MAT2A as Key Regulator and Therapeutic Target in MLLr Leukemogenesis

 , , and
, , and 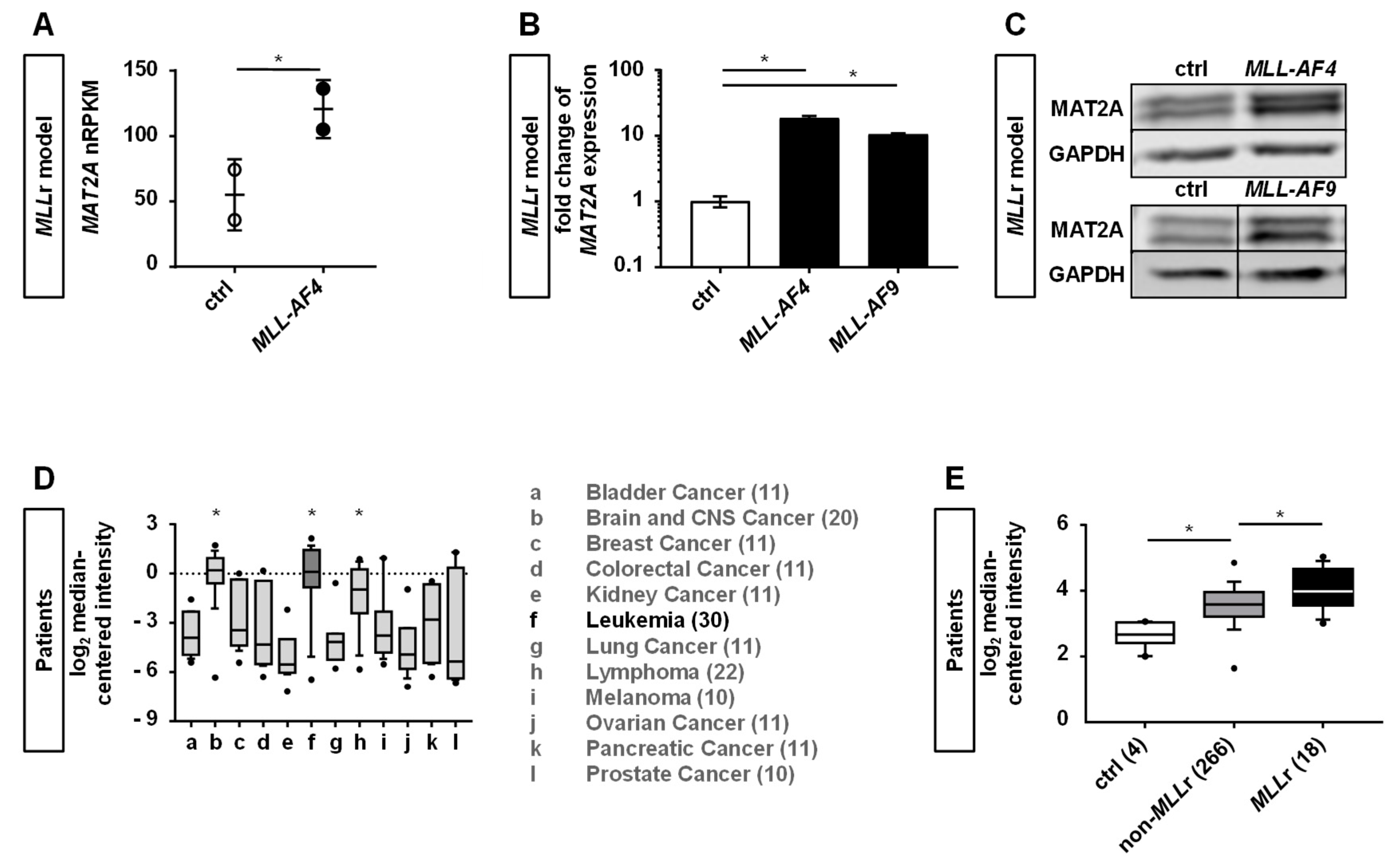{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Human CRISPR/Cas9-MLLr Model Reveals MAT2A as a Possible Target in MLLr Leukemia
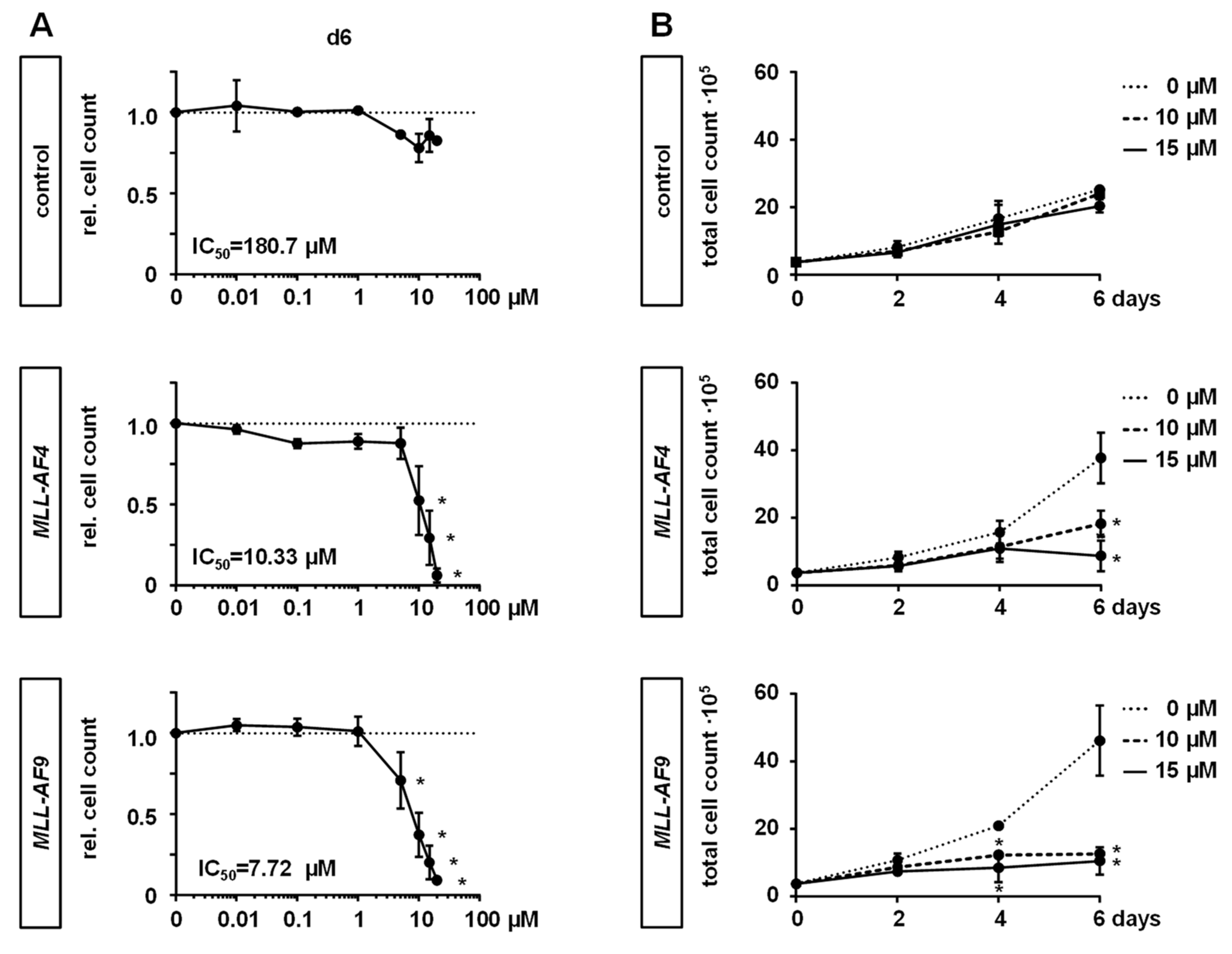
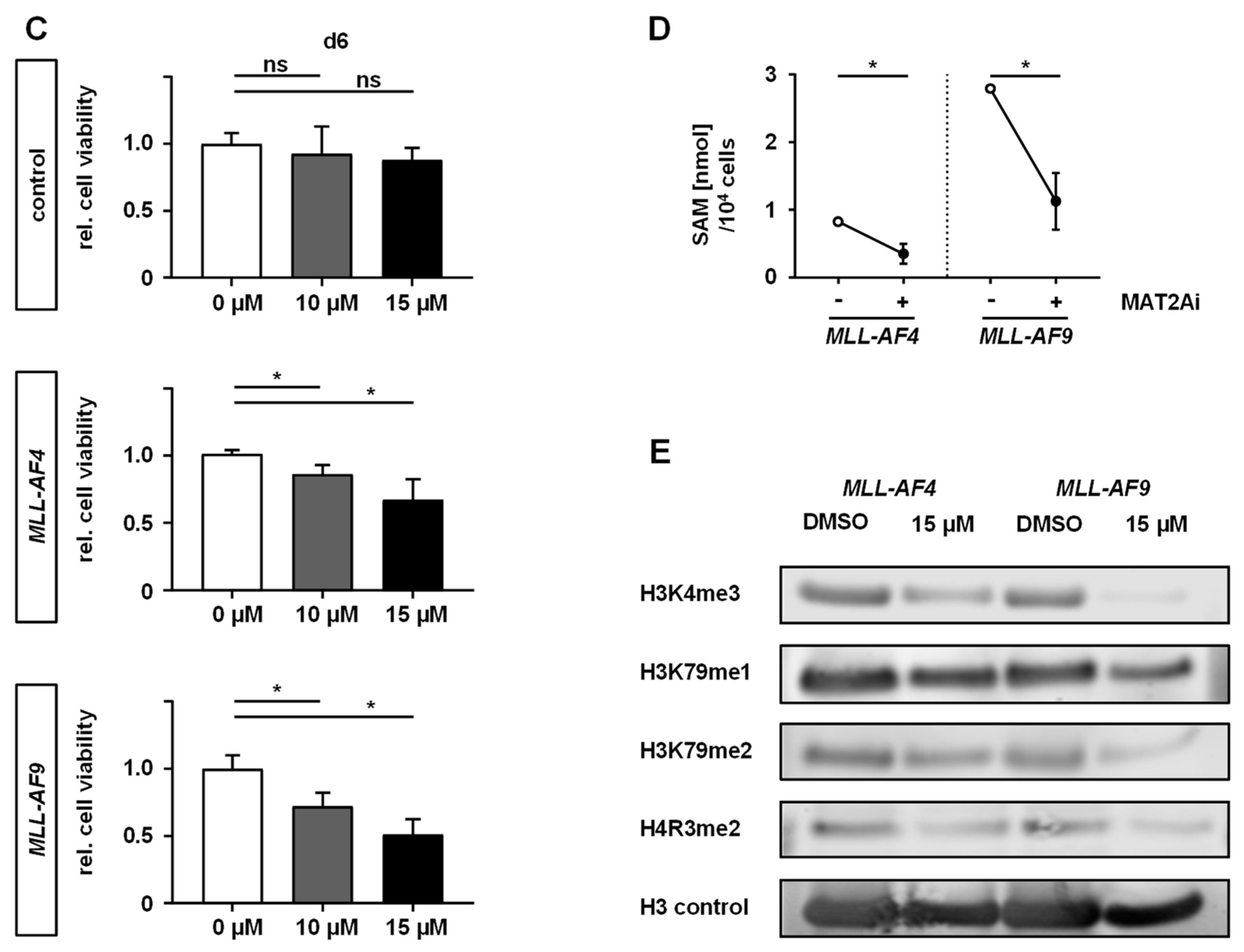
2.2. MAT2A Inhibition Impairs Proliferation and Viability of MLLr Cells and Is Associated with Decreased Global Histone Methylation
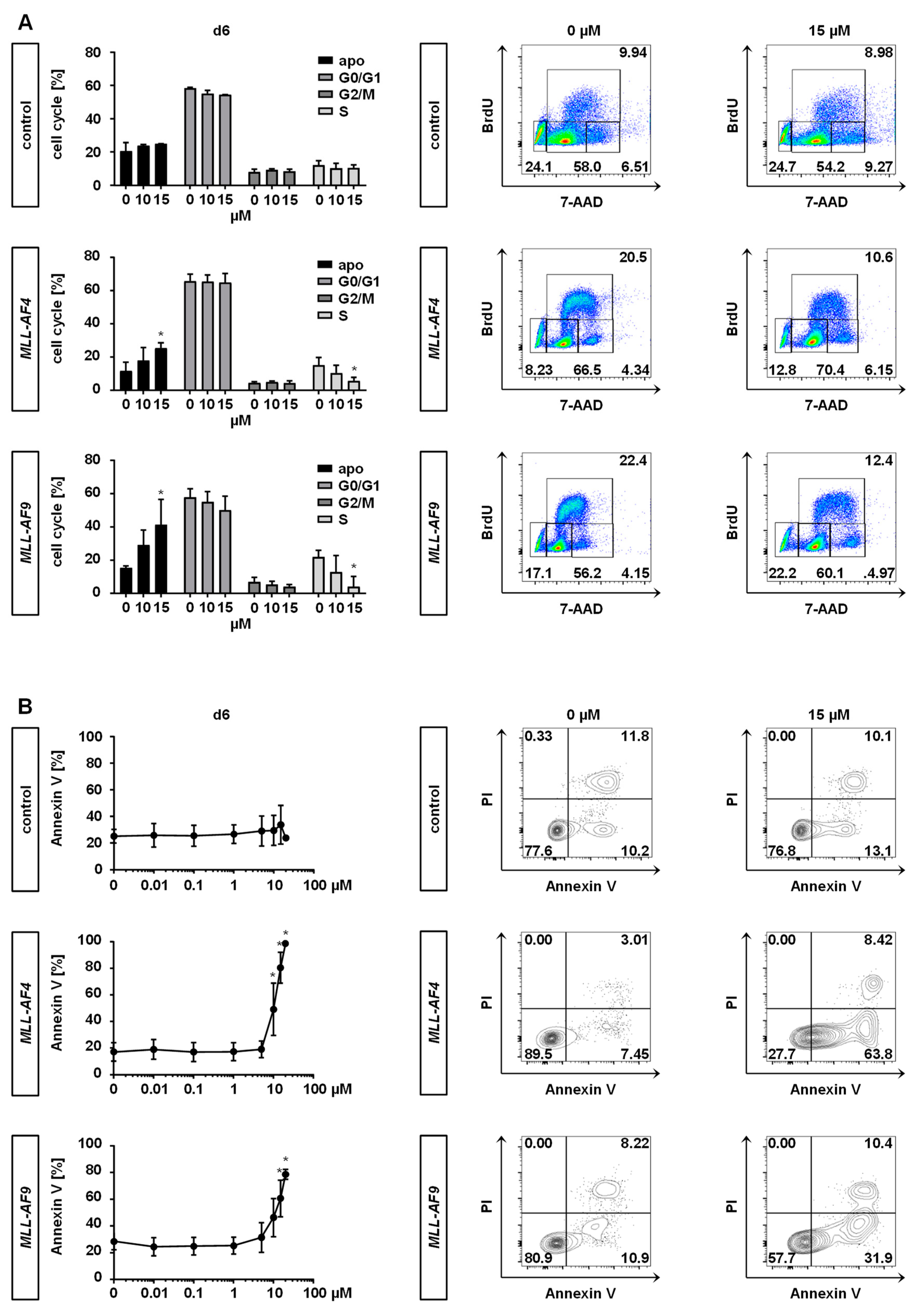
2.3. Inhibition of MAT2A Restrains Cell Cycle and Forces Maturation and Apoptosis of MLLr Cells
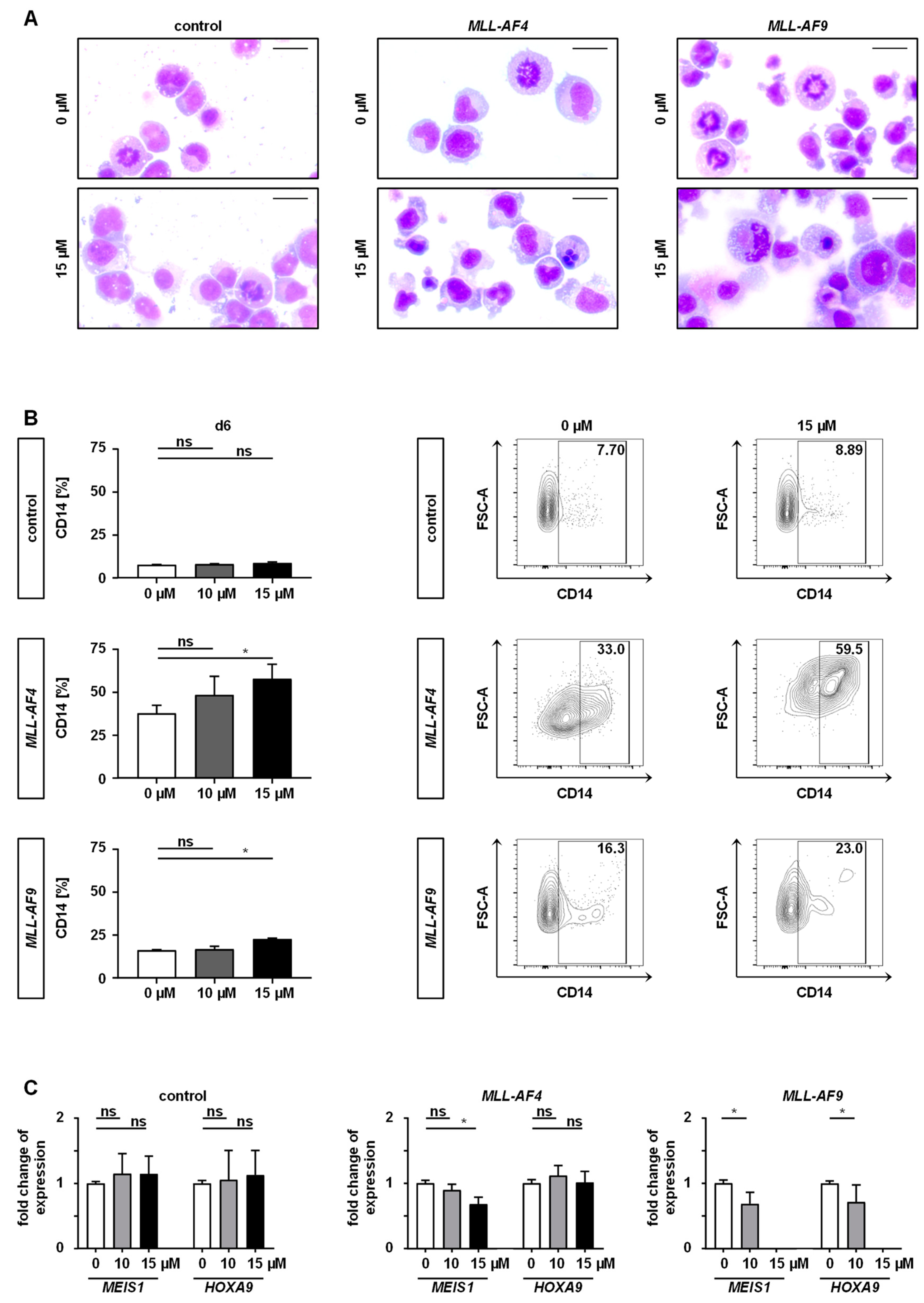
2.4. Downstream Effects of MAT2A Inhibition on Gene Expression
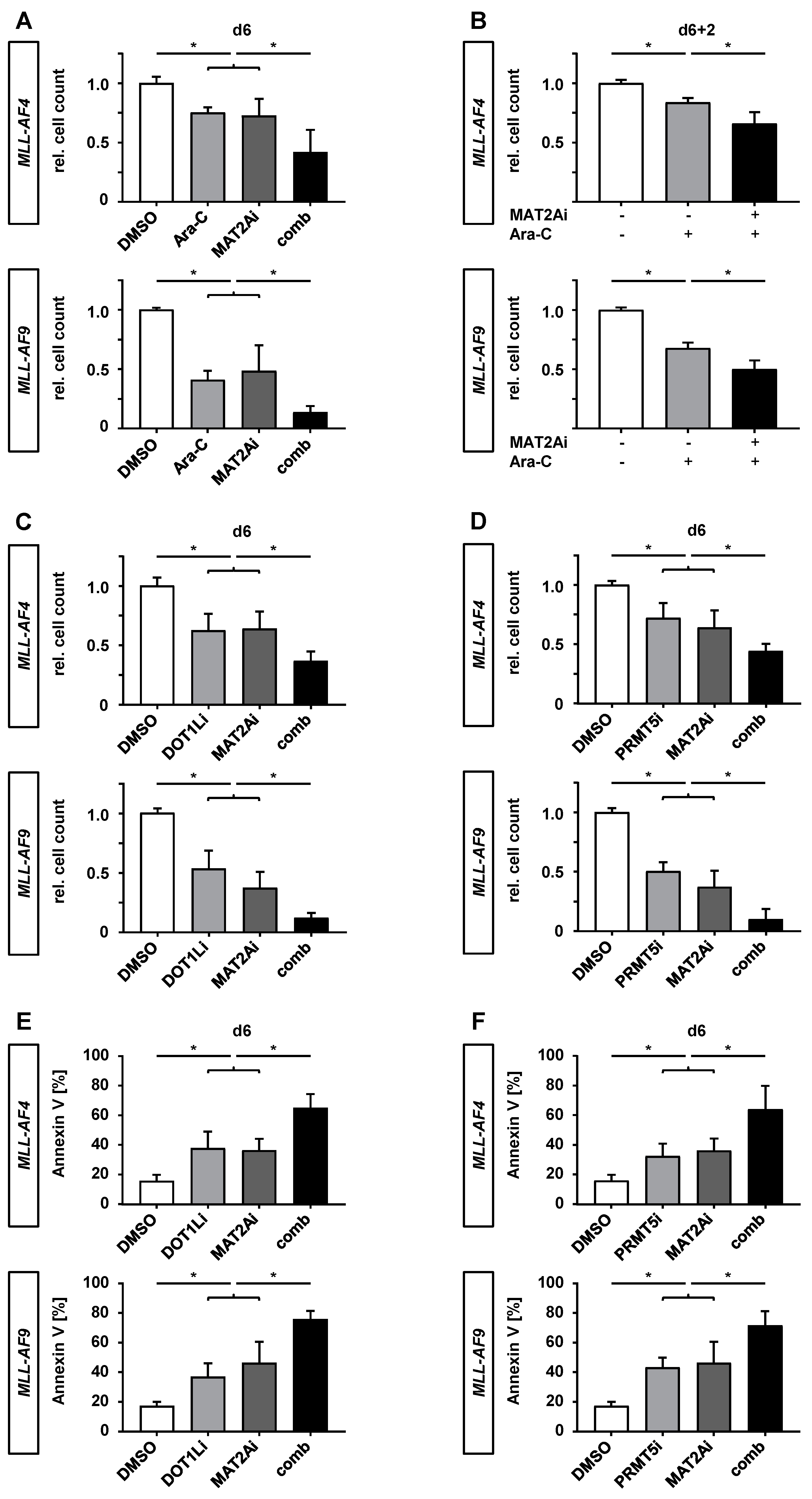
2.5. PF-9366 Sensitizes MLLr Cells to Chemotherapy and Acts Synergistically in Combination with Targeted Therapies
3. Discussion
4. Materials and Methods
4.1. Human CRISPR/Cas9-MLLr Model
4.2. Cell Culture
4.3. Quantitative Reverse Transcriptase-PCR (RT-qPCR)
4.4. Western Blot
4.5. Compound Inhibition Assays
4.6. Flow Cytometry- and Microscopy-Based Determination of Cell Counts
4.7. Viability Assay
4.8. BrdU Cell Cycle and Apoptosis Analyses
4.9. Analysis of Cell Differentiation
4.10. RNA Sequencing
4.11. siRNA Knock-Down Experiments
4.12. Histone Methylation Analysis
4.13. SAM Quantification
4.14. Statistical Analyses
4.15. Data Sharing Statement
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Meyer, C.; Burmeister, T.; Groger, D.; Tsaur, G.; Fechina, L.; Renneville, A.; Sutton, R.; Venn, N.C.; Emerenciano, M.; Pombo-de-Oliveira, M.S.; et al. The MLL recombinome of acute leukemias in 2017. Leukemia 2018, 32, 273–284. [Google Scholar] [CrossRef]
- Gaussmann, A.; Wenger, T.; Eberle, I.; Bursen, A.; Bracharz, S.; Herr, I.; Dingermann, T.; Marschalek, R. Combined effects of the two reciprocal t(4;11) fusion proteins MLL.AF4 and AF4.MLL confer resistance to apoptosis, cell cycling capacity and growth transformation. Oncogene 2007, 26, 3352–3363. [Google Scholar] [CrossRef] [Green Version]
- Kerbel, R.S. Human tumor xenografts as predictive preclinical models for anticancer drug activity in humans: Better than commonly perceived-but they can be improved. Cancer Biol. Ther. 2003, 2, S134–S139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruserud, O.; Gjertsen, B.T.; Foss, B.; Huang, T.S. New strategies in the treatment of acute myelogenous leukemia (AML): In vitro culture of aml cells—The present use in experimental studies and the possible importance for future therapeutic approaches. Stem Cells 2001, 19, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Manabe, A.; Coustan-Smith, E.; Behm, F.G.; Raimondi, S.C.; Campana, D. Bone marrow-derived stromal cells prevent apoptotic cell death in B-lineage acute lymphoblastic leukemia. Blood 1992, 79, 2370–2377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Secker, K.A.; Keppeler, H.; Duerr-Stoerzer, S.; Schmid, H.; Schneidawind, D.; Hentrich, T.; Schulze-Hentrich, J.M.; Mankel, B.; Fend, F.; Schneidawind, C. Inhibition of DOT1L and PRMT5 promote synergistic anti-tumor activity in a human MLL leukemia model induced by CRISPR/Cas9. Oncogene 2019, 38, 7181–7195. [Google Scholar] [CrossRef] [PubMed]
- Bottiglieri, T. S-Adenosyl-L-methionine (SAMe): From the bench to the bedside--molecular basis of a pleiotrophic molecule. Am. J. Clin. Nutr. 2002, 76, 1151S–1157S. [Google Scholar] [CrossRef] [Green Version]
- Timp, W.; Feinberg, A.P. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat. Rev. Cancer 2013, 13, 497–510. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Sun, W.M.; Hwang, J.J.; Stain, S.C.; Lu, S.C. Changes in S-adenosylmethionine synthetase in human liver cancer: Molecular characterization and significance. Hepatology 1996, 24, 1090–1097. [Google Scholar] [CrossRef]
- Simile, M.M.; Peitta, G.; Tomasi, M.L.; Brozzetti, S.; Feo, C.F.; Porcu, A.; Cigliano, A.; Calvisi, D.F.; Feo, F.; Pascale, R.M. MicroRNA-203 impacts on the growth, aggressiveness and prognosis of hepatocellular carcinoma by targeting MAT2A and MAT2B genes. Oncotarget 2019, 10, 2835–2854. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Wu, D.; Wang, S.; Wang, Z. MAT2B expression correlates with poor prognosis in triple-negative breast cancer. Cancer Manag. Res. 2019, 11, 5501–5511. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Xia, M.; Lin, M.; Yang, H.; Kuhlenkamp, J.; Li, T.; Sodir, N.M.; Chen, Y.H.; Josef-Lenz, H.; Laird, P.W.; et al. Role of methionine adenosyltransferase 2A and S-adenosylmethionine in mitogen-induced growth of human colon cancer cells. Gastroenterology 2007, 133, 207–218. [Google Scholar] [CrossRef]
- Jani, T.S.; Gobejishvili, L.; Hote, P.T.; Barve, A.S.; Joshi-Barve, S.; Kharebava, G.; Suttles, J.; Chen, T.; McClain, C.J.; Barve, S. Inhib61ition of methionine adenosyltransferase II induces FasL expression, Fas-DISC formation and caspase-8-dependent apoptotic death in T leukemic cells. Cell Res. 2009, 19, 358–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halim, A.B.; LeGros, H.L., Jr.; Chamberlin, M.E.; Geller, A.; Kotb, M. Distinct patterns of protein binding to the MAT2A promoter in normal and leukemic T cells. Biochim. Biophys. Acta 2001, 1540, 32–42. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.Y.; Liu, D.P.; Liang, C.C. New insight into the molecular mechanisms of MLL-associated leukemia. Leukemia 2005, 19, 183–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stumpel, D.J.; Schneider, P.; van Roon, E.H.; Boer, J.M.; de Lorenzo, P.; Valsecchi, M.G.; de Menezes, R.X.; Pieters, R.; Stam, R.W. Specific promoter methylation identifies different subgroups of MLL-rearranged infant acute lymphoblastic leukemia, influences clinical outcome, and provides therapeutic options. Blood 2009, 114, 5490–5498. [Google Scholar] [CrossRef] [PubMed]
- Schafer, E.; Irizarry, R.; Negi, S.; McIntyre, E.; Small, D.; Figueroa, M.E.; Melnick, A.; Brown, P. Promoter hypermethylation in MLL-r infant acute lymphoblastic leukemia: Biology and therapeutic targeting. Blood 2010, 115, 4798–4809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, C.L.; Kaiser, S.E.; Bolanos, B.; Nowlin, D.; Grantner, R.; Karlicek-Bryant, S.; Feng, J.L.; Jenkinson, S.; Freeman-Cook, K.; Dann, S.G.; et al. Targeting S-adenosylmethionine biosynthesis with a novel allosteric inhibitor of Mat2A. Nat. Chem. Biol. 2017, 13, 785–792. [Google Scholar] [CrossRef]
- Ramaswamy, S.; Tamayo, P.; Rifkin, R.; Mukherjee, S.; Yeang, C.H.; Angelo, M.; Ladd, C.; Reich, M.; Latulippe, E.; Mesirov, J.P.; et al. Multiclass cancer diagnosis using tumor gene expression signatures. Proc. Natl. Acad. Sci. USA 2001, 98, 15149–15154. [Google Scholar] [CrossRef] [Green Version]
- Coustan-Smith, E.; Song, G.; Clark, C.; Key, L.; Liu, P.; Mehrpooya, M.; Stow, P.; Su, X.; Shurtleff, S.; Pui, C.H.; et al. New markers for minimal residual disease detection in acute lymphoblastic leukemia. Blood 2011, 117, 6267–6276. [Google Scholar] [CrossRef]
- Armstrong, S.A.; Staunton, J.E.; Silverman, L.B.; Pieters, R.; den Boer, M.L.; Minden, M.D.; Sallan, S.E.; Lander, E.S.; Golub, T.R.; Korsmeyer, S.J. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat. Genet. 2002, 30, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Lavallee, V.P.; Baccelli, I.; Krosl, J.; Wilhelm, B.; Barabe, F.; Gendron, P.; Boucher, G.; Lemieux, S.; Marinier, A.; Meloche, S.; et al. The transcriptomic landscape and directed chemical interrogation of MLL-rearranged acute myeloid leukemias. Nat. Genet. 2015, 47, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.G.; Kennedy, A.; Zhou, X.; Radtke, I.; Phillips, L.A.; Shurtleff, S.A.; Downing, J.R. Pediatric acute myeloid leukemia with NPM1 mutations is characterized by a gene expression profile with dysregulated HOX gene expression distinct from MLL-rearranged leukemias. Leukemia 2007, 21, 2000–2009. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.A.; Faryabi, R.B.; Ergen, A.V.; Day, A.M.; Malhowski, A.; Canela, A.; Onozawa, M.; Lee, J.E.; Callen, E.; Gutierrez-Martinez, P.; et al. DNA-damage-induced differentiation of leukaemic cells as an anti-cancer barrier. Nature 2014, 514, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Wong, P.; Iwasaki, M.; Somervaille, T.C.; Ficara, F.; Carico, C.; Arnold, C.; Chen, C.Z.; Cleary, M.L. The miR-17-92 microRNA polycistron regulates MLL leukemia stem cell potential by modulating p21 expression. Cancer Res. 2010, 70, 3833–3842. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, S.; Liu, F.; Veazey, K.J.; Gao, G.; Das, P.; Neves, L.F.; Lin, K.; Zhong, Y.; Lu, Y.; Giuliani, V.; et al. Genetic deletion or small-molecule inhibition of the arginine methyltransferase PRMT5 exhibit anti-tumoral activity in mouse models of MLL-rearranged AML. Leukemia 2018, 32, 499–509. [Google Scholar] [CrossRef]
- van Lochem, E.G.; van der Velden, V.H.; Wind, H.K.; te Marvelde, J.G.; Westerdaal, N.A.; van Dongen, J.J. Immunophenotypic differentiation patterns of normal hematopoiesis in human bone marrow: Reference patterns for age-related changes and disease-induced shifts. Cytom. B Clin. Cytom. 2004, 60, 1–13. [Google Scholar] [CrossRef]
- Boyd, K.E.; Xiao, Y.Y.; Fan, K.; Poholek, A.; Copeland, N.G.; Jenkins, N.A.; Perkins, A.S. Sox4 cooperates with Evi1 in AKXD-23 myeloid tumors via transactivation of proviral LTR. Blood 2006, 107, 733–741. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.F.; Huang, Y.; Hou, J.K.; Yuan, T.T.; Zhou, C.X.; Zhang, J.; Chen, G.Q. The downregulation of onzin expression by PKCepsilon-ERK2 signaling and its potential role in AML cell differentiation. Leukemia 2010, 24, 544–551. [Google Scholar] [CrossRef] [Green Version]
- Zeng, C.; Xu, Y.; Xu, L.; Yu, X.; Cheng, J.; Yang, L.; Chen, S.; Li, Y. Inhibition of long non-coding RNA NEAT1 impairs myeloid differentiation in acute promyelocytic leukemia cells. BMC Cancer 2014, 14, 693. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Piao, H.L.; Kim, B.J.; Yao, F.; Han, Z.; Wang, Y.; Xiao, Z.; Siverly, A.N.; Lawhon, S.E.; Ton, B.N.; et al. Long noncoding RNA MALAT1 suppresses breast cancer metastasis. Nat. Genet. 2018, 50, 1705–1715. [Google Scholar] [CrossRef] [PubMed]
- Bernt, K.M.; Zhu, N.; Sinha, A.U.; Vempati, S.; Faber, J.; Krivtsov, A.V.; Feng, Z.; Punt, N.; Daigle, A.; Bullinger, L.; et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell 2011, 20, 66–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceroi, A.; Masson, D.; Roggy, A.; Roumier, C.; Chague, C.; Gauthier, T.; Philippe, L.; Lamarthee, B.; Angelot-Delettre, F.; Bonnefoy, F.; et al. LXR agonist treatment of blastic plasmacytoid dendritic cell neoplasm restores cholesterol efflux and triggers apoptosis. Blood 2016, 128, 2694–2707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautier, E.L.; Westerterp, M.; Bhagwat, N.; Cremers, S.; Shih, A.; Abdel-Wahab, O.; Lutjohann, D.; Randolph, G.J.; Levine, R.L.; Tall, A.R.; et al. HDL and Glut1 inhibition reverse a hypermetabolic state in mouse models of myeloproliferative disorders. J. Exp. Med. 2013, 210, 339–353. [Google Scholar] [CrossRef] [Green Version]
- Bartz, F.; Kern, L.; Erz, D.; Zhu, M.; Gilbert, D.; Meinhof, T.; Wirkner, U.; Erfle, H.; Muckenthaler, M.; Pepperkok, R.; et al. Identification of cholesterol-regulating genes by targeted RNAi screening. Cell Metab. 2009, 10, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, K.T.; Wang, L.M.; Wallen, C.A.; Childers, S.R.; Cline, J.M.; Keng, P.C.; Mach, R.H. Sigma-2 receptors as a biomarker of proliferation in solid tumours. Br. J. Cancer 2000, 82, 1223–1232. [Google Scholar] [CrossRef]
- Mach, R.H.; Zeng, C.; Hawkins, W.G. The sigma2 receptor: A novel protein for the imaging and treatment of cancer. J. Med. Chem. 2013, 56, 7137–7160. [Google Scholar] [CrossRef] [Green Version]
- Moparthi, S.B.; Arbman, G.; Wallin, A.; Kayed, H.; Kleeff, J.; Zentgraf, H.; Sun, X.F. Expression of MAC30 protein is related to survival and biological variables in primary and metastatic colorectal cancers. Int. J. Oncol. 2007, 30, 91–95. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.T.; Han, C.; Sun, Y.M.; Chen, Z.H.; Fang, K.; Huang, W.; Sun, L.Y.; Zeng, Z.C.; Luo, X.Q.; Chen, Y.Q. Activation of the Lysosome-Associated Membrane Protein LAMP5 by DOT1L Serves as a Bodyguard for MLL Fusion Oncoproteins to Evade Degradation in Leukemia. Clin. Cancer Res. 2019, 25, 2795–2808. [Google Scholar] [CrossRef] [Green Version]
- Tavor, S.; Petit, I.; Porozov, S.; Avigdor, A.; Dar, A.; Leider-Trejo, L.; Shemtov, N.; Deutsch, V.; Naparstek, E.; Nagler, A.; et al. CXCR4 regulates migration and development of human acute myelogenous leukemia stem cells in transplanted NOD/SCID mice. Cancer Res. 2004, 64, 2817–2824. [Google Scholar] [CrossRef] [Green Version]
- Sison, E.A.; Magoon, D.; Li, L.; Annesley, C.E.; Romagnoli, B.; Douglas, G.J.; Tuffin, G.; Zimmermann, J.; Brown, P. POL5551, a novel and potent CXCR4 antagonist, enhances sensitivity to chemotherapy in pediatric ALL. Oncotarget 2015, 6, 30902–30918. [Google Scholar] [CrossRef] [PubMed]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Basavapathruni, A.; Jin, L.; Boriack-Sjodin, P.A.; Allain, C.J.; Klaus, C.R.; Raimondi, A.; Scott, M.P.; et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 2013, 122, 1017–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stumpel, D.J.; Schneider, P.; Pieters, R.; Stam, R.W. The potential of clofarabine in MLL-rearranged infant acute lymphoblastic leukaemia. Eur. J. Cancer 2015, 51, 2008–2021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombardini, J.B.; Sufrin, J.R. Chemotherapeutic potential of methionine analogue inhibitors of tumor-derived methionine adenosyltransferases. Biochem. Pharmacol. 1983, 32, 489–495. [Google Scholar] [CrossRef]
- Greco, C.M.; Powell, H.C.; Garrett, R.S.; Lampert, P.W. Cycloleucine encephalopathy. Neuropathol. Appl. Neurobiol. 1980, 6, 349–360. [Google Scholar] [CrossRef]
- Lu, S.C.; Mato, J.M. S-adenosylmethionine in liver health, injury, and cancer. Physiol. Rev. 2012, 92, 1515–1542. [Google Scholar] [CrossRef] [Green Version]
- De La Rosa, J.; Ostrowski, J.; Hryniewicz, M.M.; Kredich, N.M.; Kotb, M.; LeGros, H.L., Jr.; Valentine, M.; Geller, A.M. Chromosomal localization and catalytic properties of the recombinant alpha subunit of human lymphocyte methionine adenosyltransferase. J. Biol. Chem. 1995, 270, 21860–21868. [Google Scholar] [CrossRef] [Green Version]
- Mato, J.M.; Lu, S.C. Role of S-adenosyl-L-methionine in liver health and injury. Hepatology 2007, 45, 1306–1312. [Google Scholar] [CrossRef]
- Cao, F.; Townsend, E.C.; Karatas, H.; Xu, J.; Li, L.; Lee, S.; Liu, L.; Chen, Y.; Ouillette, P.; Zhu, J.; et al. Targeting MLL1 H3K4 methyltransferase activity in mixed-lineage leukemia. Mol. Cell 2014, 53, 247–261. [Google Scholar] [CrossRef] [Green Version]
- Yvan-Charvet, L.; Pagler, T.; Gautier, E.L.; Avagyan, S.; Siry, R.L.; Han, S.; Welch, C.L.; Wang, N.; Randolph, G.J.; Snoeck, H.W.; et al. ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science 2010, 328, 1689–1693. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Yip, L.Y.; Lee, J.H.J.; Wu, Z.; Chew, H.Y.; Chong, P.K.W.; Teo, C.C.; Ang, H.Y.; Peh, K.L.E.; Yuan, J.; et al. Methionine is a metabolic dependency of tumor-initiating cells. Nat. Med. 2019, 25, 825–837. [Google Scholar] [CrossRef] [PubMed]
- Serio, J.; Ropa, J.; Chen, W.; Mysliwski, M.; Saha, N.; Chen, L.; Wang, J.; Miao, H.; Cierpicki, T.; Grembecka, J.; et al. The PAF complex regulation of Prmt5 facilitates the progression and maintenance of MLL fusion leukemia. Oncogene 2018, 37, 450–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, Y.; Feng, Q.; Lin, Y.; Jiang, Q.; Li, Y.; Coffield, V.M.; Su, L.; Xu, G.; Zhang, Y. hDOT1L links histone methylation to leukemogenesis. Cell 2005, 121, 167–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, C.; Hofmann, J.; Burmeister, T.; Groger, D.; Park, T.S.; Emerenciano, M.; Pombo de Oliveira, M.; Renneville, A.; Villarese, P.; Macintyre, E.; et al. The MLL recombinome of acute leukemias in 2013. Leukemia 2013, 27, 2165–2176. [Google Scholar] [CrossRef]
- Langer, T.; Metzler, M.; Reinhardt, D.; Viehmann, S.; Borkhardt, A.; Reichel, M.; Stanulla, M.; Schrappe, M.; Creutzig, U.; Ritter, J.; et al. Analysis of t(9;11) chromosomal breakpoint sequences in childhood acute leukemia: Almost identical MLL breakpoints in therapy-related AML after treatment without etoposides. Genes Chromosomes Cancer 2003, 36, 393–401. [Google Scholar] [CrossRef]
- Reichel, M.; Gillert, E.; Angermuller, S.; Hensel, J.P.; Heidel, F.; Lode, M.; Leis, T.; Biondi, A.; Haas, O.A.; Strehl, S.; et al. Biased distribution of chromosomal breakpoints involving the MLL gene in infants versus children and adults with t(4;11) ALL. Oncogene 2001, 20, 2900–2907. [Google Scholar] [CrossRef] [Green Version]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Majer, C.R.; Sneeringer, C.J.; Song, J.; Johnston, L.D.; Scott, M.P.; Smith, J.J.; Xiao, Y.; et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer cell 2011, 20, 53–65. [Google Scholar] [CrossRef] [Green Version]
- Chan-Penebre, E.; Kuplast, K.G.; Majer, C.R.; Boriack-Sjodin, P.A.; Wigle, T.J.; Johnston, L.D.; Rioux, N.; Munchhof, M.J.; Jin, L.; Jacques, S.L.; et al. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat. Chem. Biol. 2015, 11, 432–437. [Google Scholar] [CrossRef]
- Duncan, K.W.; Rioux, N.; Boriack-Sjodin, P.A.; Munchhof, M.J.; Reiter, L.A.; Majer, C.R.; Jin, L.; Johnston, L.D.; Chan-Penebre, E.; Kuplast, K.G.; et al. Structure and Property Guided Design in the Identification of PRMT5 Tool Compound EPZ015666. ACS Med. Chem. Lett. 2016, 7, 162–166. [Google Scholar] [CrossRef] [Green Version]
- Buechele, C.; Breese, E.H.; Schneidawind, D.; Lin, C.H.; Jeong, J.; Duque-Afonso, J.; Wong, S.H.; Smith, K.S.; Negrin, R.S.; Porteus, M.; et al. MLL leukemia induction by genome editing of human CD34+ hematopoietic cells. Blood 2015, 126, 1683–1694. [Google Scholar] [CrossRef] [Green Version]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware Aligner. 2014. Available online: https://www.osti.gov/servlets/purl/1241166 (accessed on 24 May 2020).
- Andrew, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 24 May 2020).
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Secker, K.-A.; Bloechl, B.; Keppeler, H.; Duerr-Stoerzer, S.; Schmid, H.; Schneidawind, D.; Jeong, J.; Hentrich, T.; Schulze-Hentrich, J.M.; Schneidawind, C. MAT2A as Key Regulator and Therapeutic Target in MLLr Leukemogenesis. Cancers 2020, 12, 1342. https://doi.org/10.3390/cancers12051342
Secker K-A, Bloechl B, Keppeler H, Duerr-Stoerzer S, Schmid H, Schneidawind D, Jeong J, Hentrich T, Schulze-Hentrich JM, Schneidawind C. MAT2A as Key Regulator and Therapeutic Target in MLLr Leukemogenesis. Cancers. 2020; 12(5):1342. https://doi.org/10.3390/cancers12051342
Chicago/Turabian StyleSecker, Kathy-Ann, Bianca Bloechl, Hildegard Keppeler, Silke Duerr-Stoerzer, Hannes Schmid, Dominik Schneidawind, Johan Jeong, Thomas Hentrich, Julia M. Schulze-Hentrich, and Corina Schneidawind. 2020. "MAT2A as Key Regulator and Therapeutic Target in MLLr Leukemogenesis" Cancers 12, no. 5: 1342. https://doi.org/10.3390/cancers12051342
APA StyleSecker, K.-A., Bloechl, B., Keppeler, H., Duerr-Stoerzer, S., Schmid, H., Schneidawind, D., Jeong, J., Hentrich, T., Schulze-Hentrich, J. M., & Schneidawind, C. (2020). MAT2A as Key Regulator and Therapeutic Target in MLLr Leukemogenesis. Cancers, 12(5), 1342. https://doi.org/10.3390/cancers12051342