Ibrutinib Resistance Mechanisms and Treatment Strategies for B-Cell Lymphomas

 , , and
, , and
Abstract
:1. Introduction
2. Complexities of Ibrutinib Resistance Development
2.1. Genetic Causes of Ibrutinib Resistance
2.2. Non-Genetic Causes of Ibrutinib Resistance
2.3. TME and Ibrutinib Resistance
2.4. Cancer Stem Cells (CSCs) and Ibrutinib Resistance
3. Strategies to Overcome Ibrutinib Resistance
3.1. New Generations of BTK Inhibitors
3.2. Strategies to Target Secondary BTKC481S or PLCG2mut
3.3. Third-Generation of BTK Inhibitors
3.4. BTK-PROTAC
4. Acquired Ibrutinib-Resistance and Therapeutic Approaches
4.1. PI3K/Akt/mTOR Pathway
4.2. Reduced BTK Expression after Chronic Ibrutinib Treatment
4.3. BCL2 Signaling
4.4. Bromodomain and Extraterminal Domain-Containing Proteins (BETs) Inhibitors
4.5. MALT1 Inhibition
4.6. IRAK4 Inhibition
4.7. SYK Inhibition
4.8. Chromatin Modifiers
4.9. Ibrutinib in Combination with CD20 Targeting Immunotherapy
5. Therapies Targeting Ibrutinib-Resistant CSCs
6. Chimeric Antigen Receptor (CAR) T-Cell Therapy with Ibrutinib for Combination Treatment
7. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Valla, K.; Flowers, C.R.; Koff, J.L. Targeting the B cell receptor pathway in non-Hodgkin lymphoma. Expert Opin. Investig Drugs 2018, 27, 513–522. [Google Scholar] [CrossRef]
- Pal Singh, S.; Dammeijer, F.; Hendriks, R.W. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol. Cancer Ther. 2018, 17, 57. [Google Scholar] [CrossRef] [PubMed]
- Duhren-von Minden, M.; Ubelhart, R.; Schneider, D.; Wossning, T.; Bach, M.P.; Buchner, M.; Hofmann, D.; Surova, E.; Follo, M.; Kohler, F.; et al. Chronic lymphocytic leukaemia is driven by antigen-independent cell-autonomous signalling. Nature 2012, 489, 309–312. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A.; Chiorazzi, N. B cell receptor signaling in chronic lymphocytic leukemia. Trends Immunol. 2013, 34, 592–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, R.M.; Phelan, J.D.; Wilson, W.H.; Staudt, L.M. Pathogenic B-cell receptor signaling in lymphoid malignancies: New insights to improve treatment. Immunol. Rev. 2019, 291, 190–213. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Lee, S.; Shah, T.; Yin, C.; Barth, M.; Miles, R.R.; Ayello, J.; Morris, E.; Harrison, L.; Van de Ven, C.; et al. Ibrutinib significantly inhibited Bruton’s tyrosine kinase (BTK) phosphorylation, in-vitro proliferation and enhanced overall survival in a preclinical Burkitt lymphoma (BL) model. Oncoimmunology 2019, 8, e1512455. [Google Scholar] [CrossRef]
- Ponader, S.; Chen, S.S.; Buggy, J.J.; Balakrishnan, K.; Gandhi, V.; Wierda, W.G.; Keating, M.J.; O’Brien, S.; Chiorazzi, N.; Burger, J.A. The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood 2012, 119, 1182–1189. [Google Scholar] [CrossRef]
- de Claro, R.A.; McGinn, K.M.; Verdun, N.; Lee, S.L.; Chiu, H.J.; Saber, H.; Brower, M.E.; Chang, C.J.; Pfuma, E.; Habtemariam, B.; et al. FDA Approval: Ibrutinib for Patients with Previously Treated Mantle Cell Lymphoma and Previously Treated Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2015, 21, 3586–3590. [Google Scholar] [CrossRef] [Green Version]
- Pleyer, C.; Wiestner, A.; Sun, C. Immunological changes with kinase inhibitor therapy for chronic lymphocytic leukemia. Leuk. Lymphoma 2018, 59, 2792–2800. [Google Scholar] [CrossRef]
- Chang, B.Y.; Francesco, M.; De Rooij, M.F.; Magadala, P.; Steggerda, S.M.; Huang, M.M.; Kuil, A.; Herman, S.E.; Chang, S.; Pals, S.T.; et al. Egress of CD19(+)CD5(+) cells into peripheral blood following treatment with the Bruton tyrosine kinase inhibitor ibrutinib in mantle cell lymphoma patients. Blood 2013, 122, 2412–2424. [Google Scholar] [CrossRef]
- Schmidl, C.; Vladimer, G.I.; Rendeiro, A.F.; Schnabl, S.; Krausgruber, T.; Taubert, C.; Krall, N.; Pemovska, T.; Araghi, M.; Snijder, B.; et al. Combined chemosensitivity and chromatin profiling prioritizes drug combinations in CLL. Nat. Chem. Biol. 2019, 15, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, J.; Hirayama, A.V.; Purushe, J.; Hay, K.A.; Lymp, J.; Li, D.; Yeung, C.; Sheih, A.; Pender, B.S.; Hawkins, R.M.; et al. Feasibility and efficacy of CD19-targeted CAR-T cells with concurrent ibrutinib for CLL after ibrutinib failure. Blood 2020. [Google Scholar] [CrossRef] [PubMed]
- Amin, N.A.; Balasubramanian, S.; Saiya-Cork, K.; Shedden, K.; Hu, N.; Malek, S.N. Cell-Intrinsic Determinants of Ibrutinib-Induced Apoptosis in Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2017, 23, 1049–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maddocks, K.J.; Ruppert, A.S.; Lozanski, G.; Heerema, N.A.; Zhao, W.; Abruzzo, L.; Lozanski, A.; Davis, M.; Gordon, A.; Smith, L.L.; et al. Etiology of Ibrutinib Therapy Discontinuation and Outcomes in Patients With Chronic Lymphocytic Leukemia. JAMA Oncol. 2015, 1, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Shah, B.; Zhao, X.; Silva, A.S.; Shain, K.H.; Tao, J. Resistance to Ibrutinib in B Cell Malignancies: One Size Does Not Fit All. Trends Cancer 2018, 4, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Furman, R.R.; Cheng, S.; Lu, P.; Setty, M.; Perez, A.R.; Guo, A.; Racchumi, J.; Xu, G.; Wu, H.; Ma, J.; et al. Ibrutinib resistance in chronic lymphocytic leukemia. N. Engl. J. Med. 2014, 370, 2352–2354. [Google Scholar] [CrossRef] [Green Version]
- Woyach, J.A.; Furman, R.R.; Liu, T.M.; Ozer, H.G.; Zapatka, M.; Ruppert, A.S.; Xue, L.; Li, D.H.; Steggerda, S.M.; Versele, M.; et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N. Engl. J. Med. 2014, 370, 2286–2294. [Google Scholar] [CrossRef] [Green Version]
- Lampson, B.L.; Brown, J.R. Are BTK and PLCG2 mutations necessary and sufficient for ibrutinib resistance in chronic lymphocytic leukemia? Expert Rev. Hematol. 2018, 11, 185–194. [Google Scholar] [CrossRef]
- Ahn, I.E.; Underbayev, C.; Albitar, A.; Herman, S.E.; Tian, X.; Maric, I.; Arthur, D.C.; Wake, L.; Pittaluga, S.; Yuan, C.M.; et al. Clonal evolution leading to ibrutinib resistance in chronic lymphocytic leukemia. Blood 2017, 129, 1469–1479. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Tsakmaklis, N.; Yang, G.; Chen, J.G.; Liu, X.; Demos, M.; Kofides, A.; Patterson, C.J.; Meid, K.; Gustine, J.; et al. Acquired mutations associated with ibrutinib resistance in Waldenstrom macroglobulinemia. Blood 2017, 129, 2519–2525. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, R.; Wright, G.W.; Huang, D.W.; Johnson, C.A.; Phelan, J.D.; Wang, J.Q.; Roulland, S.; Kasbekar, M.; Young, R.M.; Shaffer, A.L.; et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2018, 378, 1396–1407. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Phelan, J.D.; Wright, G.W.; Haupl, B.; Huang, D.W.; Shaffer, A.L., 3rd; Young, R.M.; Wang, Z.; Zhao, H.; Yu, X.; et al. Regulation of B cell receptor-dependent NF-kappaB signaling by the tumor suppressor KLHL14. Proc. Natl. Acad. Sci. USA 2020, 117, 6092–6102. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Chan, Y.C.; Tam, C.S.; Hunter, T.; Vassiliadis, D.; Teh, C.E.; Thijssen, R.; Yeh, P.; Wong, S.Q.; Ftouni, S.; et al. Dynamic molecular monitoring reveals that SWI-SNF mutations mediate resistance to ibrutinib plus venetoclax in mantle cell lymphoma. Nat. Med. 2019, 25, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; de Miranda, N.F.; Chen, L.; Wasik, A.M.; Mansouri, L.; Jurczak, W.; Galazka, K.; Dlugosz-Danecka, M.; Machaczka, M.; Zhang, H.; et al. Genetic heterogeneity in primary and relapsed mantle cell lymphomas: Impact of recurrent CARD11 mutations. Oncotarget 2016, 7, 38180–38190. [Google Scholar] [CrossRef]
- Zhang, L.; Yao, Y.; Zhang, S.; Liu, Y.; Guo, H.; Ahmed, M.; Bell, T.; Zhang, H.; Han, G.; Lorence, E.; et al. Metabolic reprogramming toward oxidative phosphorylation identifies a therapeutic target for mantle cell lymphoma. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Bartlett, N.L.; Costello, B.A.; LaPlant, B.R.; Ansell, S.M.; Kuruvilla, J.G.; Reeder, C.B.; Thye, L.S.; Anderson, D.M.; Krysiak, K.; Ramirez, C.; et al. Single-agent ibrutinib in relapsed or refractory follicular lymphoma: A phase 2 consortium trial. Blood 2018, 131, 182–190. [Google Scholar] [CrossRef] [Green Version]
- Jimenez, C.; Chan, G.G.; Xu, L.; Tsakmaklis, N.; Kofides, A.; Demos, M.G.; Chen, J.; Liu, X.; Munshi, M.; Yang, G.; et al. Genomic evolution of ibrutinib-resistant clones in Waldenstrom macroglobulinaemia. Br. J. Haematol. 2020. [Google Scholar] [CrossRef]
- Treon, S.P.; Tripsas, C.K.; Meid, K.; Warren, D.; Varma, G.; Green, R.; Argyropoulos, K.V.; Yang, G.; Cao, Y.; Xu, L.; et al. Ibrutinib in previously treated Waldenstrom’s macroglobulinemia. N. Engl. J. Med. 2015, 372, 1430–1440. [Google Scholar] [CrossRef] [Green Version]
- Kanagal-Shamanna, R.; Jain, P.; Patel, K.P.; Routbort, M.; Bueso-Ramos, C.; Alhalouli, T.; Khoury, J.D.; Luthra, R.; Ferrajoli, A.; Keating, M.; et al. Targeted multigene deep sequencing of Bruton tyrosine kinase inhibitor-resistant chronic lymphocytic leukemia with disease progression and Richter transformation. Cancer 2019, 125, 559–574. [Google Scholar] [CrossRef] [Green Version]
- Fiorcari, S.; Maffei, R.; Audrito, V.; Martinelli, S.; Ten Hacken, E.; Zucchini, P.; Grisendi, G.; Potenza, L.; Luppi, M.; Burger, J.A.; et al. Ibrutinib modifies the function of monocyte/macrophage population in chronic lymphocytic leukemia. Oncotarget 2016, 7, 65968–65981. [Google Scholar] [CrossRef]
- Sharma, S.; Galanina, N.; Guo, A.; Lee, J.; Kadri, S.; Van Slambrouck, C.; Long, B.; Wang, W.; Ming, M.; Furtado, L.V.; et al. Identification of a structurally novel BTK mutation that drives ibrutinib resistance in CLL. Oncotarget 2016, 7, 68833–68841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadri, S.; Lee, J.; Fitzpatrick, C.; Galanina, N.; Sukhanova, M.; Venkataraman, G.; Sharma, S.; Long, B.; Petras, K.; Theissen, M.; et al. Clonal evolution underlying leukemia progression and Richter transformation in patients with ibrutinib-relapsed CLL. Blood Adv. 2017, 1, 715–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burger, J.A.; Landau, D.A.; Taylor-Weiner, A.; Bozic, I.; Zhang, H.; Sarosiek, K.; Wang, L.; Stewart, C.; Fan, J.; Hoellenriegel, J.; et al. Clonal evolution in patients with chronic lymphocytic leukaemia developing resistance to BTK inhibition. Nat. Commun. 2016, 7, 11589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, W.H.; Young, R.M.; Schmitz, R.; Yang, Y.; Pittaluga, S.; Wright, G.; Lih, C.J.; Williams, P.M.; Shaffer, A.L.; Gerecitano, J.; et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat. Med. 2015, 21, 922–926. [Google Scholar] [CrossRef]
- Phelan, J.D.; Young, R.M.; Webster, D.E.; Roulland, S.; Wright, G.W.; Kasbekar, M.; Shaffer, A.L., 3rd; Ceribelli, M.; Wang, J.Q.; Schmitz, R.; et al. A multiprotein supercomplex controlling oncogenic signalling in lymphoma. Nature 2018, 560, 387–391. [Google Scholar] [CrossRef]
- Chiron, D.; Di Liberto, M.; Martin, P.; Huang, X.; Sharman, J.; Blecua, P.; Mathew, S.; Vijay, P.; Eng, K.; Ali, S.; et al. Cell-cycle reprogramming for PI3K inhibition overrides a relapse-specific C481S BTK mutation revealed by longitudinal functional genomics in mantle cell lymphoma. Cancer Discov. 2014, 4, 1022–1035. [Google Scholar] [CrossRef] [Green Version]
- Rahal, R.; Frick, M.; Romero, R.; Korn, J.M.; Kridel, R.; Chan, F.C.; Meissner, B.; Bhang, H.E.; Ruddy, D.; Kauffmann, A.; et al. Pharmacological and genomic profiling identifies NF-kappaB-targeted treatment strategies for mantle cell lymphoma. Nat. Med. 2014, 20, 87–92. [Google Scholar] [CrossRef]
- Vidal-Crespo, A.; Rodriguez, V.; Matas-Cespedes, A.; Lee, E.; Rivas-Delgado, A.; Gine, E.; Navarro, A.; Bea, S.; Campo, E.; Lopez-Guillermo, A.; et al. The Bruton tyrosine kinase inhibitor CC-292 shows activity in mantle cell lymphoma and synergizes with lenalidomide and NIK inhibitors depending on nuclear factor-kappaB mutational status. Haematologica 2017, 102, e447–e451. [Google Scholar] [CrossRef] [Green Version]
- Diop, F.; Moia, R.; Favini, C.; Spaccarotella, E.; De Paoli, L.; Bruscaggin, A.; Spina, V.; Terzi-di-Bergamo, L.; Arruga, F.; Tarantelli, C.; et al. Biological and clinical implications of BIRC3 mutations in chronic lymphocytic leukemia. Haematologica 2020, 105, 448–456. [Google Scholar] [CrossRef] [Green Version]
- Lenz, G.; Balasubramanian, S.; Goldberg, J.; Rizo, A.; Schaffer, M.; Phelps, C.; Rule, S.; Dreyling, M.H. Sequence variants in patients with primary and acquired resistance to ibrutinib in the phase 3 MCL3001 (RAY) trial. J. Clin. Oncol. 2016, 34 (Suppl. 15), 7570. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, W.S.; Ryu, K.; Kim, S.J.; Park, C. CD79B limits response of diffuse large B cell lymphoma to ibrutinib. Leuk. Lymphoma 2016, 57, 1413–1422. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Zhang, L.L.; Wu, W.; Guo, H.; Li, Y.; Sukhanova, M.; Venkataraman, G.; Huang, S.; Zhang, H.; Alikhan, M.; et al. Activation of MYC, a bona fide client of HSP90, contributes to intrinsic ibrutinib resistance in mantle cell lymphoma. Blood Adv. 2018, 2, 2039–2051. [Google Scholar] [CrossRef] [PubMed]
- Rauert-Wunderlich, H.; Rudelius, M.; Berberich, I.; Rosenwald, A. CD40L mediated alternative NFkappaB-signaling induces resistance to BCR-inhibitors in patients with mantle cell lymphoma. Cell Death Dis. 2018, 9, 86. [Google Scholar] [CrossRef] [Green Version]
- Hershkovitz-Rokah, O.; Pulver, D.; Lenz, G.; Shpilberg, O. Ibrutinib resistance in mantle cell lymphoma: Clinical, molecular and treatment aspects. Br. J. Haematol. 2018, 181, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.S.A.L. CD40L-CD40 signaling on B-cell lymphoma response to BTK inhibitors. In Proceedings of the AACR 107th Annual Meeting, New Orleans, LA, USA, 16–20 April 2016. [Google Scholar] [CrossRef]
- Ming, M.; Wu, W.; Xie, B.; Sukhanova, M.; Wang, W.; Kadri, S.; Sharma, S.; Lee, J.; Shacham, S.; Landesman, Y.; et al. XPO1 Inhibitor Selinexor Overcomes Intrinsic Ibrutinib Resistance in Mantle Cell Lymphoma via Nuclear Retention of IkappaB. Mol. Cancer Ther. 2018, 17, 2564–2574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herishanu, Y.; Perez-Galan, P.; Liu, D.; Biancotto, A.; Pittaluga, S.; Vire, B.; Gibellini, F.; Njuguna, N.; Lee, E.; Stennett, L.; et al. The lymph node microenvironment promotes B-cell receptor signaling, NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood 2011, 117, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Nwabo Kamdje, A.H.; Kamga, P.T.; Simo, R.T.; Vecchio, L.; Seke Etet, P.F.; Muller, J.M.; Bassi, G.; Lukong, E.; Goel, R.K.; Amvene, J.M.; et al. Mesenchymal stromal cells’ role in tumor microenvironment: Involvement of signaling pathways. Cancer Biol. Med. 2017, 14, 129–141. [Google Scholar] [CrossRef]
- Ten Hacken, E.; Burger, J.A. Microenvironment interactions and B-cell receptor signaling in Chronic Lymphocytic Leukemia: Implications for disease pathogenesis and treatment. Biochim. Biophys. Acta 2016, 1863, 401–413. [Google Scholar] [CrossRef]
- Wu, L.; Zhang, Y.Z.; Xia, B.; Li, X.W.; Yuan, T.; Tian, C.; Zhao, H.F.; Yu, Y.; Sotomayor, E. Ibrutinib inhibits mesenchymal Stem cells-mediated drug resistance in diffuse large B-cell lymphoma. Zhonghua Xue Ye Xue Za Zhi 2017, 38, 1036–1042. [Google Scholar] [CrossRef]
- Carabia, J.; Carpio, C.; Abrisqueta, P.; Jimenez, I.; Purroy, N.; Calpe, E.; Palacio, C.; Bosch, F.; Crespo, M. Microenvironment regulates the expression of miR-21 and tumor suppressor genes PTEN, PIAS3 and PDCD4 through ZAP-70 in chronic lymphocytic leukemia. Sci. Rep. 2017, 7, 12262. [Google Scholar] [CrossRef]
- Trimarco, V.; Ave, E.; Facco, M.; Chiodin, G.; Frezzato, F.; Martini, V.; Gattazzo, C.; Lessi, F.; Giorgi, C.A.; Visentin, A.; et al. Cross-talk between chronic lymphocytic leukemia (CLL) tumor B cells and mesenchymal stromal cells (MSCs): Implications for neoplastic cell survival. Oncotarget 2015, 6, 42130–42149. [Google Scholar] [CrossRef] [PubMed]
- Byrd, J.C.; Furman, R.R.; Coutre, S.E.; Flinn, I.W.; Burger, J.A.; Blum, K.A.; Grant, B.; Sharman, J.P.; Coleman, M.; Wierda, W.G.; et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 2013, 369, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Best, O.G.; Mulligan, S.P.; Christopherson, R.I. Ibrutinib and idelalisib block immunophenotypic changes associated with the adhesion and activation of CLL cells in the tumor microenvironment. Leuk. Lymphoma 2018, 59, 1927–1937. [Google Scholar] [CrossRef] [PubMed]
- de Rooij, M.F.; Kuil, A.; Geest, C.R.; Eldering, E.; Chang, B.Y.; Buggy, J.J.; Pals, S.T.; Spaargaren, M. The clinically active BTK inhibitor PCI-32765 targets B-cell receptor- and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia. Blood 2012, 119, 2590–2594. [Google Scholar] [CrossRef] [PubMed]
- Ping, L.; Ding, N.; Shi, Y.; Feng, L.; Li, J.; Liu, Y.; Lin, Y.; Shi, C.; Wang, X.; Pan, Z.; et al. The Bruton’s tyrosine kinase inhibitor ibrutinib exerts immunomodulatory effects through regulation of tumor-infiltrating macrophages. Oncotarget 2017, 8, 39218–39229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niemann, C.U.; Herman, S.E.; Maric, I.; Gomez-Rodriguez, J.; Biancotto, A.; Chang, B.Y.; Martyr, S.; Stetler-Stevenson, M.; Yuan, C.M.; Calvo, K.R.; et al. Disruption of in vivo Chronic Lymphocytic Leukemia Tumor-Microenvironment Interactions by Ibrutinib--Findings from an Investigator-Initiated Phase II Study. Clin. Cancer Res. 2016, 22, 1572–1582. [Google Scholar] [CrossRef] [Green Version]
- Kondo, K.; Shaim, H.; Thompson, P.A.; Burger, J.A.; Keating, M.; Estrov, Z.; Harris, D.; Kim, E.; Ferrajoli, A.; Daher, M.; et al. Ibrutinib modulates the immunosuppressive CLL microenvironment through STAT3-mediated suppression of regulatory B-cell function and inhibition of the PD-1/PD-L1 pathway. Leukemia 2018, 32, 960–970. [Google Scholar] [CrossRef]
- Long, M.; Beckwith, K.; Do, P.; Mundy, B.L.; Gordon, A.; Lehman, A.M.; Maddocks, K.J.; Cheney, C.; Jones, J.A.; Flynn, J.M.; et al. Ibrutinib treatment improves T cell number and function in CLL patients. J. Clin. Invest. 2017, 127, 3052–3064. [Google Scholar] [CrossRef] [PubMed]
- Rendeiro, A.F.; Krausgruber, T.; Fortelny, N.; Zhao, F.; Penz, T.; Farlik, M.; Schuster, L.C.; Nemc, A.; Tasnady, S.; Reti, M.; et al. Chromatin mapping and single-cell immune profiling define the temporal dynamics of ibrutinib response in CLL. Nat. Commun. 2020, 11, 577. [Google Scholar] [CrossRef] [Green Version]
- Xia, B.; Tian, C.; Guo, S.; Zhang, L.; Zhao, D.; Qu, F.; Zhao, W.; Wang, Y.; Wu, X.; Da, W. c-Myc plays part in drug resistance mediated by bone marrow stromal cells in acute myeloid leukemia. Leuk. Res. 2015, 39, 92–99. [Google Scholar] [CrossRef]
- Moyo, T.K.; Wilson, C.S.; Moore, D.J.; Eischen, C.M. Myc enhances B-cell receptor signaling in precancerous B cells and confers resistance to Btk inhibition. Oncogene 2017, 36, 4653–4661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kokabee, L.; Wang, X.; Sevinsky, C.J.; Wang, W.L.; Cheu, L.; Chittur, S.V.; Karimipoor, M.; Tenniswood, M.; Conklin, D.S. Bruton’s tyrosine kinase is a potential therapeutic target in prostate cancer. Cancer Biol. 2015, 16, 1604–1615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerra, D.A.P.; Paiva, A.E.; Sena, I.F.G.; Azevedo, P.O.; Silva, W.N.; Mintz, A.; Birbrair, A. Targeting glioblastoma-derived pericytes improves chemotherapeutic outcome. Angiogenesis 2018, 21, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Elkholy, K.H.; Wani, N.A.; Li, D.; Hu, P.; Barajas, J.M.; Yu, L.; Zhang, X.; Jacob, S.T.; Khan, W.N.; et al. Ibrutinib Potentiates Antihepatocarcinogenic Efficacy of Sorafenib by Targeting EGFR in Tumor Cells and BTK in Immune Cells in the Stroma. Mol. Cancer Ther. 2020, 19, 384–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Lwin, T.; Silva, A.; Shah, B.; Tao, J.; Fang, B.; Zhang, L.; Fu, K.; Bi, C.; Li, J.; et al. Unification of de novo and acquired ibrutinib resistance in mantle cell lymphoma. Nat. Commun. 2017, 8, 14920. [Google Scholar] [CrossRef]
- Guan, J.; Huang, D.; Yakimchuk, K.; Okret, S. p110alpha Inhibition Overcomes Stromal Cell-Mediated Ibrutinib Resistance in Mantle Cell Lymphoma. Mol. Cancer Ther. 2018, 17, 1090–1100. [Google Scholar] [CrossRef] [Green Version]
- Rudelius, M.; Rosenfeldt, M.T.; Leich, E.; Rauert-Wunderlich, H.; Solimando, A.G.; Beilhack, A.; Ott, G.; Rosenwald, A. Inhibition of focal adhesion kinase overcomes resistance of mantle cell lymphoma to ibrutinib in the bone marrow microenvironment. Haematologica 2018, 103, 116–125. [Google Scholar] [CrossRef] [Green Version]
- Guo, A.; Lu, P.; Coffey, G.; Conley, P.; Pandey, A.; Wang, Y.L. Dual SYK/JAK inhibition overcomes ibrutinib resistance in chronic lymphocytic leukemia: Cerdulatinib, but not ibrutinib, induces apoptosis of tumor cells protected by the microenvironment. Oncotarget 2017, 8, 12953–12967. [Google Scholar] [CrossRef] [Green Version]
- Boissard, F.; Fournie, J.J.; Quillet-Mary, A.; Ysebaert, L.; Poupot, M. Nurse-like cells mediate ibrutinib resistance in chronic lymphocytic leukemia patients. Blood Cancer J. 2015, 5, e355. [Google Scholar] [CrossRef] [Green Version]
- Medina, D.J.; Abass-Shereef, J.; Walton, K.; Goodell, L.; Aviv, H.; Strair, R.K.; Budak-Alpdogan, T. Cobblestone-area forming cells derived from patients with mantle cell lymphoma are enriched for CD133+ tumor-initiating cells. PLoS ONE 2014, 9, e91042. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Ayala, P.; Wang, M.; Fayad, L.; Katz, R.L.; Romaguera, J.; Caraway, N.; Neelapu, S.S.; Kwak, L.W.; Simmons, P.J.; et al. Prospective isolation of clonogenic mantle cell lymphoma-initiating cells. Stem. Cell Res. 2010, 5, 212–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, H.J.; Chen, Z.; McCarty, N. Stem-like tumor cells confer drug resistant properties to mantle cell lymphoma. Leuk. Lymphoma 2011, 52, 1066–1079. [Google Scholar] [CrossRef]
- Mathur, R.; Sehgal, L.; Braun, F.K.; Berkova, Z.; Romaguerra, J.; Wang, M.; Rodriguez, M.A.; Fayad, L.; Neelapu, S.S.; Samaniego, F. Targeting Wnt pathway in mantle cell lymphoma-initiating cells. J. Hematol. Oncol. 2015, 8, 63. [Google Scholar] [CrossRef] [Green Version]
- Wei, X.; Yang, S.; Pu, X.; He, S.; Yang, Z.; Sheng, X.; Meng, X.; Chen, X.; Jin, L.; Chen, W.; et al. Tumor-associated macrophages increase the proportion of cancer Stem. cells in lymphoma by secreting pleiotrophin. Am. J. Transl. Res. 2019, 11, 6393–6402. [Google Scholar] [PubMed]
- Lee, M.R.; Ju, H.J.; Kim, B.S.; Ko, Y.H.; Kim, W.S.; Kim, S.J. Isolation of side population cells in B-cell non-Hodgkin’s lymphomas. Acta Haematol. 2013, 129, 10–17. [Google Scholar] [CrossRef]
- Lee, C.G.; Das, B.; Lin, T.L.; Grimes, C.; Zhang, X.; Lavezzi, T.; Huang, L.; Cole, J.; Yau, L.; Li, L. A rare fraction of drug-resistant follicular lymphoma cancer Stem cells interacts with follicular dendritic cells to maintain tumourigenic potential. Br. J. Haematol. 2012, 158, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Li, Y.; Zhang, K.; Zhang, X.; Huang, Y.; Xu, M.; Li, S.; Guan, X.; Yang, T.; Liu, Z.; et al. Cancer Stem Cells of Diffuse Large B Cell Lymphoma Are Not Enriched in the CD45(+)CD19(-) cells but in the ALDH(high) Cells. J. Cancer 2020, 11, 142–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zucha, M.A.; Wu, A.T.; Lee, W.H.; Wang, L.S.; Lin, W.W.; Yuan, C.C.; Yeh, C.T. Bruton’s tyrosine kinase (Btk) inhibitor ibrutinib suppresses stem-like traits in ovarian cancer. Oncotarget 2015, 6, 13255–13268. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Su, Y.K.; Lin, C.M.; Chao, T.Y.; Huang, S.P.; Huynh, T.T.; Jan, H.J.; Whang-Peng, J.; Chiou, J.F.; Wu, A.T.; et al. Preclinical investigation of ibrutinib, a Bruton’s kinase tyrosine (Btk) inhibitor, in suppressing glioma tumorigenesis and Stem cell phenotypes. Oncotarget 2016, 7, 69961–69975. [Google Scholar] [CrossRef] [Green Version]
- Tai, P.A.; Liu, Y.L.; Wen, Y.T.; Lin, C.M.; Huynh, T.T.; Hsiao, M.; Wu, A.T.H.; Wei, L. The Development and Applications of a Dual Optical Imaging System for Studying Glioma Stem Cells. Mol. Imaging 2019, 18, 1536012119870899. [Google Scholar] [CrossRef]
- Zhou, W.; Chen, C.; Shi, Y.; Wu, Q.; Gimple, R.C.; Fang, X.; Huang, Z.; Zhai, K.; Ke, S.Q.; Ping, Y.F.; et al. Targeting Glioma Stem Cell-Derived Pericytes Disrupts the Blood-Tumor Barrier and Improves Chemotherapeutic Efficacy. Cell Stem. Cell 2017, 21, 591–603.e594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Shi, J.; Gu, Z.; Salama, M.E.; Das, S.; Wendlandt, E.; Xu, H.; Huang, J.; Tao, Y.; Hao, M.; et al. Bruton tyrosine kinase is a therapeutic target in stem-like cells from multiple myeloma. Cancer Res. 2015, 75, 594–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barf, T.; Covey, T.; Izumi, R.; van de Kar, B.; Gulrajani, M.; van Lith, B.; van Hoek, M.; de Zwart, E.; Mittag, D.; Demont, D.; et al. Acalabrutinib (ACP-196): A Covalent Bruton Tyrosine Kinase Inhibitor with a Differentiated Selectivity and In Vivo Potency Profile. J. Pharm. Exp. 2017, 363, 240–252. [Google Scholar] [CrossRef] [PubMed]
- Chi, J.; Park, J.; Saif, M.W. Ibrutinib-Induced Vasculitis in a Patient with Metastatic Colon Cancer Treated in Combination with Cetuximab. Case Rep. Oncol. Med. 2020, 2020, 6154213. [Google Scholar] [CrossRef]
- Brown, J.R.; Moslehi, J.; O’Brien, S.; Ghia, P.; Hillmen, P.; Cymbalista, F.; Shanafelt, T.D.; Fraser, G.; Rule, S.; Kipps, T.J.; et al. Characterization of atrial fibrillation adverse events reported in ibrutinib randomized controlled registration trials. Haematologica 2017, 102, 1796–1805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guha, A.; Derbala, M.H.; Zhao, Q.; Wiczer, T.E.; Woyach, J.A.; Byrd, J.C.; Awan, F.T.; Addison, D. Ventricular Arrhythmias Following Ibrutinib Initiation for Lymphoid Malignancies. J. Am. Coll Cardiol. 2018, 72, 697–698. [Google Scholar] [CrossRef]
- Salem, J.E.; Manouchehri, A.; Bretagne, M.; Lebrun-Vignes, B.; Groarke, J.D.; Johnson, D.B.; Yang, T.; Reddy, N.M.; Funck-Brentano, C.; Brown, J.R.; et al. Cardiovascular Toxicities Associated With Ibrutinib. J. Am. Coll Cardiol. 2019, 74, 1667–1678. [Google Scholar] [CrossRef]
- McMullen, J.R.; Boey, E.J.; Ooi, J.Y.; Seymour, J.F.; Keating, M.J.; Tam, C.S. Ibrutinib increases the risk of atrial fibrillation, potentially through inhibition of cardiac PI3K-Akt signaling. Blood 2014, 124, 3829–3830. [Google Scholar] [CrossRef] [Green Version]
- Roeker, L.E.; Sarraf Yazdy, M.; Rhodes, J.; Goodfriend, J.; Narkhede, M.; Carver, J.; Mato, A. Hypertension in Patients Treated With Ibrutinib for Chronic Lymphocytic Leukemia. JAMA Netw. Open 2019, 2, e1916326. [Google Scholar] [CrossRef]
- Shatzel, J.J.; Olson, S.R.; Tao, D.L.; McCarty, O.J.T.; Danilov, A.V.; DeLoughery, T.G. Ibrutinib-associated bleeding: Pathogenesis, management and risk reduction strategies. J. Thromb. Haemost 2017, 15, 835–847. [Google Scholar] [CrossRef]
- Nicolson, P.L.R.; Hughes, C.E.; Watson, S.; Nock, S.H.; Hardy, A.T.; Watson, C.N.; Montague, S.J.; Clifford, H.; Huissoon, A.P.; Malcor, J.D.; et al. Inhibition of Btk by Btk-specific concentrations of ibrutinib and acalabrutinib delays but does not block platelet aggregation mediated by glycoprotein VI. Haematologica 2018, 103, 2097–2108. [Google Scholar] [CrossRef] [PubMed]
- Stephens, D.M.; Byrd, J.C. How I manage ibrutinib intolerance and complications in patients with chronic lymphocytic leukemia. Blood 2019, 133, 1298–1307. [Google Scholar] [CrossRef] [PubMed]
- Ball, S.; Das, A.; Vutthikraivit, W.; Edwards, P.J.; Hardwicke, F.; Short, N.J.; Borthakur, G.; Maiti, A. Risk of Infection Associated With Ibrutinib in Patients With B-Cell Malignancies: A Systematic Review and Meta-analysis of Randomized Controlled Trials. Clin. Lymphoma Myeloma Leuk. 2020, 20, 87–97.e85. [Google Scholar] [CrossRef] [PubMed]
- Borge, M.; Belen Almejun, M.; Podaza, E.; Colado, A.; Fernandez Grecco, H.; Cabrejo, M.; Bezares, R.F.; Giordano, M.; Gamberale, R. Ibrutinib impairs the phagocytosis of rituximab-coated leukemic cells from chronic lymphocytic leukemia patients by human macrophages. Haematologica 2015, 100, e140–e142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohrt, H.E.; Sagiv-Barfi, I.; Rafiq, S.; Herman, S.E.; Butchar, J.P.; Cheney, C.; Zhang, X.; Buggy, J.J.; Muthusamy, N.; Levy, R.; et al. Ibrutinib (PCI-32765) Antagonizes Rituximab-Dependent NK-Cell Mediated Cytotoxicity. Blood 2014, 123, 1957–1960. [Google Scholar] [CrossRef] [Green Version]
- Owen, C.; Berinstein, N.L.; Christofides, A.; Sehn, L.H. Review of Bruton tyrosine kinase inhibitors for the treatment of relapsed or refractory mantle cell lymphoma. Curr Oncol. 2019, 26, e233–e240. [Google Scholar] [CrossRef] [Green Version]
- Awan, F.T.; Schuh, A.; Brown, J.R.; Furman, R.R.; Pagel, J.M.; Hillmen, P.; Stephens, D.M.; Woyach, J.; Bibikova, E.; Charuworn, P.; et al. Acalabrutinib monotherapy in patients with chronic lymphocytic leukemia who are intolerant to ibrutinib. Blood Adv. 2019, 3, 1553–1562. [Google Scholar] [CrossRef] [Green Version]
- Ghia, P.P.A.; Wach, M.; Lysak, D.; Kozak, T.; Simkovic, M. Acalabrutinib vs rituximab plus idelalisib (IdR) or bendamustine (BR) by investigator choice in relapsed/refractory (RR) chronic lymphocytic leukemia: Phase 3 ASCEND study. Hematol Oncol. 2019, 37, 86–87. [Google Scholar] [CrossRef] [Green Version]
- Sharman, J.P.M.B.V.; Fogliatto, L.M.; Herishanu, Y.; Munir, T.; Walewska, R. phase 3 study of acalabrutinib combined with obinutuzumab (O) or alone vs O plus chlorambucil (Clb) in patients (Pts) with treatment-naive chronic lymphocytic leukemia. Blood 2019, 134 (Suppl. 1), 31. [Google Scholar] [CrossRef]
- Tam, C.S.; Trotman, J.; Opat, S.; Burger, J.A.; Cull, G.; Gottlieb, D.; Harrup, R.; Johnston, P.B.; Marlton, P.; Munoz, J.; et al. Phase 1 study of the selective BTK inhibitor zanubrutinib in B-cell malignancies and safety and efficacy evaluation in CLL. Blood 2019, 134, 851–859. [Google Scholar] [CrossRef] [Green Version]
- Tam, C.S.; Robak, T.; Ghia, P.; Kahl, B.S.; Walker, P.; Janowski, W.; Simpson, D.; Shadman, M.; Ganly, P.S.; Laurenti, L.; et al. Efficacy and safety of zanubrutinib in patients with treatment-naive chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL) with Del(17p): Initial results from Arm C of the Sequoia (BGB-3111-304) Trial. Blood 2019, 134 (Suppl. 1), 499. [Google Scholar] [CrossRef]
- Zou, Y.X.; Zhu, H.Y.; Li, X.T.; Xia, Y.; Miao, K.R.; Zhao, S.S.; Wu, Y.J.; Wang, L.; Xu, W.; Li, J.Y. The impacts of zanubrutinib on immune cells in patients with chronic lymphocytic leukemia/small lymphocytic lymphoma. Hematol. Oncol. 2019, 37, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Aalipour, A.; Advani, R.H. Bruton’s tyrosine kinase inhibitors and their clinical potential in the treatment of B-cell malignancies: Focus on ibrutinib. Adv. Hematol. 2014, 5, 121–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woyach, J.A.; Ruppert, A.S.; Guinn, D.; Lehman, A.; Blachly, J.S.; Lozanski, A.; Heerema, N.A.; Zhao, W.; Coleman, J.; Jones, D.; et al. BTK(C481S)-Mediated Resistance to Ibrutinib in Chronic Lymphocytic Leukemia. J. Clin. Oncol. 2017, 35, 1437–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woyach, J.A.; Smucker, K.; Smith, L.L.; Lozanski, A.; Zhong, Y.; Ruppert, A.S.; Lucas, D.; Williams, K.; Zhao, W.; Rassenti, L.; et al. Prolonged lymphocytosis during ibrutinib therapy is associated with distinct molecular characteristics and does not indicate a suboptimal response to therapy. Blood 2014, 123, 1810–1817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Liu, C.; Tsui, S.T.; Liu, D. Second-generation inhibitors of Bruton tyrosine kinase. J. Hematol. Oncol. 2016, 9, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Lee, G.S.; Brady, J.; Datta, S.; Katan, M.; Sheikh, A.; Martins, M.S.; Bunney, T.D.; Santich, B.H.; Moir, S.; et al. A hypermorphic missense mutation in PLCG2, encoding phospholipase Cgamma2, causes a dominantly inherited autoinflammatory disease with immunodeficiency. Am. J. Hum. Genet. 2012, 91, 713–720. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.M.; Woyach, J.A.; Zhong, Y.; Lozanski, A.; Lozanski, G.; Dong, S.; Strattan, E.; Lehman, A.; Zhang, X.; Jones, J.A.; et al. Hypermorphic mutation of phospholipase C, gamma2 acquired in ibrutinib-resistant CLL confers BTK independency upon B-cell receptor activation. Blood 2015, 126, 61–68. [Google Scholar] [CrossRef] [Green Version]
- Walliser, C.; Hermkes, E.; Schade, A.; Wiese, S.; Deinzer, J.; Zapatka, M.; Desire, L.; Mertens, D.; Stilgenbauer, S.; Gierschik, P. The Phospholipase Cgamma2 Mutants R665W and L845F Identified in Ibrutinib-resistant Chronic Lymphocytic Leukemia Patients Are Hypersensitive to the Rho GTPase Rac2 Protein. J. Biol. Chem. 2016, 291, 22136–22148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, D.; Woyach, J.A.; Zhao, W.; Caruthers, S.; Tu, H.; Coleman, J.; Byrd, J.C.; Johnson, A.J.; Lozanski, G. PLCG2 C2 domain mutations co-occur with BTK and PLCG2 resistance mutations in chronic lymphocytic leukemia undergoing ibrutinib treatment. Leukemia 2017, 31, 1645–1647. [Google Scholar] [CrossRef] [PubMed]
- Reiff, S.D.; Mantel, R.; Smith, L.L.; Greene, J.T.; Muhowski, E.M.; Fabian, C.A.; Goettl, V.M.; Tran, M.; Harrington, B.K.; Rogers, K.A.; et al. The BTK Inhibitor ARQ 531 Targets Ibrutinib-Resistant CLL and Richter Transformation. Cancer Discov. 2018, 8, 1300–1315. [Google Scholar] [CrossRef] [PubMed]
- Woyach, J.F.I.; Stephens, D.M. A phase 1 dose escalation study of ARQ 531 in selected patients with relapsed or refractory hematologic malignancies. Blood 2018, 132 (Suppl. 1), 3136. [Google Scholar] [CrossRef]
- Elgamal, O.A.; Mehmood, A.; Jeon, J.Y.; Carmichael, B.; Lehman, A.; Orwick, S.J.; Truxall, J.; Goettl, V.M.; Wasmuth, R.; Tran, M.; et al. Preclinical efficacy for a novel tyrosine kinase inhibitor, ArQule 531 against acute myeloid leukemia. J. Hematol. Oncol. 2020, 13, 8. [Google Scholar] [CrossRef] [PubMed]
- Brandhuber, B.G.; Smith, S. LOXO-305, a next generation reversible BTK inhibitor, for overcoming acquired resistance to irreversible BTK inhibitors. Clin. Lymphoma Myeloma Leuk. 2018, 18 (Suppl. 1), S216. [Google Scholar] [CrossRef]
- Binnerts, M.E.O.K.; Hopkins, B.T. SNS-062 is a potent noncovalent BTK inhibitor with comparable activity against wild type BTK and BTK with an acquired resistance mutation. Mol. Cancer Ther. 2015, 14 (Suppl. 2), C186. [Google Scholar] [CrossRef]
- Neuman, L.L.W.R.; Arnold, D. First-in-human phase 1a study of the safety, pharmacokinetics, and pharmacodynamics of the noncovalent Bruton tyrosine kinase (BTK) inhibitor SNS-062 in healthy subjects. Blood 2016, 128, 2032. [Google Scholar] [CrossRef]
- Gui, F.; Jiang, J.; He, Z.; Li, L.; Li, Y.; Deng, Z.; Lu, Y.; Wu, X.; Chen, G.; Su, J.; et al. A non-covalent inhibitor XMU-MP-3 overrides ibrutinib-resistant Btk(C481S) mutation in B-cell malignancies. Br. J. Pharm. 2019, 176, 4491–4509. [Google Scholar] [CrossRef]
- Byrd, J.C.; Smith, S.; Wagner-Johnston, N.; Sharman, J.; Chen, A.I.; Advani, R.; Augustson, B.; Marlton, P.; Renee Commerford, S.; Okrah, K.; et al. First-in-human phase 1 study of the BTK inhibitor GDC-0853 in relapsed or refractory B-cell NHL and CLL. Oncotarget 2018, 9, 13023–13035. [Google Scholar] [CrossRef] [Green Version]
- Johnson, A.R.; Kohli, P.B.; Katewa, A.; Gogol, E.; Belmont, L.D.; Choy, R.; Penuel, E.; Burton, L.; Eigenbrot, C.; Yu, C.; et al. Battling Btk Mutants With Noncovalent Inhibitors That Overcome Cys481 and Thr474 Mutations. Acs Chem. Biol. 2016, 11, 2897–2907. [Google Scholar] [CrossRef]
- Smith, P.F.; Krishnarajah, J.; Nunn, P.A.; Hill, R.J.; Karr, D.; Tam, D.; Masjedizadeh, M.; Funk, J.O.; Gourlay, S.G. A phase I trial of PRN1008, a novel reversible covalent inhibitor of Bruton’s tyrosine kinase, in healthy volunteers. Br. J. Clin. Pharm. 2017, 83, 2367–2376. [Google Scholar] [CrossRef]
- Lee, S.K.; Xing, J.; Catlett, I.M.; Adamczyk, R.; Griffies, A.; Liu, A.; Murthy, B.; Nowak, M. Safety, pharmacokinetics, and pharmacodynamics of BMS-986142, a novel reversible BTK inhibitor, in healthy participants. Eur. J. Clin. Pharm. 2017, 73, 689–698. [Google Scholar] [CrossRef] [Green Version]
- Jain, N.; Hartert, K.; Tadros, S.; Fiskus, W.; Havranek, O.; Ma, M.C.J.; Bouska, A.; Heavican, T.; Kumar, D.; Deng, Q.; et al. Targetable genetic alterations of TCF4 (E2-2) drive immunoglobulin expression in diffuse large B cell lymphoma. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Fiskus, W.; Qian, Y.; Rajapakshe, K.; Raina, K.; Coleman, K.G.; Crew, A.P.; Shen, A.; Saenz, D.T.; Mill, C.P.; et al. BET protein proteolysis targeting chimera (PROTAC) exerts potent lethal activity against mantle cell lymphoma cells. Leukemia 2018, 32, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Qian, Y.; Altieri, M.; Dong, H.; Wang, J.; Raina, K.; Hines, J.; Winkler, J.D.; Crew, A.P.; Coleman, K.; et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol. 2015, 22, 755–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raina, K.; Lu, J.; Qian, Y.; Altieri, M.; Gordon, D.; Rossi, A.M.; Wang, J.; Chen, X.; Dong, H.; Siu, K.; et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 7124–7129. [Google Scholar] [CrossRef] [Green Version]
- Lai, A.C.; Toure, M.; Hellerschmied, D.; Salami, J.; Jaime-Figueroa, S.; Ko, E.; Hines, J.; Crews, C.M. Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew Chem. Int. Ed. Engl. 2016, 55, 807–810. [Google Scholar] [CrossRef] [Green Version]
- Bondeson, D.P.; Smith, B.E.; Burslem, G.M.; Buhimschi, A.D.; Hines, J.; Jaime-Figueroa, S.; Wang, J.; Hamman, B.D.; Ishchenko, A.; Crews, C.M. Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell Chem. Biol. 2018, 25, 78–87.e75. [Google Scholar] [CrossRef] [Green Version]
- Burslem, G.M.; Smith, B.E.; Lai, A.C.; Jaime-Figueroa, S.; McQuaid, D.C.; Bondeson, D.P.; Toure, M.; Dong, H.; Qian, Y.; Wang, J.; et al. The Advantages of Targeted Protein Degradation Over Inhibition: An RTK Case Study. Cell Chem. Biol. 2018, 25, 67–77.e63. [Google Scholar] [CrossRef]
- Yang, K.; Song, Y.; Xie, H.; Wu, H.; Wu, Y.T.; Leisten, E.D.; Tang, W. Development of the first small molecule histone deacetylase 6 (HDAC6) degraders. Bioorganic Med. Chem. Lett. 2018, 28, 2493–2497. [Google Scholar] [CrossRef]
- Huang, H.T.; Dobrovolsky, D.; Paulk, J.; Yang, G.; Weisberg, E.L.; Doctor, Z.M.; Buckley, D.L.; Cho, J.H.; Ko, E.; Jang, J.; et al. A Chemoproteomic Approach to Query the Degradable Kinome Using a Multi-kinase Degrader. Cell Chem. Biol. 2018, 25, 88–99.e86. [Google Scholar] [CrossRef] [Green Version]
- Buhimschi, A.D.; Armstrong, H.A.; Toure, M.; Jaime-Figueroa, S.; Chen, T.L.; Lehman, A.M.; Woyach, J.A.; Johnson, A.J.; Byrd, J.C.; Crews, C.M. Targeting the C481S Ibrutinib-Resistance Mutation in Bruton’s Tyrosine Kinase Using PROTAC-Mediated Degradation. Biochemistry 2018, 57, 3564–3575. [Google Scholar] [CrossRef]
- Sun, Y.; Zhao, X.; Ding, N.; Gao, H.; Wu, Y.; Yang, Y.; Zhao, M.; Hwang, J.; Song, Y.; Liu, W.; et al. PROTAC-induced BTK degradation as a novel therapy for mutated BTK C481S induced ibrutinib-resistant B-cell malignancies. Cell Res. 2018, 28, 779–781. [Google Scholar] [CrossRef] [PubMed]
- Jaime-Figueroa, S.; Buhimschi, A.D.; Toure, M.; Hines, J.; Crews, C.M. Design, synthesis and biological evaluation of Proteolysis Targeting Chimeras (PROTACs) as a BTK degraders with improved pharmacokinetic properties. Bioorganic Med. Chem. Lett. 2020, 30, 126877. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wang, J.; Yao, X.; Zheng, W.; Mao, Y.; Lan, T.; Wang, L.; Sun, Y.; Zhang, X.; Zhao, Q.; et al. A chemical approach for global protein knockdown from mice to non-human primates. Cell Discov. 2019, 5, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Ding, N.; Song, Y.; Yang, Z.; Liu, W.; Zhu, J.; Rao, Y. Degradation of Bruton’s tyrosine kinase mutants by PROTACs for potential treatment of ibrutinib-resistant non-Hodgkin lymphomas. Leukemia 2019, 33, 2105–2110. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Nomie, K.; Zhang, H.; Bell, T.; Pham, L.; Kadri, S.; Segal, J.; Li, S.; Zhou, S.; Santos, D.; et al. B-Cell Lymphoma Patient-Derived Xenograft Models Enable Drug Discovery and Are a Platform for Personalized Therapy. Clin. Cancer Res. 2017, 23, 4212–4223. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, I.; Li, Y.; Sharma, A.; Zhu, H.; Bodo, J.; Xu, W.; Hsi, E.D.; Hill, B.T.; Almasan, A. Resistance to BTK inhibition by ibrutinib can be overcome by preventing FOXO3a nuclear export and PI3K/AKT activation in B-cell lymphoid malignancies. Cell Death Dis. 2019, 10, 924. [Google Scholar] [CrossRef]
- Jain, N.H.O.; Singh, R.K.; Khashab, T.; Shirazi, F. View ORCID ProfileLalit Sehgal, Felipe Samaniego. Overcoming ibrutinib resistance by targeting phosphatidylinositol-3-kinase signaling in diffuse large B-cell lymphoma. Biorxiv 2019. [Google Scholar] [CrossRef]
- Davis, R.E.; Ngo, V.N.; Lenz, G.; Tolar, P.; Young, R.M.; Romesser, P.B.; Kohlhammer, H.; Lamy, L.; Zhao, H.; Yang, Y.; et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 2010, 463, 88–92. [Google Scholar] [CrossRef] [Green Version]
- Lenz, G.; Davis, R.E.; Ngo, V.N.; Lam, L.; George, T.C.; Wright, G.W.; Dave, S.S.; Zhao, H.; Xu, W.; Rosenwald, A.; et al. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science 2008, 319, 1676–1679. [Google Scholar] [CrossRef]
- Ngo, V.N.; Young, R.M.; Schmitz, R.; Jhavar, S.; Xiao, W.; Lim, K.H.; Kohlhammer, H.; Xu, W.; Yang, Y.; Zhao, H.; et al. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011, 470, 115–119. [Google Scholar] [CrossRef] [Green Version]
- Paul, J.; Soujon, M.; Wengner, A.M.; Zitzmann-Kolbe, S.; Sturz, A.; Haike, K.; Keng Magdalene, K.H.; Tan, S.H.; Lange, M.; Tan, S.Y.; et al. Simultaneous Inhibition of PI3Kdelta and PI3Kalpha Induces ABC-DLBCL Regression by Blocking BCR-Dependent and -Independent Activation of NF-kappaB and AKT. Cancer Cell 2017, 31, 64–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, C.; Zhang, R.; Zhang, S.; Lee, W.; Huang, S.; Liu, Y.; Leeming, A.; McIntosh, J.M.; Jain, P.; Nomie, K.; et al. Targeting PI3K and PLK1 to Overcome Ibrutinib-Venetoclax Resistance in Mantle Cell Lymphoma. Blood 2019, 134 (Suppl. 1), 4062. [Google Scholar] [CrossRef]
- Cervantes-Gomez, F.; Kumar Patel, V.; Bose, P.; Keating, M.J.; Gandhi, V. Decrease in total protein level of Bruton’s tyrosine kinase during ibrutinib therapy in chronic lymphocytic leukemia lymphocytes. Leukemia 2016, 30, 1803–1804. [Google Scholar] [CrossRef] [PubMed]
- Holmes, K.B.; Sadreev, I.I.; Rawstron, A.C.; Munir, T.; Westhead, D.R.; Hillmen, P.; Lefevre, P.F. Ibrutinib induces chromatin reorganisation of chronic lymphocytic leukaemia cells. Oncogenesis 2019, 8, 32. [Google Scholar] [CrossRef] [PubMed]
- Tomita, A. Genetic and Epigenetic Modulation of CD20 Expression in B-Cell Malignancies: Molecular Mechanisms and Significance to Rituximab Resistance. J. Clin. Exp. Hematop 2016, 56, 89–99. [Google Scholar] [CrossRef] [Green Version]
- Vandenberg, C.J.; Cory, S. ABT-199, a new Bcl-2-specific BH3 mimetic, has in vivo efficacy against aggressive Myc-driven mouse lymphomas without provoking thrombocytopenia. Blood 2013, 121, 2285–2288. [Google Scholar] [CrossRef]
- Kuo, H.P.; Ezell, S.A.; Schweighofer, K.J.; Cheung, L.W.K.; Hsieh, S.; Apatira, M.; Sirisawad, M.; Eckert, K.; Hsu, S.J.; Chen, C.T.; et al. Combination of Ibrutinib and ABT-199 in Diffuse Large B-Cell Lymphoma and Follicular Lymphoma. Mol. Cancer Ther. 2017, 16, 1246–1256. [Google Scholar] [CrossRef] [Green Version]
- Paulus, A.; Akhtar, S.; Yousaf, H.; Manna, A.; Paulus, S.M.; Bashir, Y.; Caulfield, T.R.; Kuranz-Blake, M.; Chitta, K.; Wang, X.; et al. Waldenstrom macroglobulinemia cells devoid of BTK(C481S) or CXCR4(WHIM-like) mutations acquire resistance to ibrutinib through upregulation of Bcl-2 and AKT resulting in vulnerability towards venetoclax or MK2206 treatment. Blood Cancer J. 2017, 7, e565. [Google Scholar] [CrossRef]
- Deng, J.; Isik, E.; Fernandes, S.M.; Brown, J.R.; Letai, A.; Davids, M.S. Bruton’s tyrosine kinase inhibition increases BCL-2 dependence and enhances sensitivity to venetoclax in chronic lymphocytic leukemia. Leukemia 2017, 31, 2075–2084. [Google Scholar] [CrossRef]
- Axelrod, M.; Ou, Z.; Brett, L.K.; Zhang, L.; Lopez, E.R.; Tamayo, A.T.; Gordon, V.; Ford, R.J.; Williams, M.E.; Pham, L.V.; et al. Combinatorial drug screening identifies synergistic co-targeting of Bruton’s tyrosine kinase and the proteasome in mantle cell lymphoma. Leukemia 2014, 28, 407–410. [Google Scholar] [CrossRef]
- Jain, N.; Keating, M.; Thompson, P.; Ferrajoli, A.; Burger, J.; Borthakur, G.; Takahashi, K.; Estrov, Z.; Fowler, N.; Kadia, T.; et al. Ibrutinib and Venetoclax for First-Line Treatment of CLL. N. Engl. J. Med. 2019, 380, 2095–2103. [Google Scholar] [CrossRef] [PubMed]
- Eide, C.A.; Kurtz, S.E.; Kaempf, A.; Long, N.; Agarwal, A.; Tognon, C.E.; Mori, M.; Druker, B.J.; Chang, B.H.; Danilov, A.V.; et al. Simultaneous kinase inhibition with ibrutinib and BCL2 inhibition with venetoclax offers a therapeutic strategy for acute myeloid leukemia. Leukemia 2020. [Google Scholar] [CrossRef] [PubMed]
- Jayappa, K.D.; Portell, C.A.; Gordon, V.L.; Capaldo, B.J.; Bekiranov, S.; Axelrod, M.J.; Brett, L.K.; Wulfkuhle, J.D.; Gallagher, R.I.; Petricoin, E.F.; et al. Microenvironmental agonists generate de novo phenotypic resistance to combined ibrutinib plus venetoclax in CLL and MCL. Blood Adv. 2017, 1, 933–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceribelli, M.; Kelly, P.N.; Shaffer, A.L.; Wright, G.W.; Xiao, W.; Yang, Y.; Mathews Griner, L.A.; Guha, R.; Shinn, P.; Keller, J.M.; et al. Blockade of oncogenic IkappaB kinase activity in diffuse large B-cell lymphoma by bromodomain and extraterminal domain protein inhibitors. Proc. Natl. Acad. Sci. USA 2014, 111, 11365–11370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, B.; Shah, B.; Fiskus, W.; Qi, J.; Rajapakshe, K.; Coarfa, C.; Li, L.; Devaraj, S.G.; Sharma, S.; Zhang, L.; et al. Synergistic activity of BET protein antagonist-based combinations in mantle cell lymphoma cells sensitive or resistant to ibrutinib. Blood 2015, 126, 1565–1574. [Google Scholar] [CrossRef]
- Kim, E.; Ten Hacken, E.; Sivina, M.; Clarke, A.; Thompson, P.A.; Jain, N.; Ferrajoli, A.; Estrov, Z.; Keating, M.J.; Wierda, W.G.; et al. The BET inhibitor GS-5829 targets chronic lymphocytic leukemia cells and their supportive microenvironment. Leukemia 2019. [Google Scholar] [CrossRef]
- Fontan, L.; Qiao, Q.; Hatcher, J.M.; Casalena, G.; Us, I.; Teater, M.; Durant, M.; Du, G.; Xia, M.; Bilchuk, N.; et al. Specific covalent inhibition of MALT1 paracaspase suppresses B cell lymphoma growth. J. Clin. Invest. 2018, 128, 4397–4412. [Google Scholar] [CrossRef] [Green Version]
- Saba, N.S.; Wong, D.H.; Tanios, G.; Iyer, J.R.; Lobelle-Rich, P.; Dadashian, E.L.; Liu, D.; Fontan, L.; Flemington, E.K.; Nichols, C.M.; et al. MALT1 Inhibition Is Efficacious in Both Naive and Ibrutinib-Resistant Chronic Lymphocytic Leukemia. Cancer Res. 2017, 77, 7038–7048. [Google Scholar] [CrossRef] [Green Version]
- Dai, B.; Grau, M.; Juilland, M.; Klener, P.; Horing, E.; Molinsky, J.; Schimmack, G.; Aukema, S.M.; Hoster, E.; Vogt, N.; et al. B-cell receptor-driven MALT1 activity regulates MYC signaling in mantle cell lymphoma. Blood 2017, 129, 333–346. [Google Scholar] [CrossRef] [Green Version]
- Kelly, P.N.; Romero, D.L.; Yang, Y.; Shaffer, A.L., 3rd; Chaudhary, D.; Robinson, S.; Miao, W.; Rui, L.; Westlin, W.F.; Kapeller, R.; et al. Selective interleukin-1 receptor-associated kinase 4 inhibitors for the treatment of autoimmune disorders and lymphoid malignancy. J. Exp. Med. 2015, 212, 2189–2201. [Google Scholar] [CrossRef]
- Delvecchio, V.S.; Sana, I.; Mantione, M.E.; Vilia, M.G.; Ranghetti, P.; Rovida, A.; Angelillo, P.; Scarfo, L.; Ghia, P.; Muzio, M. Interleukin-1 receptor-associated kinase 4 inhibitor interrupts toll-like receptor signalling and sensitizes chronic lymphocytic leukaemia cells to apoptosis. Br. J. Haematol. 2020. [Google Scholar] [CrossRef]
- Schaffer, M.; Chaturvedi, S.; Davis, C.; Aquino, R.; Stepanchick, E.; Versele, M.; Liu, Y.; Yang, J.; Lu, R.; Balasubramanian, S. Identification of potential ibrutinib combinations in hematological malignancies using a combination high-throughput screen. Leuk. Lymphoma 2018, 59, 931–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degorce, S.L.; Anjum, R.; Bloecher, A.; Carbajo, R.J.; Dillman, K.S.; Drew, L.; Halsall, C.T.; Lenz, E.M.; Lindsay, N.A.; Mayo, M.F.; et al. Discovery of a Series of 5-Azaquinazolines as Orally Efficacious IRAK4 Inhibitors Targeting MyD88(L265P) Mutant Diffuse Large B Cell Lymphoma. J. Med. Chem. 2019, 62, 9918–9930. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Bai, G.; Ning, Y.; Cai, S.; Zhang, T.; Song, P.; Zhou, J.; Duan, W.; Ding, J.; Xie, H.; et al. Design and synthesis of Imidazo[1,2-b]pyridazine IRAK4 inhibitors for the treatment of mutant MYD88 L265P diffuse large B-cell lymphoma. Eur. J. Med. Chem. 2020, 190, 112092. [Google Scholar] [CrossRef] [PubMed]
- Buchner, M.; Fuchs, S.; Prinz, G.; Pfeifer, D.; Bartholome, K.; Burger, M.; Chevalier, N.; Vallat, L.; Timmer, J.; Gribben, J.G.; et al. Spleen tyrosine kinase is overexpressed and represents a potential therapeutic target in chronic lymphocytic leukemia. Cancer Res. 2009, 69, 5424–5432. [Google Scholar] [CrossRef] [Green Version]
- Munshi, M.; Liu, X.; Chen, J.G.; Xu, L.; Tsakmaklis, N.; Demos, M.G.; Kofides, A.; Guerrera, M.L.; Jimenez, C.; Chan, G.G.; et al. SYK is activated by mutated MYD88 and drives pro-survival signaling in MYD88 driven B-cell lymphomas. Blood Cancer J. 2020, 10, 12. [Google Scholar] [CrossRef] [Green Version]
- Purroy, N.; Carabia, J.; Abrisqueta, P.; Egia, L.; Aguilo, M.; Carpio, C.; Palacio, C.; Crespo, M.; Bosch, F. Inhibition of BCR signaling using the Syk inhibitor TAK-659 prevents stroma-mediated signaling in chronic lymphocytic leukemia cells. Oncotarget 2017, 8, 742–756. [Google Scholar] [CrossRef] [Green Version]
- Sermer, D.; Pasqualucci, L.; Wendel, H.G.; Melnick, A.; Younes, A. Emerging epigenetic-modulating therapies in lymphoma. Nat. Rev. Clin. Oncol. 2019, 16, 494–507. [Google Scholar] [CrossRef]
- Garding, A.; Bhattacharya, N.; Claus, R.; Ruppel, M.; Tschuch, C.; Filarsky, K.; Idler, I.; Zucknick, M.; Caudron-Herger, M.; Oakes, C.; et al. Epigenetic upregulation of lncRNAs at 13q14.3 in leukemia is linked to the In Cis downregulation of a gene cluster that targets NF-kB. Plos Genet. 2013, 9, e1003373. [Google Scholar] [CrossRef]
- Wolf, C.; Garding, A.; Filarsky, K.; Bahlo, J.; Robrecht, S.; Becker, N.; Zucknick, M.; Rouhi, A.; Weigel, A.; Claus, R.; et al. NFATC1 activation by DNA hypomethylation in chronic lymphocytic leukemia correlates with clinical staging and can be inhibited by ibrutinib. Int. J. Cancer 2018, 142, 322–333. [Google Scholar] [CrossRef] [Green Version]
- Maharaj, K.; Powers, J.J.; Achille, A.; Deng, S.; Fonseca, R.; Pabon-Saldana, M.; Quayle, S.N.; Jones, S.S.; Villagra, A.; Sotomayor, E.M.; et al. Silencing of HDAC6 as a therapeutic target in chronic lymphocytic leukemia. Blood Adv. 2018, 2, 3012–3024. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.J.; Cai, L.M.; Qian, Z.J.; Wang, C.Y.; Sun, N.; Sun, X.H.; Huang, H.; Guo, W.J.; Lin, H.Y.; Yao, R.X. Increased histone deacetylase 6 expression serves as a favorable prognostic factor for diffuse large B-cell lymphoma. Onco Targets Ther. 2017, 10, 5129–5136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.; Zeng, D.; Zhang, H.; Bell, T.; Yao, J.; Liu, Y.; Huang, S.; Li, C.J.; Lorence, E.; Zhou, S.; et al. Dual inhibition of PI3K signaling and histone deacetylation halts proliferation and induces lethality in mantle cell lymphoma. Oncogene 2019, 38, 1802–1814. [Google Scholar] [CrossRef] [PubMed]
- Mondello, P.; Brea, E.J.; De Stanchina, E.; Toska, E.; Chang, A.Y.; Fennell, M.; Seshan, V.; Garippa, R.; Scheinberg, D.A.; Baselga, J.; et al. Panobinostat acts synergistically with ibrutinib in diffuse large B cell lymphoma cells with MyD88 L265P mutations. JCI Insight 2017, 2, e90196. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Kim, G.W.; Kwon, S.H. The HDAC6-selective inhibitor is effective against non-Hodgkin lymphoma and synergizes with ibrutinib in follicular lymphoma. Mol. Carcinog 2019, 58, 944–956. [Google Scholar] [CrossRef] [PubMed]
- Brach, D.; Johnston-Blackwell, D.; Drew, A.; Lingaraj, T.; Motwani, V.; Warholic, N.M.; Feldman, I.; Plescia, C.; Smith, J.J.; Copeland, R.A.; et al. EZH2 Inhibition by Tazemetostat Results in Altered Dependency on B-cell Activation Signaling in DLBCL. Mol. Cancer Ther. 2017, 16, 2586–2597. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Chitnis, N.; Nakagawa, H.; Kita, Y.; Natsugoe, S.; Yang, Y.; Li, Z.; Wasik, M.; Klein-Szanto, A.J.; Rustgi, A.K.; et al. PRMT5 is required for lymphomagenesis triggered by multiple oncogenic drivers. Cancer Discov. 2015, 5, 288–303. [Google Scholar] [CrossRef] [Green Version]
- Sloan, S.; Brown, F.; Chung, J.H.; Prouty, A.; Wheeler, E.; Harrington, B.K.; Brooks, E.; Yilmaz, A.S.; Ozer, H.G.; Byrd, J.C.; et al. Targeting PRMT5 to Circumvent Acquired Ibrutinib Resistance in Mantle Cell Lymphoma. Blood 2019, 134 (Suppl. 1), 4065. [Google Scholar] [CrossRef]
- Zhou, X.; Steinhardt, M.J.; Dull, J.; Krummenast, F.; Danhof, S.; Meckel, K.; Nickel, K.; Grathwohl, D.; Leicht, H.B.; Rosenwald, A.; et al. Obinutuzumab and venetoclax induced complete remission in a patient with ibrutinib-resistant non-nodal leukemic mantle cell lymphoma. Eur. J. Haematol. 2020, 104, 352–355. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Tedeschi, A.; Trotman, J.; Garcia-Sanz, R.; Macdonald, D.; Leblond, V.; Mahe, B.; Herbaux, C.; Tam, C.; Orsucci, L.; et al. Phase 3 Trial of Ibrutinib plus Rituximab in Waldenstrom’s Macroglobulinemia. N. Engl. J. Med. 2018, 378, 2399–2410. [Google Scholar] [CrossRef]
- Correction to Lancet. Lancet Oncol. 2019, 20, e10. [CrossRef] [Green Version]
- Woyach, J.A.; Ruppert, A.S.; Heerema, N.A.; Zhao, W.; Booth, A.M.; Ding, W.; Bartlett, N.L.; Brander, D.M.; Barr, P.M.; Rogers, K.A.; et al. Ibrutinib Regimens versus Chemoimmunotherapy in Older Patients with Untreated CLL. N. Engl. J. Med. 2018, 379, 2517–2528. [Google Scholar] [CrossRef]
- Burger, J.A.; Sivina, M.; Jain, N.; Kim, E.; Kadia, T.; Estrov, Z.; Nogueras-Gonzalez, G.M.; Huang, X.; Jorgensen, J.; Li, J.; et al. Randomized trial of ibrutinib vs ibrutinib plus rituximab in patients with chronic lymphocytic leukemia. Blood 2019, 133, 1011–1019. [Google Scholar] [CrossRef] [Green Version]
- Skarzynski, M.; Niemann, C.U.; Lee, Y.S.; Martyr, S.; Maric, I.; Salem, D.; Stetler-Stevenson, M.; Marti, G.E.; Calvo, K.R.; Yuan, C.; et al. Interactions between Ibrutinib and Anti-CD20 Antibodies: Competing Effects on the Outcome of Combination Therapy. Clin. Cancer Res. 2016, 22, 86–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, X.W.; Wang, H.Q.; Ban, W.W.; Chang, Z.; Chen, H.Z.; Jia, L.; Liu, F.T. Novel HDAC inhibitor Chidamide synergizes with Rituximab to inhibit diffuse large B-cell lymphoma tumour growth by upregulating CD20. Cell Death Dis. 2020, 11, 20. [Google Scholar] [CrossRef] [PubMed]
- Moreno, C.; Greil, R.; Demirkan, F.; Tedeschi, A.; Anz, B.; Larratt, L.; Simkovic, M.; Samoilova, O.; Novak, J.; Ben-Yehuda, D.; et al. Ibrutinib plus obinutuzumab versus chlorambucil plus obinutuzumab in first-line treatment of chronic lymphocytic leukaemia (iLLUMINATE): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2019, 20, 43–56. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Trotman, J.; Tedeschi, A.; Matous, J.V.; Macdonald, D.; Tam, C.; Tournilhac, O.; Ma, S.; Oriol, A.; Heffner, L.T.; et al. Ibrutinib for patients with rituximab-refractory Waldenstrom’s macroglobulinaemia (iNNOVATE): An open-label substudy of an international, multicentre, phase 3 trial. Lancet Oncol. 2017, 18, 241–250. [Google Scholar] [CrossRef]
- Chanan-Khan, A.; Cramer, P.; Demirkan, F.; Fraser, G.; Silva, R.S.; Grosicki, S.; Pristupa, A.; Janssens, A.; Mayer, J.; Bartlett, N.L.; et al. Ibrutinib combined with bendamustine and rituximab compared with placebo, bendamustine, and rituximab for previously treated chronic lymphocytic leukaemia or small lymphocytic lymphoma (HELIOS): A randomised, double-blind, phase 3 study. Lancet Oncol. 2016, 17, 200–211. [Google Scholar] [CrossRef]
- Wang, M.L.; Lee, H.; Chuang, H.; Wagner-Bartak, N.; Hagemeister, F.; Westin, J.; Fayad, L.; Samaniego, F.; Turturro, F.; Oki, Y.; et al. Ibrutinib in combination with rituximab in relapsed or refractory mantle cell lymphoma: A single-centre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 48–56. [Google Scholar] [CrossRef]
- Gong, J.J.; Yin, Q.S.; Li, M.J.; Ai, H.; Wang, Q.; Chen, L.; Wei, X.D.; Song, Y.P. Ibrutinib combined with CAR-T cells in the treatment of del (17p) chronic lymphocytic leukemia with BCL-2 inhibitor resistance: A case report and literature review. Zhonghua Xue Ye Xue Za Zhi 2019, 40, 750–754. [Google Scholar] [CrossRef]
- Jordan Gauthier, A.V.H.; Kevin, A.H.; Li, D.; Lymp, J.; Sheih, A.; Purushe, J.; Barbara, S.; Pender, R.M.H.; Aesha, V.; Tinh-Doan, P.; et al. Comparison of Efficacy and Toxicity of CD19-Specific Chimeric Antigen Receptor T-Cells Alone or in Combination with Ibrutinib for Relapsed and/or Refractory CLL. Blood 2018, 132 (Suppl. 1), 299. [Google Scholar] [CrossRef]
- Teshima, K.; Nara, M.; Watanabe, A.; Ito, M.; Ikeda, S.; Hatano, Y.; Oshima, K.; Seto, M.; Sawada, K.; Tagawa, H. Dysregulation of BMI1 and microRNA-16 collaborate to enhance an anti-apoptotic potential in the side population of refractory mantle cell lymphoma. Oncogene 2014, 33, 2191–2203. [Google Scholar] [CrossRef] [Green Version]
- Maeda, A.; Nishida, Y.; Weetall, M.; Cao, L.; Branstrom, A.; Ishizawa, J.; Nii, T.; Schober, W.D.; Abe, Y.; Matsue, K.; et al. Targeting of BMI-1 expression by the novel small molecule PTC596 in mantle cell lymphoma. Oncotarget 2018, 9, 28547–28560. [Google Scholar] [CrossRef] [Green Version]
- Luanpitpong, S.; Poohadsuan, J.; Samart, P.; Kiratipaiboon, C.; Rojanasakul, Y.; Issaragrisil, S. Reactive oxygen species mediate cancer stem-like cells and determine bortezomib sensitivity via Mcl-1 and Zeb-1 in mantle cell lymphoma. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3739–3753. [Google Scholar] [CrossRef] [PubMed]
- Sircar, A.; Chowdhury, S.M.; Hart, A.; Bell, W.C.; Singh, S.; Sehgal, L.; Epperla, N. Impact and Intricacies of Bone Marrow Microenvironment in B-cell Lymphomas: From Biology to Therapy. Int. J. Mol. Sci. 2020, 21, 904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, N.N.; Fry, T.J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019, 16, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Fraietta, J.A.; Beckwith, K.A.; Patel, P.R.; Ruella, M.; Zheng, Z.; Barrett, D.M.; Lacey, S.F.; Melenhorst, J.J.; McGettigan, S.E.; Cook, D.R.; et al. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood 2016, 127, 1117–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geyer, M.B.; Riviere, I.; Senechal, B.; Wang, X.; Wang, Y.; Purdon, T.J.; Hsu, M.; Devlin, S.M.; Palomba, M.L.; Halton, E.; et al. Safety and tolerability of conditioning chemotherapy followed by CD19-targeted CAR T cells for relapsed/refractory CLL. JCI Insight 2019, 5. [Google Scholar] [CrossRef]
- Turtle, C.J.; Hay, K.A.; Hanafi, L.A.; Li, D.; Cherian, S.; Chen, X.; Wood, B.; Lozanski, A.; Byrd, J.C.; Heimfeld, S.; et al. Durable Molecular Remissions in Chronic Lymphocytic Leukemia Treated With CD19-Specific Chimeric Antigen Receptor-Modified T Cells After Failure of Ibrutinib. J. Clin. Oncol. 2017, 35, 3010–3020. [Google Scholar] [CrossRef]
- Ruella, M.; Kenderian, S.S.; Shestova, O.; Fraietta, J.A.; Qayyum, S.; Zhang, Q.; Maus, M.V.; Liu, X.; Nunez-Cruz, S.; Klichinsky, M.; et al. The Addition of the BTK Inhibitor Ibrutinib to Anti-CD19 Chimeric Antigen Receptor T Cells (CART19) Improves Responses against Mantle Cell Lymphoma. Clin. Cancer Res. 2016, 22, 2684–2696. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.S.; Johnstone, T.G.; Baturevych, A.; Hause, R.J.; Ragan, S.P.; Clouser, C.R.; Jones, J.C.; Ponce, R.; Krejsa, C.M.; Salmon, R.A.; et al. Antitumor Potency of an Anti-CD19 Chimeric Antigen Receptor T-Cell Therapy, Lisocabtagene Maraleucel in Combination With Ibrutinib or Acalabrutinib. J. Immunother 2020. [Google Scholar] [CrossRef] [PubMed]
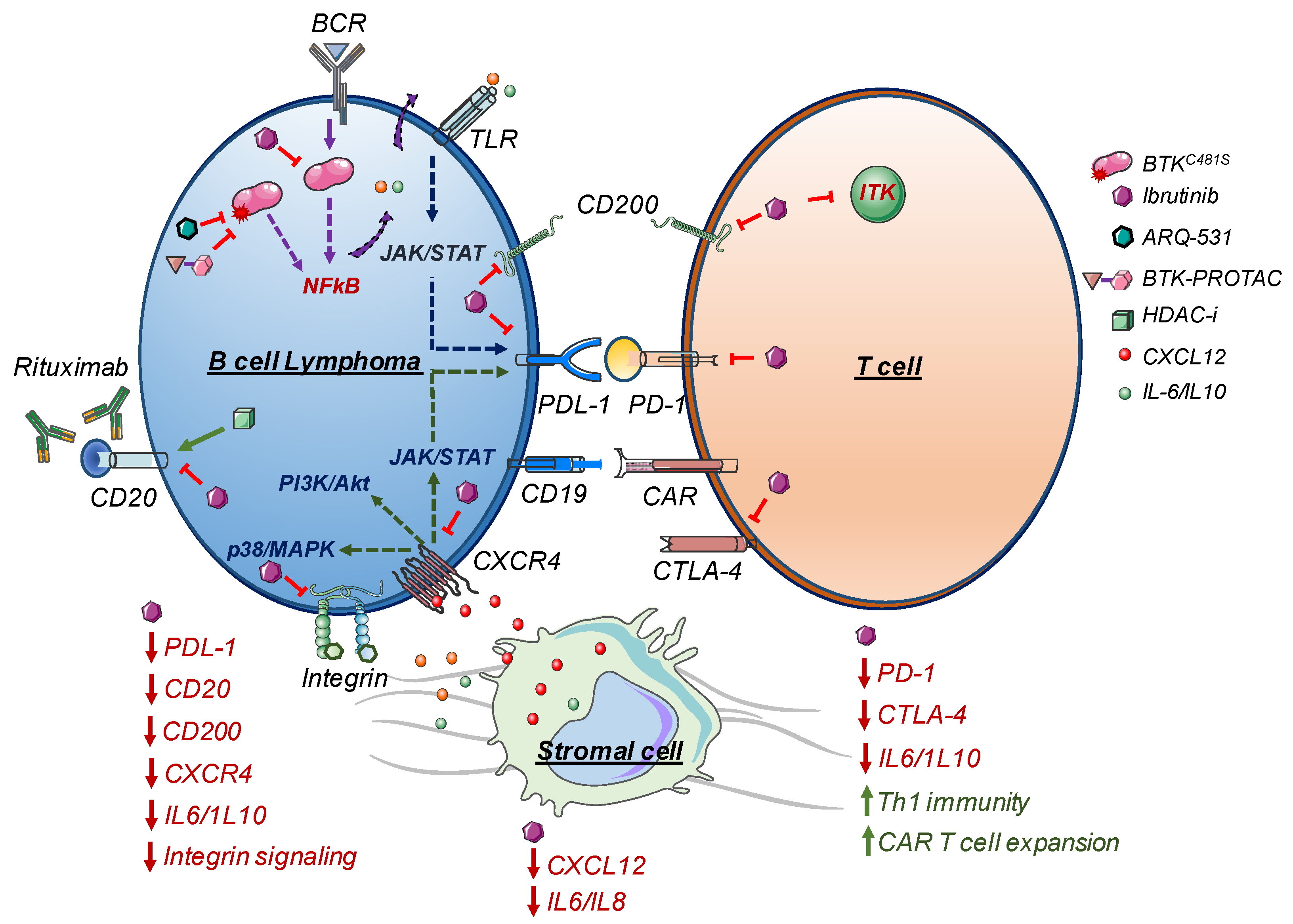

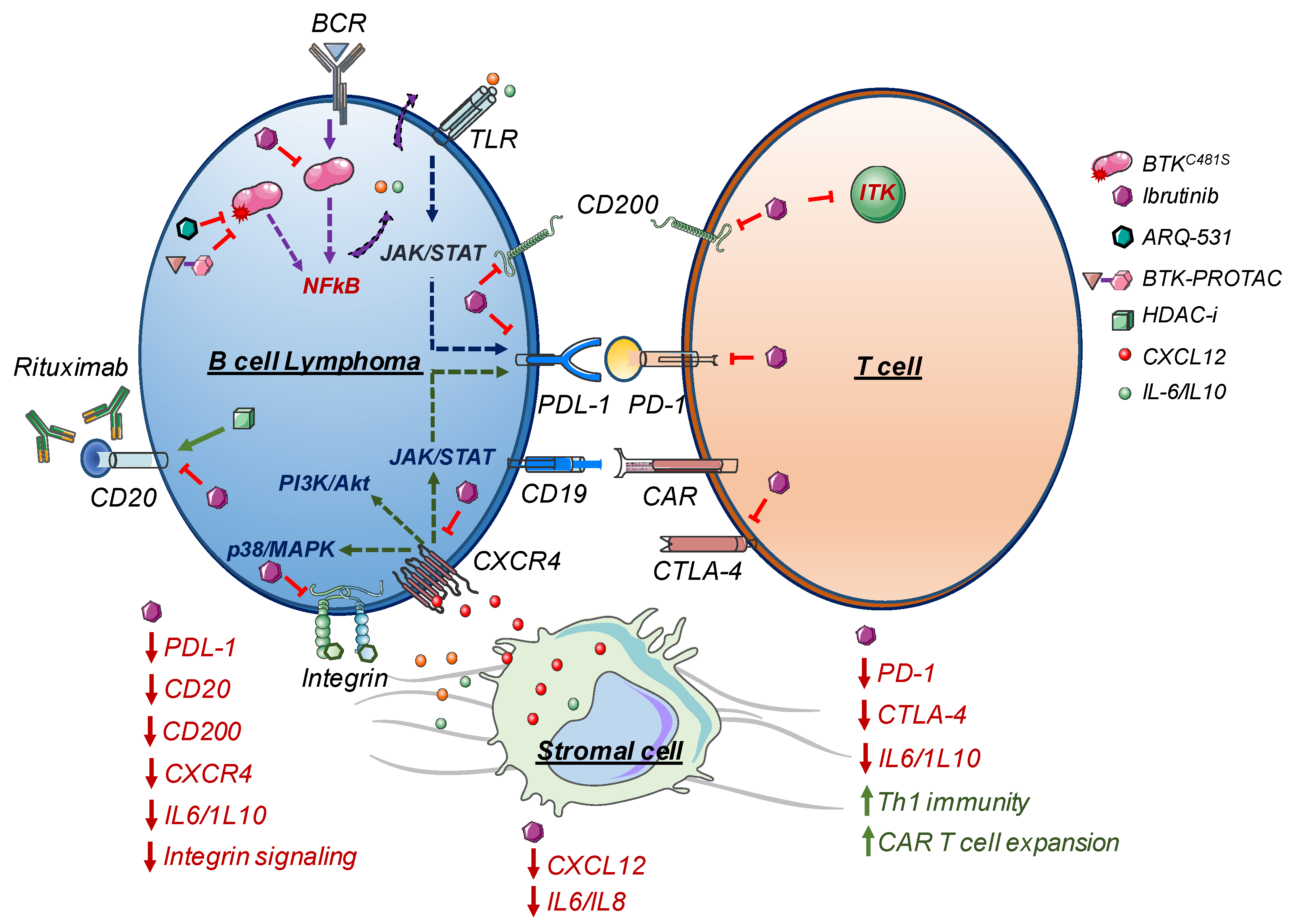{kind=link}
| Study | Method | Major Findings | Size | Reference |
|---|---|---|---|---|
| DLBCL | WES and transcriptomics | Inactivating mutation of KLHL14, enriched in 29.6% tumors of MYD88L265P-CD79B subtype | 574 (biopsy) | Schmitz [21] Jaewoo [22] |
| MCL | WES, and TDS | 9p21.1–p24.3 loss and/or mutations in components of SWI–SNF chromatin-remodelling complex | 24 (R/R) | Aggarwal [23] |
| WES on IS and IR | CARD11 mutation in 5.5% of cases | 13 | Chenglin [24] | |
| WES on 7 IS, 7 IR | Changes in DNA copy number alteration, broad deletions of 6q, 9p, and chromosome 13 | 37 | Zhang [25] | |
| FL | TDS panel of 140 genes, on pre-ibrutinib treatment | CARD11 (16%) and predicted resistance to ibrutinib (NCT01849263) | 31 (biopsy) | Bartlett [26] |
| WM | WES on ibrutinib progressed tumors | Homozygous loss of chr; 6q and 8q at baseline (33% and 66%), at progression (60% and 80%) in tumor of MYD88L265P | 5 (biopsy) | Jimenez [27] |
| AS-PCR for MYD88 and CXCR4 mutation followed by ibrutinib response | Major response rate; MYD88L265PCXCR4WT (91.2%), MYD88L265PCXCR4WHIM (61.9%); Clinical Trial (NCT01614821) | 63 (biopsy) | Treon [28] | |
| CLL | TDS, for mutations in 29 genes | Mutation in TP53, SF3B1, and CARD11 genes | 11 (paired) | Shamanna [29] |
| WES and SNP 6.0 array profiling | Acquired or increased status of del17p/TP53 mutation in three out of five ibrutinib-resistant cases. | 48 (paired) | Amin [13] | |
| WES and TDS | Chr;8p del with additional driver mutations (EP300, MLL2 and EIF2A) | 5 | Burger [30] | |
| A hybrid capture or SNV for panel with 1200 or 1212 CAG | BTKT316A mutation confer activation of PLCG2 | 1 and 9 | Sharma [31], Kadri [32]. |
| Compound | FDA Approved | Date of FDA | Study | Outcome | Adverse Events | CT Identifier |
|---|---|---|---|---|---|---|
| Ibrutinib (Imbruvica); Janssen Biotech, Inc | With Obinutuzumab for TN CLL | 28 Jan,2019 | Phase-3, TN 229 CLL | 30-month PFS 79% (95% CI 70–85). | Grade 3–4, neutropenia, thrombocytopenia | NCT02264574 (PCYC-1130) |
| With Rituximab for WM | 27 Aug,2018 | Phase-3, 150 WM | 30-month PFS 82% (HR,0.20; P < 0.001) | diarrhea, arthralgia; AF: (12% with ibrutinib vs. 1% with Rituximab) | NCT02165397 (PCYC-1127) | |
| GVDH | 2 Aug,2017 | Phase-2, 42 GVHD | 13.9-month ORR 67% | fatigue, diarrhea, muscle spasms, nausea, bruising | NCT02195869 (PCYC-1129-CA) | |
| Acalabrutinib (ACP-196, (Calquence); AstraZeneca | With Obinutuzumab or monotherapy | 21 Nov,2019 | Phase-3, 535 TN CLL | HR, 95% CI, 0.006–0.17; P < 0.0001) | neutropenia, 31% vs. 11%, in acalabrutinib plus obinutuzumab vs. acalabrutinib | ELEVATE-TN (ACE-CL-007) |
| Phase-3, 306 R/R CLL | HR, 0.31; 95% CI, 0.20–0.49; P < 0.0001 | Calquence vs. other group AF: 5% vs. 3%; bleeding: 26% vs. 8%. | ASCEND (ACE-CL-309) | |||
| R/R MCL, one prior therapy | 31 Oct,2017 | Phase-2, 124 MCL | 15·2-months ORR 80% | Grade 1–2 myalgia (21%), diarrhea (31%); Grade 3–4 neutropenia (10%) | NCT02213926 (ACE-LY-004) | |
| Zanubrutinib (Brukinsa); BeiGene Ltd. | MCL, one prior therapy | 14 Nov,2019 | Phase-2, 86 R/R MCL | 18.4-month ORR 84% | Any grade, neutropenia (31.4%), URTI (29.1%), rash (29.1%). | NCT03206970 (BGB-3111-206) |
| Phase-1/2, TN 32 MCL | 18.8-month ORR 84% | Grade ≥ 3 (≥5%) were neutropenia, pneumonia, thrombocytopenia, and leukopenia | NCT02343120 (BGB-3111-AU-003) |
| BTK-PROTAC | Potency/Efficacy | E3 protein-ligand | PROTAC-Structure | Reference |
|---|---|---|---|---|
| MT-802 | >99% degradation at 250 nM conc., more potent than ibrutinib, not suitable for in-vivo studies. | CRBN (C5) |  | Buhimschi [132] |
| SJF620 | Equivalent potency to MT-802, can be used for in-vivo studies. | CRBN (lenalidomide analog) | 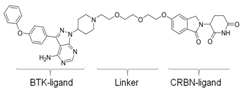 | Figueroa [134] |
| P13I | 89% BTK degradation at 100nM. | CRBN (pomalidomide) | 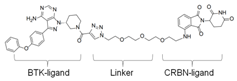 | Sun [133] |
| L18I | Improved solubility vs. P13I in PBS | CRBN (lenalidomide) | 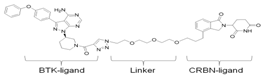 | Sun [136] |
| CJH-005-067 | Efficient degradation of BTK at 100 nM conc., bosutinib-based | CRBN (pomalidomide) | 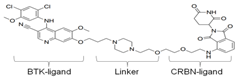 | Huang [131] |
| DD-04-015 | Efficient degradation of BTK at 100 nM conc., RN486-based | CRBN (pomalidomide) | 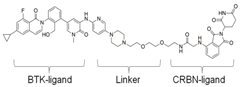 | Huang [131] |
| Study | Patient | Size (M-Age) | Regimen | Outcome | Adverse Events | CT Identifier |
|---|---|---|---|---|---|---|
| Phase-3; Moreno [188] | CLL | 229 (65) | Obinutuzumab-IB vs. obinutuzumab- chlorambucil | 30-month PFS 79% vs. 31% | Serious adverse events: 58% vs. 35% | NCT02264574 (iLLUMINATE) |
| Phase-2; Burger [185] | CLL | 208 (65) | Rituximab-IB vs. IB | 36-month PFS 86.9% vs. 86% | Grade 3/4 TEAE: 65% vs. 64% | NCT02007044 |
| Phase-3; Woyach [184] | CLL | 547 (≥65) | BM-Rituximab vs IB-Rituximab vs. IB | 38-month OS –not significant | Grade 3-5 hematological adverse events: 61% vs. 39% vs. 41% | NCT01886872 |
| Phase-3; Meletios [182] | WM | 150 (69) | Rituximab-IB vs. Rituximab | 30-month PFS 82% vs. 28% | AF: (12% vs. 1%) Hypertension: (13% vs. 4%) | NCT02165397 |
| Phase-3; Meletios [189] | WM (Rituximab refractory) | 31 (67) | IB | 18-month PFS 86% OS 97% | Grade-3 events: Neutropenia-13%, Hypertension-10%. | NCT02165397 (iNNOVATE) |
| Phase-3; Khan [190] | CLL | 578 (≥18) | BM-rituximab vs. BM-Rituximab-IB | 18-month PFS 24% vs. 79% | Grade 3-4 neutropenia: 51% vs. 54% | NCT01611090 (HELIOS) |
| Phase-2; Wang [191] | MCL (R/R) | 50 (67) | Rituximab-IB | 16.5-month PR 44% CR 44% | Grade 3 AF: 12% Grade 4 neutropenia: 1 patient | NCT01880567 |
| Gauthier [12] | CLL (R/R) with del (17p) | 19 | CD19 CAR-T-cell-IB | 4-week ORR 83% | Lower CRS after addition of IB | - |
| Gong [192] | CLL R/R to venetoclax, del(17p) | 1 | CD19 CAR-T-cell-IB | 1-month CR, negligible MRD | Grade 1 CRS | - |
| Phase-1/2; Gauthier [193] | CLL (IB-resistant) | 43 | JCAR014-Cy-Flu-IB vs. JCAR014-Cy-Flu | 4-week ORR 88% vs. 56% | No difference in grade ≥3 cytopenias, similar grade ≥1 CRS | NCT01865617 |
| Phase-1/2; multicenter study | CLL/SLL | 200 | JCAR017 or JCAR017-IB | on-going | - | NCT03331198 |
| Phase-1 | Multiple B-cell malignancies | 274 | JCAR017 | on-going | - | NCT02631044 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
George, B.; Mullick Chowdhury, S.; Hart, A.; Sircar, A.; Singh, S.K.; Nath, U.K.; Mamgain, M.; Singhal, N.K.; Sehgal, L.; Jain, N. Ibrutinib Resistance Mechanisms and Treatment Strategies for B-Cell Lymphomas. Cancers 2020, 12, 1328. https://doi.org/10.3390/cancers12051328
George B, Mullick Chowdhury S, Hart A, Sircar A, Singh SK, Nath UK, Mamgain M, Singhal NK, Sehgal L, Jain N. Ibrutinib Resistance Mechanisms and Treatment Strategies for B-Cell Lymphomas. Cancers. 2020; 12(5):1328. https://doi.org/10.3390/cancers12051328
Chicago/Turabian StyleGeorge, Bhawana, Sayan Mullick Chowdhury, Amber Hart, Anuvrat Sircar, Satish Kumar Singh, Uttam Kumar Nath, Mukesh Mamgain, Naveen Kumar Singhal, Lalit Sehgal, and Neeraj Jain. 2020. "Ibrutinib Resistance Mechanisms and Treatment Strategies for B-Cell Lymphomas" Cancers 12, no. 5: 1328. https://doi.org/10.3390/cancers12051328
APA StyleGeorge, B., Mullick Chowdhury, S., Hart, A., Sircar, A., Singh, S. K., Nath, U. K., Mamgain, M., Singhal, N. K., Sehgal, L., & Jain, N. (2020). Ibrutinib Resistance Mechanisms and Treatment Strategies for B-Cell Lymphomas. Cancers, 12(5), 1328. https://doi.org/10.3390/cancers12051328