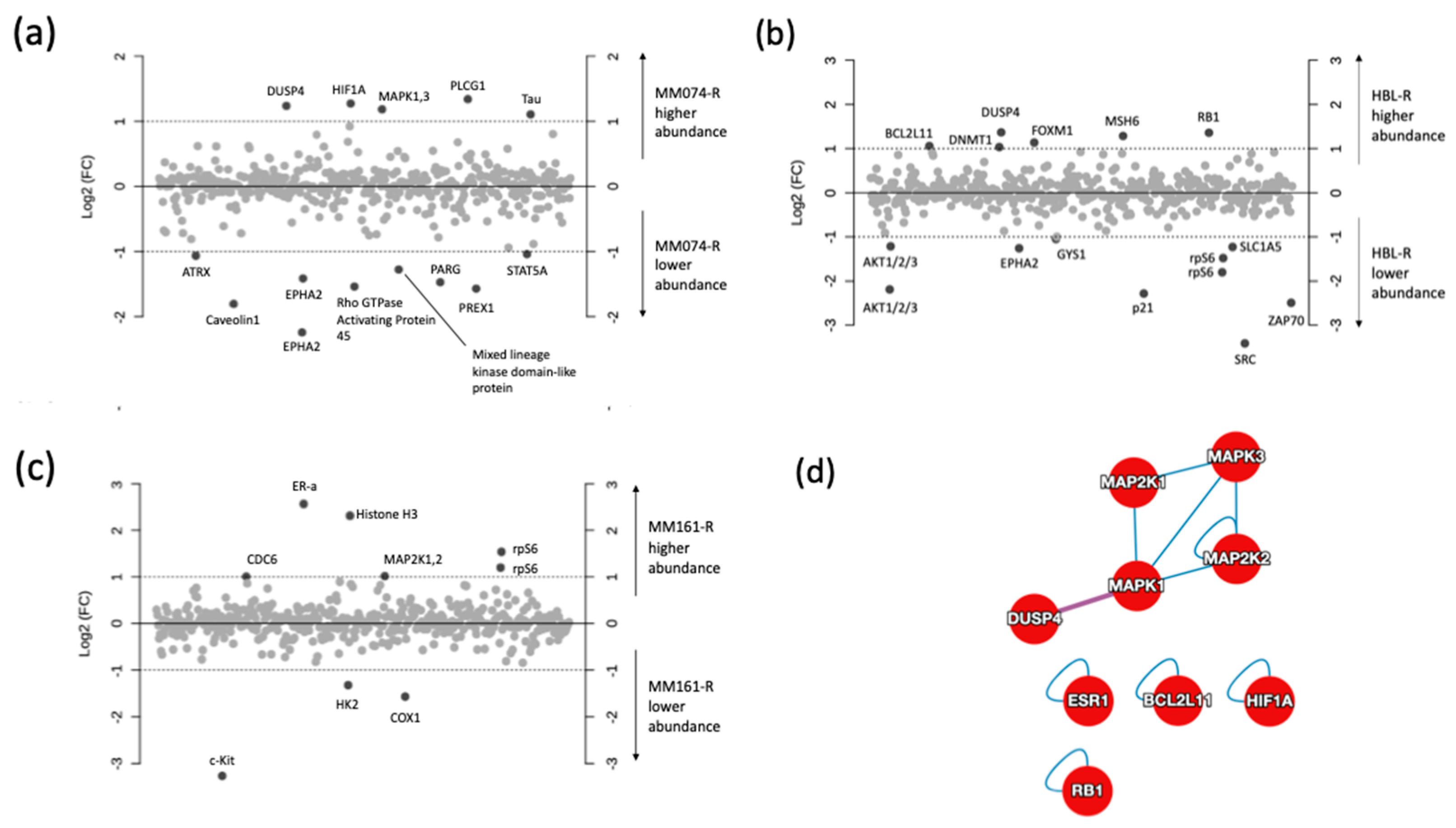
2.3. Protein Microarray and Interactome Analyses
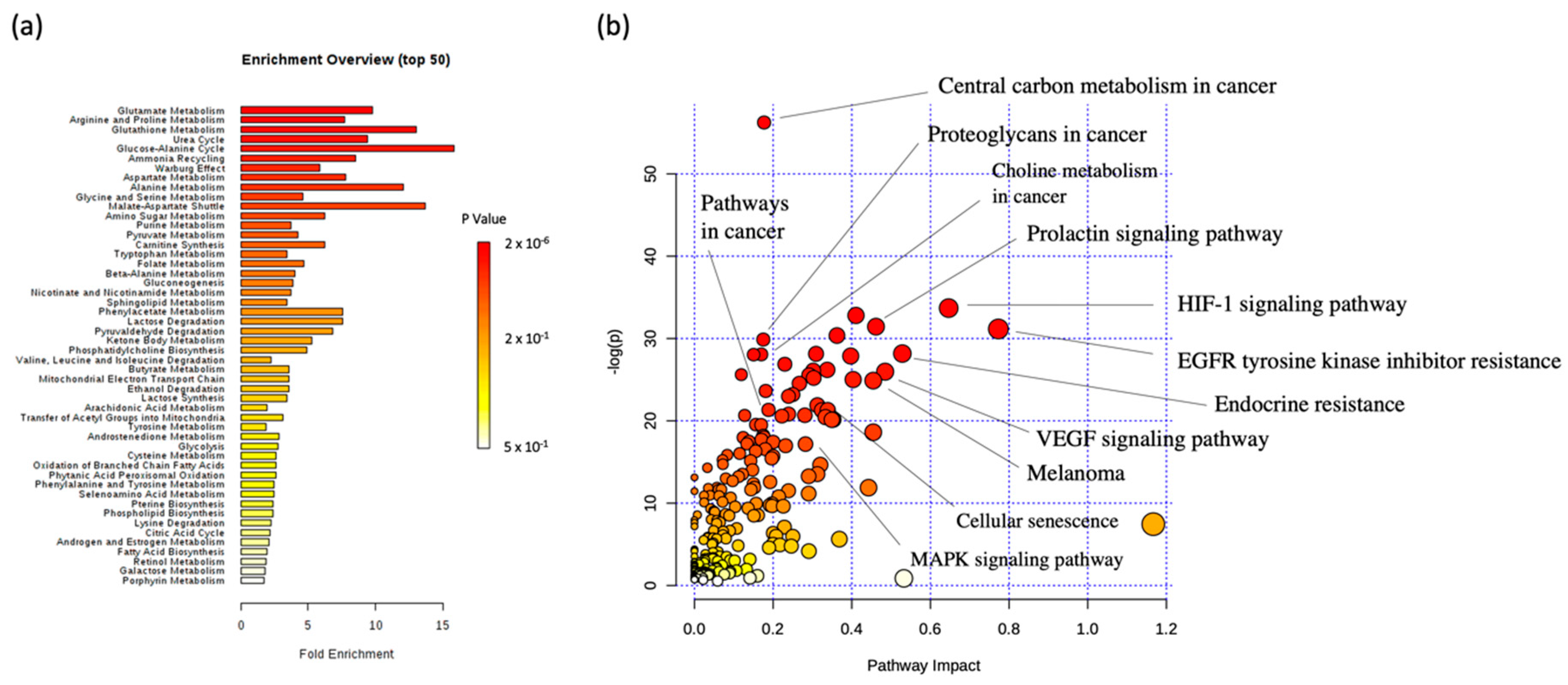
2.4. Integration of Metabonomics and Protein Microarray Data
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture
4.2. Effectors
4.3. 1H-NMR Spectroscopy
4.4. Protein Microarray
4.5. Protein–Protein Interactions (PPI)
4.6. MSEA and JOINT Pathway Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lo, J.A.; Fisher, D.E. The melanoma revolution: From UV carcinogenesis to a new era in therapeutics. Science 2014, 346, 945–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA. Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Millet, A.; Martin, A.R.; Ronco, C.; Rocchi, S.; Benhida, R. Metastatic Melanoma: Insights Into the Evolution of the Treatments and Future Challenges. Med. Res. Rev. 2017, 37, 98–148. [Google Scholar] [CrossRef] [PubMed]
- PDQ Adult Treatment Editorial Board Melanoma Treatment (PDQ®). Health Professional Version. In PDQ Cancer Information Summaries; National Cancer Institute (US): Bethesda, MD, USA, 2002. [Google Scholar]
- Davey, R.J.; van der Westhuizen, A.; Bowden, N.A. Metastatic melanoma treatment: Combining old and new therapies. Crit. Rev. Oncol. Hematol. 2016, 98, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Amaral, T.; Sinnberg, T.; Meier, F.; Krepler, C.; Levesque, M.; Niessner, H.; Garbe, C. The mitogen-activated protein kinase pathway in melanoma part I—Activation and primary resistance mechanisms to BRAF inhibition. Eur. J. Cancer Oxf. Engl. 1990 2017, 73, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westermark, U.K.; Wilhelm, M.; Frenzel, A.; Henriksson, M.A. The MYCN oncogene and differentiation in neuroblastoma. Semin. Cancer Biol. 2011, 21, 256–266. [Google Scholar] [CrossRef]
- Yuneva, M.; Zamboni, N.; Oefner, P.; Sachidanandam, R.; Lazebnik, Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J. Cell Biol. 2007, 178, 93–105. [Google Scholar] [CrossRef] [Green Version]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a Kras-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef]
- Gaglio, D.; Metallo, C.M.; Gameiro, P.A.; Hiller, K.; Danna, L.S.; Balestrieri, C.; Alberghina, L.; Stephanopoulos, G.; Chiaradonna, F. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol. Syst. Biol. 2011, 7, 523. [Google Scholar] [CrossRef]
- Shestov, A.A.; Mancuso, A.; Lee, S.-C.; Guo, L.; Nelson, D.S.; Roman, J.C.; Henry, P.-G.; Leeper, D.B.; Blair, I.A.; Glickson, J.D. Bonded Cumomer Analysis of Human Melanoma Metabolism Monitored by 13C NMR Spectroscopy of Perfused Tumor Cells. J. Biol. Chem. 2016, 291, 5157–5171. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, L.; Supuran, C.T.; Alfarouk, K.O. The Warburg Effect and the Hallmarks of Cancer. Anticancer Agents Med. Chem. 2017, 17, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Venneti, S.; Nagrath, D. Glutaminolysis: A Hallmark of Cancer Metabolism. Annu. Rev. Biomed. Eng. 2017, 19, 163–194. [Google Scholar] [CrossRef]
- Khamari, R.; Trinh, A.; Gabert, P.E.; Corazao-Rozas, P.; Riveros-Cruz, S.; Balayssac, S.; Malet-Martino, M.; Dekiouk, S.; Joncquel Chevalier Curt, M.; Maboudou, P.; et al. Glucose metabolism and NRF2 coordinate the antioxidant response in melanoma resistant to MAPK inhibitors. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Beger, R.D. A Review of Applications of Metabolomics in Cancer. Metabolites 2013, 3, 552–574. [Google Scholar] [CrossRef] [Green Version]
- Wishart, D.S.; Mandal, R.; Stanislaus, A.; Ramirez-Gaona, M. Cancer Metabolomics and the Human Metabolome Database. Metabolites 2016, 6, 10. [Google Scholar] [CrossRef] [Green Version]
- Puchades-Carrasco, L.; Pineda-Lucena, A. Metabolomics Applications in Precision Medicine: An Oncological Perspective. Curr. Top. Med. Chem. 2017, 17, 2740–2751. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Davies, J.E.; Tran, T.Q.; Reid, M.A.; Rosales, K.R.; Lowman, X.H.; Pan, M.; Moriceau, G.; Yang, Y.; Wu, J.; Lo, R.S.; et al. Vemurafenib resistance reprograms melanoma cells towards glutamine dependence. J. Transl. Med. 2015, 13, 210. [Google Scholar] [CrossRef] [Green Version]
- Corazao-Rozas, P.; Guerreschi, P.; André, F.; Gabert, P.-E.; Lancel, S.; Dekiouk, S.; Fontaine, D.; Tardivel, M.; Savina, A.; Quesnel, B.; et al. Mitochondrial oxidative phosphorylation controls cancer cell’s life and death decisions upon exposure to MAPK inhibitors. Oncotarget 2016, 7, 39473–39485. [Google Scholar] [CrossRef] [Green Version]
- Baenke, F.; Chaneton, B.; Smith, M.; Van Den Broek, N.; Hogan, K.; Tang, H.; Viros, A.; Martin, M.; Galbraith, L.; Girotti, M.R.; et al. Resistance to BRAF inhibitors induces glutamine dependency in melanoma cells. Mol. Oncol. 2016, 10, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Luebker, S.A.; Koepsell, S.A. Diverse Mechanisms of BRAF Inhibitor Resistance in Melanoma Identified in Clinical and Preclinical Studies. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, A.; Roberto, M.; Panebianco, M.; Botticelli, A.; Mazzuca, F.; Marchetti, P. Drug resistance of BRAF-mutant melanoma: Review of up-to-date mechanisms of action and promising targeted agents. Eur. J. Pharmacol. 2019, 862, 172621. [Google Scholar] [CrossRef] [PubMed]
- Najem, A.; Krayem, M.; Salès, F.; Hussein, N.; Badran, B.; Robert, C.; Awada, A.; Journe, F.; Ghanem, G.E. P53 and MITF/Bcl-2 identified as key pathways in the acquired resistance of NRAS-mutant melanoma to MEK inhibition. Eur. J. Cancer Oxf. Engl. 1990 2017, 83, 154–165. [Google Scholar] [CrossRef]
- Krayem, M.; Najem, A.; Journe, F.; Morandini, R.; Sales, F.; Awada, A.; Ghanem, G.E. Acquired resistance to BRAFi reverses senescence-like phenotype in mutant BRAF melanoma. Oncotarget 2018, 9, 31888–31903. [Google Scholar] [CrossRef]
- Najem, A.; Krayem, M.; Perdrix, A.; Kerger, J.; Awada, A.; Journe, F.; Ghanem, G. New Drug Combination Strategies in Melanoma: Current Status and Future Directions. Anticancer Res. 2017, 37, 5941–5953. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Ji, H.; Niu, X.; Yin, L.; Wang, Y.; Gu, Y.; Li, D.; Zhang, H.; Lu, M.; Zhang, F.; et al. Sodium-Dependent Glucose Transporter 1 (SGLT1) Stabled by HER2 Promotes Breast Cancer Cell Proliferation by Activation of the PI3K/Akt/mTOR Signaling Pathway in HER2+ Breast Cancer. Dis. Markers 2020, 2020, 6103542. [Google Scholar] [CrossRef]
- Locasale, J.W.; Cantley, L.C. Metabolic flux and the regulation of mammalian cell growth. Cell Metab. 2011, 14, 443–451. [Google Scholar] [CrossRef] [Green Version]
- Richtig, G.; Hoeller, C.; Kashofer, K.; Aigelsreiter, A.; Heinemann, A.; Kwong, L.N.; Pichler, M.; Richtig, E. Beyond the BRAFV600E hotspot: Biology and clinical implications of rare BRAF gene mutations in melanoma patients. Br. J. Dermatol. 2017, 177, 936–944. [Google Scholar] [CrossRef]
- Muñoz-Couselo, E.; Adelantado, E.Z.; Ortiz, C.; García, J.S.; Perez-Garcia, J. NRAS-mutant melanoma: Current challenges and future prospect. OncoTargets Ther. 2017, 10, 3941–3947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerner, B.A.; Stewart, L.A.; Horowitz, D.P.; Carvajal, R.D. Mucosal Melanoma: New Insights and Therapeutic Options for a Unique and Aggressive Disease. Oncol. Williston Park N 2017, 31, e23–e32. [Google Scholar]
- Hirschhaeuser, F.; Sattler, U.G.A.; Mueller-Klieser, W. Lactate: A Metabolic Key Player in Cancer. Cancer Res. 2011, 71, 6921–6925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marignac, V.M.; Smith, S.; Toban, N.; Bazile, M.; Aloyz, R. Resistance to Dasatinib in primary chronic lymphocytic leukemia lymphocytes involves AMPK-mediated energetic re-programming. Oncotarget 2013, 4, 2550–2566. [Google Scholar] [CrossRef] [Green Version]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 749. [Google Scholar] [CrossRef]
- Altinok, O.; Poggio, J.L.; Stein, D.E.; Bowne, W.B.; Shieh, A.C.; Snyder, N.W.; Orynbayeva, Z. Malate-aspartate shuttle promotes l-lactate oxidation in mitochondria. J. Cell. Physiol. 2020, 235, 2569–2581. [Google Scholar] [CrossRef]
- Allen, E.L.; Ulanet, D.B.; Pirman, D.; Mahoney, C.E.; Coco, J.; Si, Y.; Chen, Y.; Huang, L.; Ren, J.; Choe, S.; et al. Differential Aspartate Usage Identifies a Subset of Cancer Cells Particularly Dependent on OGDH. Cell Rep. 2016, 17, 876–890. [Google Scholar] [CrossRef] [Green Version]
- Luís, R.; Brito, C.; Pojo, M. Melanoma Metabolism: Cell Survival and Resistance to Therapy. Adv. Exp. Med. Biol. 2020, 1219, 203–223. [Google Scholar] [CrossRef]
- Haq, R.; Fisher, D.E. Biology and Clinical Relevance of the Micropthalmia Family of Transcription Factors in Human Cancer. J. Clin. Oncol. 2011, 29, 3474–3482. [Google Scholar] [CrossRef]
- Garcia-Bermudez, J.; Baudrier, L.; La, K.; Zhu, X.G.; Fidelin, J.; Sviderskiy, V.O.; Papagiannakopoulos, T.; Molina, H.; Snuderl, M.; Lewis, C.A.; et al. Aspartate is a limiting metabolite for cancer cell proliferation under hypoxia and in tumours. Nat. Cell Biol. 2018, 20, 775–781. [Google Scholar] [CrossRef]
- Muñoz-Pinedo, C.; El Mjiyad, N.; Ricci, J.-E. Cancer metabolism: Current perspectives and future directions. Cell Death Dis. 2012, 3, e248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Little, A.S.; Balmanno, K.; Sale, M.J.; Newman, S.; Dry, J.R.; Hampson, M.; Edwards, P.A.W.; Smith, P.D.; Cook, S.J. A correction to the research article titled: “Amplification of the driving oncogene, KRAS or BRAF, underpins acquired resistance to MEK1/2 inhibitors in colorectal cancer cells”. Sci. Signal. 2011, 4, er2. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.; Yacoub, N.; Mishra, R.; White, A.; Yuan, L.; Alanazi, S.; Garrett, J.T. Current Advances in the Treatment of BRAF-Mutant Melanoma. Cancers 2020, 12, 482. [Google Scholar] [CrossRef] [Green Version]
- Trunzer, K.; Pavlick, A.C.; Schuchter, L.; Gonzalez, R.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; Kim, K.B.; Weber, J.S.; et al. Pharmacodynamic effects and mechanisms of resistance to vemurafenib in patients with metastatic melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2013, 31, 1767–1774. [Google Scholar] [CrossRef]
- Saha, S.K.; Islam, S.M.R.; Abdullah-AL-Wadud, M.; Islam, S.; Ali, F.; Park, K.S. Multiomics Analysis Reveals that GLS and GLS2 Differentially Modulate the Clinical Outcomes of Cancer. J. Clin. Med. 2019, 8, 355. [Google Scholar] [CrossRef] [Green Version]
- Matés, J.M.; Segura, J.A.; Martín-Rufián, M.; Campos-Sandoval, J.A.; Alonso, F.J.; Márquez, J. Glutaminase isoenzymes as key regulators in metabolic and oxidative stress against cancer. Curr. Mol. Med. 2013, 13, 514–534. [Google Scholar] [CrossRef]
- Matés, J.M.; Campos-Sandoval, J.A.; de Santos-Jiménez, J.L.; Márquez, J. Dysregulation of glutaminase and glutamine synthetase in cancer. Cancer Lett. 2019, 467, 29–39. [Google Scholar] [CrossRef]
- Petővári, G.; Dankó, T.; Krencz, I.; Hujber, Z.; Rajnai, H.; Vetlényi, E.; Raffay, R.; Pápay, J.; Jeney, A.; Sebestyén, A. Inhibition of Metabolic Shift can Decrease Therapy Resistance in Human High-Grade Glioma Cells. Pathol. Oncol. Res. POR 2019. [Google Scholar] [CrossRef] [Green Version]
- Jacque, N.; Ronchetti, A.M.; Larrue, C.; Meunier, G.; Birsen, R.; Willems, L.; Saland, E.; Decroocq, J.; Maciel, T.T.; Lambert, M.; et al. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood 2015, 126, 1346–1356. [Google Scholar] [CrossRef] [Green Version]
- Rai, Y.; Yadav, P.; Kumari, N.; Kalra, N.; Bhatt, A.N. Hexokinase II inhibition by 3-bromopyruvate sensitizes myeloid leukemic cells K-562 to anti-leukemic drug, daunorubicin. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-J.; Li, C.-F.; Ruan, D.; He, J.; Montal, E.D.; Lorenz, S.; Girnun, G.D.; Chan, C.-H. Non-proteolytic ubiquitination of Hexokinase 2 by HectH9 controls tumor metabolism and cancer stem cell expansion. Nat. Commun. 2019, 10, 2625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schepkens, C.; Dallons, M.; Dehairs, J.; Talebi, A.; Jeandriens, J.; Drossart, L.-M.; Auquier, G.; Tagliatti, V.; Swinnen, J.V.; Colet, J.-M. A New Classification Method of Metastatic Cancers Using a 1H-NMR-Based Approach: A Study Case of Melanoma, Breast, and Prostate Cancer Cell Lines. Metabolites 2019, 9, 281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iorio, E.; Caramujo, M.J.; Cecchetti, S.; Spadaro, F.; Carpinelli, G.; Canese, R.; Podo, F. Key Players in Choline Metabolic Reprograming in Triple-Negative Breast Cancer. Front. Oncol. 2016, 6, 205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, X.-M.; Hu, L.; Gu, J.; Wang, R.-Y.; Li, L.; Tang, J.; Zhang, B.-H.; Yan, X.-Z.; Zhu, Y.-J.; Hu, C.-L.; et al. Choline Kinase α Mediates Interactions Between the Epidermal Growth Factor Receptor and Mechanistic Target of Rapamycin Complex 2 in Hepatocellular Carcinoma Cells to Promote Drug Resistance and Xenograft Tumor Progression. Gastroenterology 2017, 152, 1187–1202. [Google Scholar] [CrossRef] [PubMed]
- Glunde, K.; Bhujwalla, Z.M.; Ronen, S.M. Choline metabolism in malignant transformation. Nat. Rev. Cancer 2011, 11, 835–848. [Google Scholar] [CrossRef] [Green Version]
- Mu, X.; Zhao, T.; Xu, C.; Shi, W.; Geng, B.; Shen, J.; Zhang, C.; Pan, J.; Yang, J.; Hu, S.; et al. Oncometabolite succinate promotes angiogenesis by upregulating VEGF expression through GPR91-mediated STAT3 and ERK activation. Oncotarget 2017, 8, 13174–13185. [Google Scholar] [CrossRef]
- Yang, L.; Yu, D.; Mo, R.; Zhang, J.; Hua, H.; Hu, L.; Feng, Y.; Wang, S.; Zhang, W.-Y.; Yin, N.; et al. The Succinate Receptor GPR91 Is Involved in Pressure Overload-Induced Ventricular Hypertrophy. PLoS ONE 2016, 11, e0147597. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Alesi, G.N.; Kang, S. Glutaminolysis as a target for cancer therapy. Oncogene 2016, 35, 3619–3625. [Google Scholar] [CrossRef] [Green Version]
- Thompson, R.M.; Dytfeld, D.; Reyes, L.; Robinson, R.M.; Smith, B.; Manevich, Y.; Jakubowiak, A.; Komarnicki, M.; Przybylowicz-Chalecka, A.; Szczepaniak, T.; et al. Glutaminase inhibitor CB-839 synergizes with carfilzomib in resistant multiple myeloma cells. Oncotarget 2017, 8, 35863–35876. [Google Scholar] [CrossRef] [Green Version]
- Qie, S.; Yoshida, A.; Parnham, S.; Oleinik, N.; Beeson, G.C.; Beeson, C.C.; Ogretmen, B.; Bass, A.J.; Wong, K.-K.; Rustgi, A.K.; et al. Targeting glutamine-addiction and overcoming CDK4/6 inhibitor resistance in human esophageal squamous cell carcinoma. Nat. Commun. 2019, 10, 1296. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Zhou, B.; Liu, Z.; Xu, Y.; Lu, H.; Xia, M.; Guo, E.; Shan, W.; Chen, G.; Wang, C. Blockage of glutaminolysis enhances the sensitivity of ovarian cancer cells to PI3K/mTOR inhibition involvement of STAT3 signaling. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2016, 37, 11007–11015. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, P.; Guerreschi, P.; Mortier, L.; Kluza, J. Integration of Mitochondrial Targeting for Molecular Cancer Therapeutics. Int. J. Cell Biol. 2015, 2015, 283145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colapietro, A.; Gravina, G.L.; Petragnano, F.; Fasciani, I.; Scicchitano, B.M.; Beirinckx, F.; Pujuguet, P.; Saniere, L.; Van der Aar, E.; Musio, D.; et al. Antitumorigenic Effects of Inhibiting Ephrin Receptor Kinase Signaling by GLPG1790 against Colorectal Cancer Cell Lines In Vitro and In Vivo. J. Oncol. 2020, 2020, 9342732. [Google Scholar] [CrossRef] [PubMed]
- Ishigaki, H.; Minami, T.; Morimura, O.; Kitai, H.; Horio, D.; Koda, Y.; Fujimoto, E.; Negi, Y.; Nakajima, Y.; Niki, M.; et al. EphA2 inhibition suppresses proliferation of small-cell lung cancer cells through inducing cell cycle arrest. Biochem. Biophys. Res. Commun. 2019, 519, 846–853. [Google Scholar] [CrossRef] [PubMed]
- Human Metabolome Database. Available online: https://hmdb.ca/ (accessed on 24 March 2020).
- Functional Proteomics RPPA Core Facility. Available online: https://www.mdanderson.org/research/research-resources/core-facilities/functional-proteomics-rppa-core.html (accessed on 24 March 2020).
- HuRI. Home. Available online: http://www.interactome-atlas.org/ (accessed on 24 March 2020).
- Metabo Analyst. Available online: https://www.metaboanalyst.ca/ (accessed on 24 March 2020).




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soumoy, L.; Schepkens, C.; Krayem, M.; Najem, A.; Tagliatti, V.; Ghanem, G.E.; Saussez, S.; Colet, J.-M.; Journe, F. Metabolic Reprogramming in Metastatic Melanoma with Acquired Resistance to Targeted Therapies: Integrative Metabolomic and Proteomic Analysis. Cancers 2020, 12, 1323. https://doi.org/10.3390/cancers12051323
Soumoy L, Schepkens C, Krayem M, Najem A, Tagliatti V, Ghanem GE, Saussez S, Colet J-M, Journe F. Metabolic Reprogramming in Metastatic Melanoma with Acquired Resistance to Targeted Therapies: Integrative Metabolomic and Proteomic Analysis. Cancers. 2020; 12(5):1323. https://doi.org/10.3390/cancers12051323
Chicago/Turabian StyleSoumoy, Laura, Corentin Schepkens, Mohammad Krayem, Ahmad Najem, Vanessa Tagliatti, Ghanem E. Ghanem, Sven Saussez, Jean-Marie Colet, and Fabrice Journe. 2020. "Metabolic Reprogramming in Metastatic Melanoma with Acquired Resistance to Targeted Therapies: Integrative Metabolomic and Proteomic Analysis" Cancers 12, no. 5: 1323. https://doi.org/10.3390/cancers12051323
APA StyleSoumoy, L., Schepkens, C., Krayem, M., Najem, A., Tagliatti, V., Ghanem, G. E., Saussez, S., Colet, J.-M., & Journe, F. (2020). Metabolic Reprogramming in Metastatic Melanoma with Acquired Resistance to Targeted Therapies: Integrative Metabolomic and Proteomic Analysis. Cancers, 12(5), 1323. https://doi.org/10.3390/cancers12051323