Pancreatic Cancer Associated Fibroblasts (CAF): Under-Explored Target for Pancreatic Cancer Treatment


Abstract
:1. Background
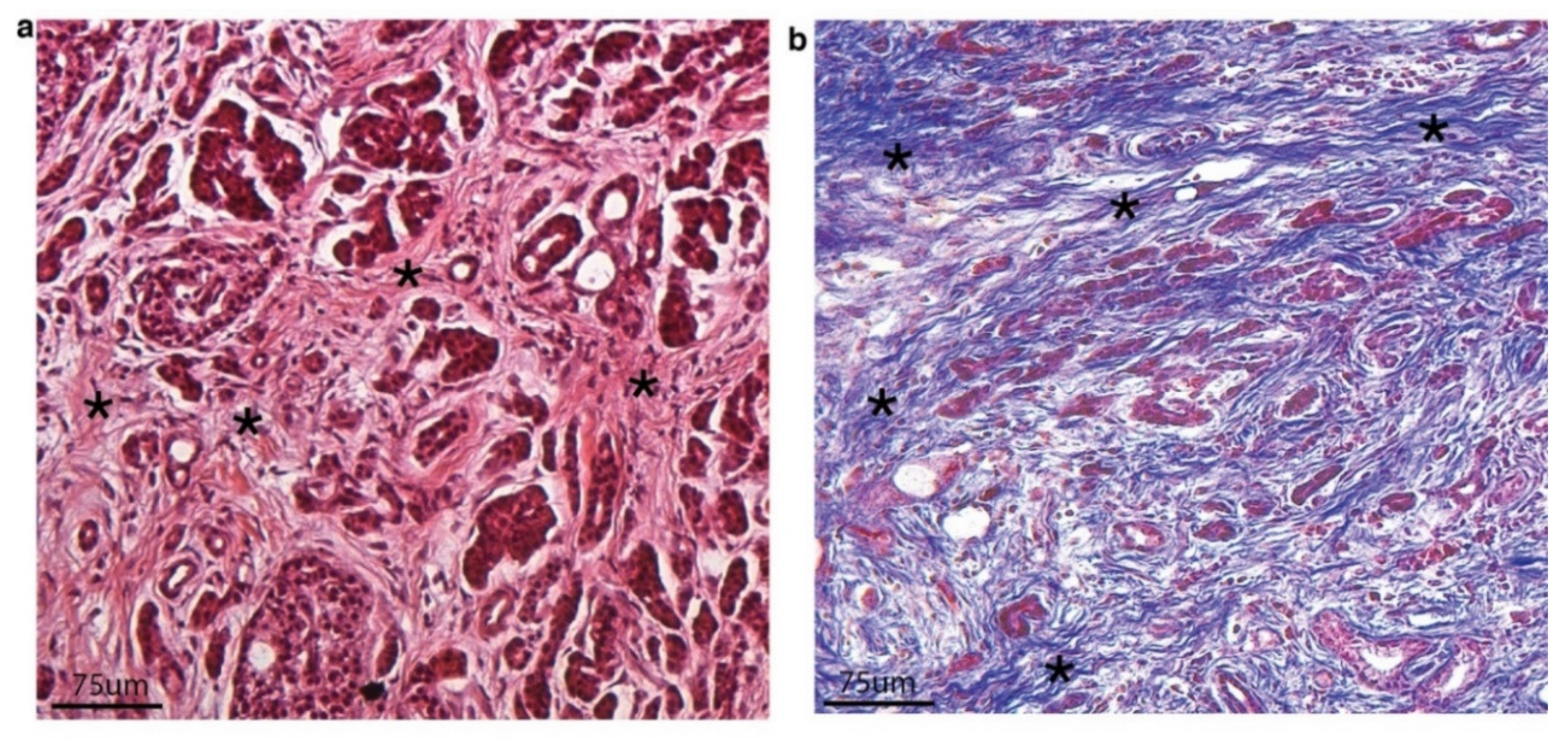
2. CAFs and Tumor Stroma/Extracellular Matrix
3. PDAC CAF Heterogeneity
4. CAFs and Tumor Progression
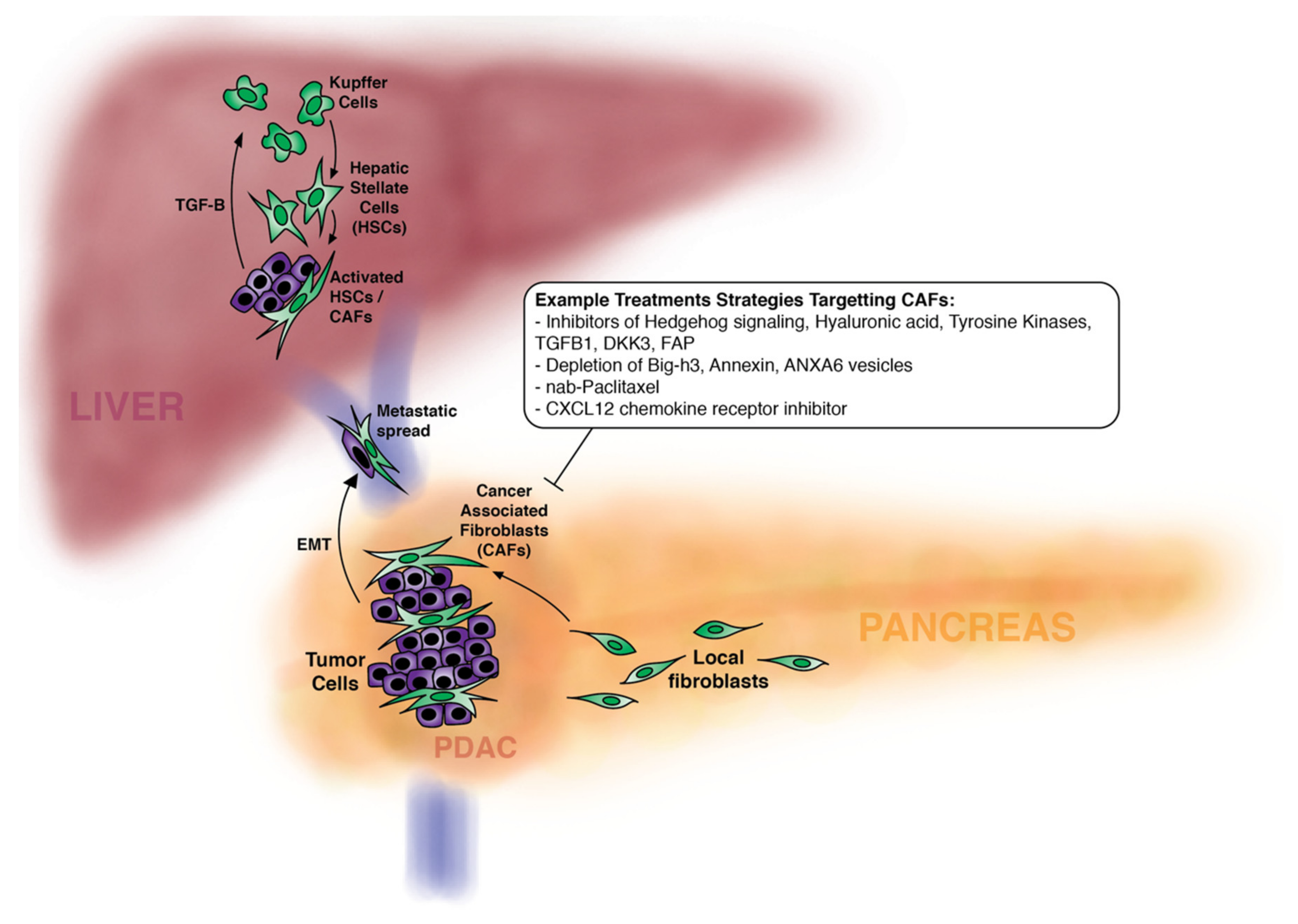
5. CAFS and PDAC Metastatic Spread
6. Modulation of CAFs to Treat Pancreatic Cancer
7. Conclusions
Funding
Conflicts of Interest
References
- American Cancer Society. Cancer Facts and Figures 2019; ACS Publications: Washington, DC, USA, 2019. [Google Scholar]
- Evan, G.I.; Hah, N.; Littlewood, T.D.; Sodir, N.M.; Campos, T.; Downes, M.; Evans, R.M. Re-engineering the Pancreas Tumor Microenvironment: A “Regenerative Program” Hacked. Clin. Cancer Res. 2017, 23, 1647–1655. [Google Scholar] [CrossRef] [Green Version]
- Waghray, M.; Yalamanchili, M.; di Magliano, M.; Simone, D. Deciphering the role of stroma in pancreatic cancer. Curr. Opin. Gastroenterol. 2013, 29, 537–543. [Google Scholar] [CrossRef] [Green Version]
- Vennin, C.; Murphy, K.J.; Morton, J.P.; Cox, T.R.; Pajic, M.; Timpson, P. Reshaping the tumor stroma for the treatment of pancreatic cancer. Gastroenterology 2018, 152, 820–838. [Google Scholar] [CrossRef] [Green Version]
- Von Ahrens, D.; Bhagat, T.D.; Nagrath, D.; Maitra, A.; Verma, A. The role of stromal cancer-associated fibroblasts in pancreatic cancer. J. Hematol. Oncol. 2017, 10, 76. [Google Scholar] [CrossRef] [Green Version]
- Brabletz, T. To differentiate or not-routes towards metastasis. Nat. Rev. Cancer 2012, 12, 425–436. [Google Scholar] [CrossRef]
- Lapshyn, H.; Bolm, L.; Kohler, I.; Werner, M.; Billmann, F.G.; Bausch, D.; Hopt, U.T.; Makowiec, F.; Wittel, U.A.; Keck, T.; et al. Histopathological tumor invasion of the mesenterico-portal vein is characterized by aggressive biology and stromal fibroblast activation. HPB Oxford 2017, 19, 67–74. [Google Scholar] [CrossRef] [Green Version]
- Sherman, M.H. Stellate Cells in Tissue Repair, Inflammation, and Cancer. Annu. Rev. Cell Dev. Biol. 2018, 34, 333–355. [Google Scholar] [CrossRef]
- Sherman, M.H.; Yu, R.T.; Tseng, T.W.; Sousa, C.M.; Liu, S.; Truitt, M.L.; He, N.; Ding, N.; Liddle, C.; Atkins, A.R.; et al. Stromal cues regulate the pancreatic cancer epigenome and metabolome. Proc. Natl. Acad. Sci. USA 2017, 114, 1129–1134. [Google Scholar] [CrossRef] [Green Version]
- Ansari, D.; Carvajo, M.; Bauden, M.; Andersson, R. Pancreatic cancer stroma: Controversies and current insights. Scand. J. Gastroenterol. 2017, 52, 641–646. [Google Scholar] [CrossRef]
- Chang, H.Y.; Sneddon, J.B.; Alizadeh, A.A.; Sood, R.; West, R.B.; Montgomery, K.; Chi, J.T.; van de Rijn, M.; Botstein, D.; Brown, P.O. Gene expression signature of fibroblast serum response predicts human cancer progression: Similarities between tumors and wounds. PLoS Biol. 2004, 2, E7. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.Y.; Chi, J.T.; Dudoit, S.; Bondre, C.; van de Rijn, M.; Botstein, D.; Brown, P.O. Diversity, topographic differentiation, and positional memory in human fibroblasts. Proc. Natl. Acad. Sci. USA 2002, 99, 12877–12882. [Google Scholar] [CrossRef] [Green Version]
- Neesse, A.; Bauer, C.A.; Ohlund, D.; Lauth, M.; Buchholz, M.; Michl, P.; Tuveson, D.A.; Gress, T.M. Stromal biology and therapy in pancreatic cancer: Ready for clinical translation. GUT 2019, 68, 159–171. [Google Scholar] [CrossRef]
- Yin, Z.; Dong, C.; Jiang, K.; Xu, Z.; Li, R.; Guo, K.; Shao, S.; Wang, L. Heterogeneity of cancer-associated fibroblasts and roles in the progression, prognosis, and therapy of hepatocellular carcinoma. J. Hematol. Oncol. 2019, 12, 101. [Google Scholar] [CrossRef]
- Heinrich, S.; Besselink, M.; Moehler, M.; van Laethem, J.L.; Ducreux, M.; Grimminger, P.; Mittler, J.; Lang, H.; Lutz, M.P.; Lesurtel, M.; et al. Opinions and use of neoadjuvant therapy for resectable, borderline resectable, and locally advanced pancreatic cancer: International survey and case-vignette study. BMC Cancer 2019, 19, 675. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, R.; Musteanu, M.; Garcia-Garcia, E.; Lopez-Casas, P.P.; Megias, D.; Guerra, C.; Munoz, M.; Quijano, Y.; Cubillo, A.; Rodriguez-Pascual, J.; et al. Stromal disrupting effects of nab-paclitaxel in pancreatic cancer. Br. J. Cancer 2013, 109, 926–933. [Google Scholar] [CrossRef] [Green Version]
- Okada, K.; Kawai, M.; Hirono, S.; Satoi, S.; Yanagimoto, H.; Ioka, T.; Miyazawa, M.; Shimizu, A.; Kitahata, Y.; Yamaue, H. Impact of treatment duration of neoadjuvant FIRINOX in patients with borderline resectable pancreatic cancer: A pilot trial. Cancer Chemother. Pharmacol. 2016, 78, 719–726. [Google Scholar] [CrossRef]
- Xu, X.; Wu, Q.; Wang, Z.; Zheng, S.; Ge, K.; Jia, C. Meta-analysis of FOLFIRINOX regimen as the first-line chemotherapy for locally advanced pancreatic cancer and borderline resectable pancvreatic cancer. Clin. Exp. Med. 2018, 19, 149–157. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of Carcinoma-Associated Fibroblasts and Fibrosis Induces Immunosuppression and Accelerates Pancreas Cancer with Diminished Survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [Green Version]
- Catenacci, D.V.; Junttila, M.R.; Karrison, T.; Bahary, N.; Horiba, M.N.; Nattam, S.R.; Marsh, R.; Wallace, J.; Kozloff, M.; Rajdev, L.; et al. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients With Metastatic Pancreatic Cancer. J. Clin. Oncol. 2015, 33, 4284–4292. [Google Scholar] [CrossRef]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Zhang, B.; Hu, Q.; Qin, Y.; Xu, W.; Liu, W.; Yu, X.; Xu, J. The impact of cancer-associated fibroblasts on major hallmarks of pancreatic cancer. Theranostics 2018, 8, 5072–5087. [Google Scholar] [CrossRef]
- Lafero, K.J.; Melstrom, L.G. The paradixical web of pancreatic cancer tumor microenvironment. Am. J. Pathol. 2019, 189, 44–57. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Han, C.; Wang, S.; Fang, P.; Ma, Z.; Xu, L.; Yin, R. Cancer-associated fibroblasts: An emerging target of anti-cancer immunotherapy. J. Hematol. Oncol. 2019, 12, 15–86. [Google Scholar] [CrossRef]
- Park, D.; Sahai, E.; Rullan, A. SnapShot: Cancer-Associated Fibroblasts. Cell 2020, 181, 481–486. [Google Scholar] [CrossRef]
- Prakash, J. Cancer-Associated Fibroblasts: Perspective in Cancer Therapy. Trends Cancer 2016, 2, 277–279. [Google Scholar] [CrossRef]
- Biffi, G.; Tuveson, D.A. Deciphering cancer fibroblasts. J. Exp. Med. 2018, 215, 2967–2968. [Google Scholar] [CrossRef] [Green Version]
- Stylianou, A.; Gkretsi, V.; Stylianopoulos, T. Transforming growth factor-beta modulates pancreatic cancer associated fibroblasts cell shape, stiffness and invasion. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 1537–1546. [Google Scholar] [CrossRef]
- Leca, J.; Martinez, S.; Lac, S.; Nigri, J.; Secq, V.; Rubis, M.; Bressy, C.; Serge, A.; Lavaut, M.N.; Dusetti, N.; et al. Cancer-associated fibroblast-derived annexin A6+ extracellular vesicles support pancreatic cancer aggressiveness. J. Clin. Investig. 2016, 126, 4140–4156. [Google Scholar] [CrossRef] [Green Version]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [Green Version]
- Drifka, C.R.; Loeffler, A.G.; Mathewson, K.; Keikhosravi, A.; Eickhoff, J.C.; Liu, Y.; Weber, S.M.; Kao, W.J.; Eliceiri, K.W. Highly aligned stromal collagen is a negative prognostic factor following pancreatic ductal adenocarcinoma resection. Oncotarget 2016, 7, 76197–76213. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wu, H.; Wang, L.; Zhang, H.; Lu, J.; Liang, Z.; Liu, T. Asporin promotes pancreatic cancer cell invasion and migration by regulating the epithelial-to-mesenchymal transition (EMT) through both autocrine and paracrine mechanisms. Cancer Lett. 2017, 398, 24–36. [Google Scholar] [CrossRef]
- Ahmadzadeh, M.; Rosenberg, S.A. TGF-beta 1 attenuates the acquisition and expression of effector function by tumor antigen-specific human memory CD8 T cells. J. Immunol. 2005, 174, 5215–5223. [Google Scholar] [CrossRef] [Green Version]
- Ligorio, M.; Sil, S.; Malagon-Lopez, J.; Nieman, L.T.; Misale, S.; Di Pilato, M.; Ebright, R.Y.; Karabacak, M.N.; Kulkarni, A.S.; Liu, A.; et al. Stromal Microenvironment Shapes the Intratumoral Architecture of Pancreatic Cancer. Cell 2019, 178, 160–175. [Google Scholar] [CrossRef]
- Wang, T.; Notta, F.; Navab, R.; Joseph, J.; Ibrahimov, E.; Xu, J.; Zhu, C.Q.; Borgida, A.; Gallinger, S.; Tsao, M.S. Senescent Carcinoma-Associated Fibroblasts Upregulate IL8 to Enhance Prometastatic Phenotypes. Mol. Cancer Res. 2017, 15, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Goehrig, D.; Nigri, J.; Samain, R.; Wu, Z.; Cappello, P.; Gabiane, G.; Zhang, X.; Zhao, Y.; Kim, I.S.; Chanal, M.; et al. Stromal protein betaig-h3 reprogrammes tumour microenvironment in pancreatic cancer. Gut 2019, 68, 693–707. [Google Scholar] [CrossRef] [Green Version]
- Kawase, T.; Yasui, Y.; Nishina, S.; Hara, Y.; Yanatori, I.; Tomiyama, Y.; Nakashima, Y.; Yoshida, K.; Kishi, F.; Nakamura, M.; et al. Fibroblast activation protein-alpha-expressing fibroblasts promote the progression of pancreatic ductal adenocarcinoma. BMC Gastroenterol. 2015, 15, 109. [Google Scholar] [CrossRef] [Green Version]
- Hu, G.; Wang, S.; Xu, F.; Ding, Q.; Chen, W.; Zhong, K.; Huang, L.; Xu, Q. Tumor-Infiltrating Podoplanin+ Fibroblasts Predict Worse Outcome in Solid Tumors. Cell Physiol. Biochem. 2018, 51, 1041–1050. [Google Scholar] [CrossRef]
- Duluc, C.; Moatassim-Billah, S.; Chalabi-Dchar, M.; Perraud, A.; Samain, R.; Breibach, F.; Gayral, M.; Cordelier, P.; Delisle, M.B.; Bousquet-Dubouch, M.P.; et al. Pharmacological targeting of the protein synthesis mTOR/4E-BP1 pathway in cancer-associated fibroblasts abrogates pancreatic tumour chemoresistance. EMBO Mol. Med. 2015, 7, 735–753. [Google Scholar] [CrossRef]
- Masamune, A.; Kikuta, K.; Watanabe, T.; Satoh, K.; Hirota, M.; Shimosegawa, T. Hypoxia stimulates pancreatic stellate cells to induce fibrosis and angiogenesis in pancreatic cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G709–G717. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Ye, H.; Li, G.; Lu, Y.; Zhou, Q.; Zheng, S.; Lin, Q.; Liu, Y.; Li, Z.; Chen, R. Cancer-associated fibroblasts promote progression and gemcitabine resistance via the SDF-1/SATB-1 pathway in pancreatic cancer. Cell Death Dis. 2018, 9, 1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunetto, E.; De Monte, L.; Balzano, G.; Camisa, B.; Laino, V.; Riba, M.; Heltai, S.; Bianchi, M.; Bordignon, C.; Falconi, M.; et al. The IL-1/IL-1 receptor axis and tumor cell released inflammasome adaptor ASC are key regulators of TSLP secretion by cancer associated fibroblasts in pancreatic cancer. J. Immunother. Cancer 2019, 7, 45. [Google Scholar] [CrossRef]
- Conklin, M.W.; Eickhoff, J.C.; Riching, K.M.; Pehlke, C.A.; Eliceiri, K.W.; Provenzano, P.P.; Friedl, A.; Keely, P.J. Aligned collagen is a prognostic signature for survival in human breast carcinoma. Am. J. Pathol. 2011, 178, 1221–1232. [Google Scholar] [CrossRef]
- Neumann, C.C.M.; von Horschelmann, E.; Reutzel-Selke, A.; Seidel, E.; Sauer, I.M.; Pratschke, J.; Bahra, M.; Schmuck, R.B. Tumor-stromal cross-talk modulating the therapeutic response in pancreatic cancer. Hepatobil. Pancreat. Dis. Int. 2018, 17, 461–472. [Google Scholar] [CrossRef]
- Mahdavi, V.; Hynes, R.O. Effects of cocultivation with transformed cells on surface proteins of normal cells. Biochim. Biophys. Acta 1978, 542, 191–208. [Google Scholar] [CrossRef]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFbeta to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef] [Green Version]
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Nielsen, M.F.B.; Mortensen, M.B.; Detlefsen, S. Typing of pancreatic cancer-associated fibroblasts identifies different subpopulations. World J. Gastroenterol. 2018, 24, 4663–4678. [Google Scholar] [CrossRef]
- Neuzillet, C.; Tijeras-Raballand, A.; Ragulan, C.; Cros, J.; Patil, Y.; Martinet, M.; Erkan, M.; Kleeff, J.; Wilson, J.; Apte, M.; et al. Inter- and intra-tumoural heterogeneity in cancer-associated fibroblasts of human pancreatic ductal adenocarcinoma. J. Pathol. 2019, 248, 51–65. [Google Scholar] [CrossRef] [Green Version]
- Whittle, M.C.; Hingorani, S.R. Fibroblasts in Pancreatic Ductal Adenocarcinoma: Biological Mechanisms and Therapeutic Targets. Gastroenterology 2019, 156, 2085–2096. [Google Scholar] [CrossRef]
- Waghray, M.; Yalamanchili, M.; Dziubinski, M.; Zeinali, M.; Erkkinen, M.; Yang, H.; Schradle, K.A.; Urs, S.; Pasca Di Magliano, M.; Welling, T.H.; et al. GM-CSF Mediates Mesenchymal-Epithelial Cross-talk in Pancreatic Cancer. Cancer Discov. 2016, 6, 886–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunderson, A.J.; Yamazaki, T.; McCarty, K.; Phillips, M.; Alice, A.; Bambina, S.; Zebertavage, L.; Friedman, D.; Cottam, B.; Newell, P.; et al. Blockade of fibroblast activation protein in combination with radiation treatment in murine models of pancreatic adenocarcinoma. PLoS ONE 2019, 14, e0211117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wormann, S.M.; Song, L.; Ai, J.; Diakopoulos, K.N.; Kurkowski, M.U.; Gorgulu, K.; Ruess, D.; Campbell, A.; Doglioni, C.; Jodrell, D.; et al. Loss of P53 Function Activates JAK2-STAT3 Signaling to Promote Pancreatic Tumor Growth, Stroma Modification, and Gemcitabine Resistance in Mice and Is Associated With Patient Survival. Gastroenterology 2016, 151, 180–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Wang, H.; Hsiao, C.H.; Chow, D.S.; Koay, E.J.; Kang, Y.; Wen, X.; Huang, Q.; Ma, Y.; Bankson, J.A.; et al. Simultaneous inhibition of hedgehog signaling and tumor proliferation remodels stroma and enhances pancreatic cancer therapy. Biomaterials 2018, 159, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, T.; Yashiro, M.; Doi, Y.; Kinoshita, H.; Morisaki, T.; Fukuoka, T.; Hasegawa, T.; Kimura, K.; Amano, R.; Hirakawa, K. Pancreatic Fibroblasts Stimulate the Motility of Pancreatic Cancer Cells through IGF1/IGF1R Signaling under Hypoxia. PLoS ONE 2016, 11, e0159912. [Google Scholar] [CrossRef] [Green Version]
- Tjomsland, V.; Niklasson, L.; Sandstrom, P.; Borch, K.; Druid, H.; Bratthall, C.; Messmer, D.; Larsson, M.; Spangeus, A. The desmoplastic stroma plays an essential role in the accumulation and modulation of infiltrated immune cells in pancreatic adenocarcinoma. Clin. Dev. Immunol. 2011, 2011, 212810. [Google Scholar] [CrossRef]
- Crawford, H.C.; Pasca di Magliano, M.; Banerjee, S. Signaling Networks That Control Cellular Plasticity in Pancreatic Tumorigenesis, Progression, and Metastasis. Gastroenterology 2019, 156, 2073–2084. [Google Scholar] [CrossRef]
- Tjomsland, V.; Spangeus, A.; Valila, J.; Sandstrom, P.; Borch, K.; Druid, H.; Falkmer, S.; Falkmer, U.; Messmer, D.; Larsson, M. Interleukin 1alpha sustains the expression of inflammatory factors in human pancreatic cancer microenvironment by targeting cancer-associated fibroblasts. Neoplasia 2011, 13, 664–675. [Google Scholar] [CrossRef] [Green Version]
- Begum, A.; McMillan, R.H.; Chang, Y.T.; Penchev, V.R.; Rajeshkumar, N.V.; Maitra, A.; Goggins, M.G.; Eshelman, J.R.; Wolfgang, C.L.; Rasheed, Z.A.; et al. Direct Interactions With Cancer-Associated Fibroblasts Lead to Enhanced Pancreatic Cancer Stem Cell Function. Pancreas 2019, 48, 329–334. [Google Scholar] [CrossRef]
- Brennen, W.N.; Isaacs, J.T.; Denmeade, S.R. Rationale Behind Targeting Fibroblast Activation Protein–Expressing Carcinoma-Associated Fibroblasts as a Novel Chemotherapeutic Strategy. Mol. Cancer Ther. 2012, 11, 257–266. [Google Scholar] [CrossRef] [Green Version]
- Fearon, D.T. The carcinoma-associated fibroblast expressing fibroblast activation protein and escape from immune surveillance. Cancer Immunol. Res. 2014, 2, 187–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roulis, M.; Kaklamanos, A.; Schernthanner, M.; Bielecki, P.; Zhao, J.; Kaffe, E.; Frommelt, L.S.; Qu, R.; Knapp, M.S.; Henriques, A.; et al. Paracrine orchestration of intestinal tumorigenesis by a mesenchymal niche. Nature 2020, 580, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Ertl, H.C. Depletion of FAP+ cells reduces immunosuppressive cells and improves metabolism and functions CD8+T cells within tumors. Oncotarget 2016, 7, 23282–23299. [Google Scholar] [CrossRef] [Green Version]
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F.; et al. CD10(+)GPR77(+) Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 2018, 172, 841–856. [Google Scholar] [CrossRef]
- Bronsert, P.; Kohler, I.; Timme, S.; Kiefer, S.; Werner, M.; Schilling, O.; Vashist, Y.; Makowiec, F.; Brabletz, T.; Hopt, U.T.; et al. Prognostic significance of Zinc finger E-box binding homeobox 1 (ZEB1) expression in cancer cells and cancer-associated fibroblasts in pancreatic head cancer. Surgery 2014, 156, 97–108. [Google Scholar] [CrossRef]
- Halbrook, C.J.; Lyssiotis, C.A. Employing metabolism to improve the diagnosis and treatment of pancreatic cancer. Cancer Cell 2017, 31, 5–19. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.M.; Hwang, K.A.; Choi, K.C. Diverse pathways of epithelial mesenchymal transition related with cancer progression and metastasis and potential effects of endocrine disrupting chemicals on epithelial mesenchymal transition process. Mol. Cell Endocrinol. 2017, 457, 103–113. [Google Scholar] [CrossRef]
- Zheng, B.; Ohuchida, K.; Chijiiwa, Y.; Zhao, M.; Mizuuchi, Y.; Cui, L.; Horioka, K.; Ohtsuka, T.; Mizumoto, K.; Oda, Y.; et al. CD146 attenuation in cancer-associated fibroblasts promotes pancreatic cancer progression. Mol. Carcinog. 2016, 55, 1560–1572. [Google Scholar] [CrossRef]
- Kadera, B.E.; Li, L.; Toste, P.A.; Wu, N.; Adams, C.; Dawson, D.W.; Donahue, T.R. MicroRNA-21 in pancreatic ductal adenocarcinoma tumor-associated fibroblasts promotes metastasis. PLoS ONE 2013, 8, e71978. [Google Scholar] [CrossRef] [Green Version]
- Jones, M.L.; Siddiqui, J.; Pienta, K.J.; Getzenberg, R.H. Circulating fibroblast-like cells in men with metastatic prostate cancer. Prostate 2013, 73, 176–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ao, Z.; Shah, S.H.; Machlin, L.M.; Parajuli, R.; Miller, P.C.; Rawal, S.; Williams, A.J.; Cote, R.J.; Lippman, M.E.; Datar, R.H.; et al. Identification of Cancer-Associated Fibroblasts in Circulating Blood from Patients with Metastatic Breast Cancer. Cancer Res. 2015, 75, 4681–4687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duda, D.G.; Duyverman, A.M.; Kohno, M.; Snuderl, M.; Steller, E.J.; Fukumura, D.; Jain, R.K. Malignant cells facilitate lung metastasis by bringing their own soil. Proc. Natl. Acad. Sci. USA 2010, 107, 21677–21682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knudsen, E.S.; Balaji, U.; Freinkman, E.; McCue, P.; Witkiewicz, A.K. Unique metabolic features of pancreatic cancer stroma: Relevance to the tumor compartment, prognosis, and invasive potential. Oncotarget 2016, 7, 78396–78411. [Google Scholar] [CrossRef] [Green Version]
- Ao, M.; Brewer, B.M.; Yang, L.; Franco Coronel, O.E.; Hayward, S.W.; Webb, D.J.; Li, D. Stretching fibroblasts remodels fibronectin and alters cancer cell migration. Sci. Rep. 2015, 5, 8334. [Google Scholar] [CrossRef] [Green Version]
- Lecka, J.; Fausther, M.; Kunzli, B.; Sevigny, J. Ticlopidine in its prodrug form is a selective inhibitor of human NTPDase1. Mediat. Inflamm. 2014, 2014, 1–8. [Google Scholar] [CrossRef]
- Aiello, N.; Brabletz, T.; Kang, Y.; Nieto, M.A.; Weinberg, R.A.; Stanger, B.Z. Upholding a role for EMT in pancreatic cancer metastasis. Nature 2017, 547, E7–E8. [Google Scholar] [CrossRef]
- Aiello, N.M.; Bajor, D.L.; Norgard, R.J.; Sahmoud, A.; Bhagwat, N.; Pham, M.N.; Cornish, T.C.; Iacobuzio-Donahue, C.A.; Vonderheide, R.H.; Stanger, B.Z. Metastatic progression is associated with dynamic changes in the local microenvironment. Nat. Commun. 2016, 7, 12819. [Google Scholar] [CrossRef]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [Green Version]
- Ramanathan, R.K.; McDonough, S.L.; Philip, P.A.; Hingorani, S.R.; Lacy, J.; Kortmansky, J.S.; Thumar, J.; Chiorean, E.G.; Shields, A.F.; Behl, D.; et al. Phase IB/II Randomized Study of FOLFIRINOX Plus Pegylated Recombinant Human Hyaluronidase Versus FOLFIRINOX Alone in Patients With Metastatic Pancreatic Adenocarcinoma: SWOG S1313. J. Clin. Oncol. 2019, 37, 1062–1069. [Google Scholar] [CrossRef]
- Doherty, G.J.; Tempero, M.; Corrie, P.G. HALO-109-301: A Phase III trial of PEGPH20 (with gemcitabine and nab-paclitaxel) in hyaluronic acid-high stage IV pancreatic cancer. Futur. Oncol. 2018, 14, 13–22. [Google Scholar] [CrossRef]
- Mpekris, F.; Papageorgis, P.; Polydorou, C.; Voutouri, C.; Kalli, M.; Pirentis, A.P.; Stylianopoulos, T. Sonic-hedgehog pathway inhibition normalizes desmoplastic tumor microenvironment to improve chemo- and nanotherapy. J. Control. Release 2017, 261, 105–112. [Google Scholar] [CrossRef] [Green Version]
- Saison-Ridinger, M.; DelGiorno, K.E.; Zhang, T.; Kraus, A.; French, R.; Jaquish, D.; Tsui, C.; Erikson, G.; Spike, B.T.; Shokhirev, M.N.; et al. Reprogramming pancreatic stellate cells via p53 activation: A putative target for pancreatic cancer therapy. PLoS ONE 2017, 12, e0189051. [Google Scholar] [CrossRef] [PubMed]
- Vaz, J.; Ansari, D.; Sasor, A.; Andersson, R. SPARC: A Potential Prognostic and Therapeutic Target in Pancreatic Cancer. Pancreas 2015, 44, 1024–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, K.M.; Horton, K.J.; Coveler, A.L.; Hingorani, S.R.; Harris, W.P. Targeting the Tumor Stroma: The Biology and Clinical Development of Pegylated Recombinant Human Hyaluronidase (PEGPH20). Curr. Oncol. Rep. 2017, 19, 47. [Google Scholar] [CrossRef] [PubMed]
- Melisi, D.; Garcia-Carbonero, R.; Macarulla, T.; Pezet, D.; Deplanque, G.; Fuchs, M.; Trojan, J.; Oettle, H.; Kozloff, M.; Cleverly, A.; et al. Galunisertib plus gemcitabine vs. gemcitabine for first-line treatment of patients with unresectable pancreatic cancer. Br. J. Cancer 2018, 119, 1208–1214. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Xiao, Z.; Li, T.; Chen, H.; Yuan, Y.; Wang, Y.A.; Hsiao, C.H.; Chow, D.S.; Overwijk, W.W.; Li, C. Stromal Modulation Reverses Primary Resistance to Immune Checkpoint Blockade in Pancreatic Cancer. ACS Nano 2018, 12, 9881–9893. [Google Scholar] [CrossRef]
- Chang, J.; Lucas, M.C.; Leonte, L.E.; Garcia-Montolio, M.; Singh, L.B.; Findlay, A.D.; Deodhar, M.; Foot, J.S.; Jarolimek, W.; Timpson, P.; et al. Pre-clinical evaluation of small molecule LOXL2 inhibitors in breast cancer. Oncotarget 2017, 8, 26066–26078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickup, M.W.; Laklai, H.; Acerbi, I.; Owens, P.; Gorska, A.E.; Chytil, A.; Aakre, M.; Weaver, V.M.; Moses, H.L. Stromally Derived Lysyl Oxidase Promotes Metastasis of Transforming Growth Factor-β Deficient Mouse Mammary Carcinomas. Cancer Res. 2013, 73, 5336–5346. [Google Scholar] [CrossRef] [Green Version]
- Kanteti, R.; Mirzapoiazova, T.; Riehm, J.J.; Dhanasingh, I.; Mambetsariev, B.; Wang, J.; Kulkarni, P.; Kaushik, G.; Seshacharyulu, P.; Ponnusamy, M.P.; et al. Focal adhesion kinase a potential therapeutic target for pancreatic cancer and malignant pleural mesothelioma. Cancer Biol. Ther. 2018, 19, 316–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Jeong, K.; Murphy, J.M.; Rodriguez, Y.A.R.; Kim, J.S.; Ahn, E.E.; Lim, S.S. FAK inhibition reduces metastasis of alpha4 integrin-expressing melanoma to lymph nodes by targeting lymphatic VCAM-1 expression. Biochem. Biophys. Res. Commun. 2019, 509, 1034–1040. [Google Scholar] [CrossRef]
- Topalovski, M.; Brekken, R.A. Matrix control of pancreatic cancer: New insights into fibronectin signaling. Cancer Lett. 2016, 381, 252–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, R.E.; Awasthi, N.; Konduri, S.; Caldwell, L.; Cafasso, D.; Schwarz, M.A. Antitumor effects of EMAP II against pancreatic cancer through inhibition of fibronectin-dependent proliferation. Cancer Biol. Ther. 2010, 9, 632–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munasinghe, A.; Malik, K.; Mohamedi, F.; Moaraf, S.; Kocher, H.; Jones, L.; Hill, N.J. Fibronectin acts as a molecular switch to determine SPARC function in pancreatic cancer. Cancer Lett. 2020, 477, 88–96. [Google Scholar] [CrossRef]
- Heeg, S.; Das, K.K.; Reichert, M.; Bakir, B.; Takano, S.; Caspers, J.; Aiello, N.M.; Wu, K.; Neesse, A.; Maitra, A.; et al. ETS-Transcription Factor ETV1 Regulates Stromal Expansion and Metastasis in Pancreatic Cancer. Gastroenterology 2016, 151, 540–553. [Google Scholar] [CrossRef] [Green Version]
- Ogier, C.; Colombo, P.E.; Bousquet, C.; Canterel-Thouennon, L.; Sicard, P.; Garambois, V.; Thomas, G.; Gaborit, N.; Jarlier, M.; Pirot, N.; et al. Targeting the NRG1/HER3 pathway in tumor cells and cancer-associated fibroblasts with an anti-neuregulin 1 antibody inhibits tumor growth in pre-clinical models of pancreatic cancer. Cancer Lett. 2018, 432, 227–236. [Google Scholar] [CrossRef]
- Morton, J.P.; Karim, S.A.; Graham, K.; Timpson, P.; Jamieson, N.; Athineos, D.; Doyle, B.; McKay, C.; Heung, M.Y.; Oien, K.A.; et al. Dasatinib inhibits the development of metastases in a mouse model of pancreatic ductal adenocarcinoma. Gastroenterology 2010, 139, 292–303. [Google Scholar] [CrossRef]
- Itkin, T.; Gur-Cohen, S.; Spencer, J.A.; Schajnovitz, A.; Ramasamy, S.K.; Kusumbe, A.P.; Ledergor, G.; Jung, Y.; Milo, I.; Poulos, M.G.; et al. Distinct bone marrow blood vessels differentially regulate haematopoiesis. Nature 2016, 532, 323–328. [Google Scholar] [CrossRef]
- Mizutani, Y.; Kobayashi, H.; Iida, T.; Asai, N.; Masamune, A.; Hara, A.; Esaki, N.; Ushida, K.; Mii, S.; Shiraki, Y.; et al. Meflin-Positive Cancer-Associated Fibroblasts Inhibit Pancreatic Carcinogenesis. Cancer Res. 2019, 79, 5367–5381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, C.; Wang, Q.; Meng, Z.; Wang, P. Cancer-Associated Fibroblasts in Pancreatic Cancer: Should They Be Deleted or Reeducated? Integr. Cancer Ther. 2018, 17, 1016–1019. [Google Scholar] [CrossRef] [PubMed]
- Kuninty, P.R.; Bojmar, L.; Tjomsland, V.; Larsson, M.; Storm, G.; Ostman, A.; Sandstrom, P.; Prakash, J. MicroRNA-199a and -214 as potential therapeutic targets in pancreatic stellate cells in pancreatic tumor. Oncotarget 2016, 7, 16396–16408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blair, A.B.; Kim, V.M.; Muth, S.T.; Saung, M.T.; Lokker, N.; Blouw, B.; Armstrong, T.D.; Jaffee, E.M.; Tsujikawa, T.; Coussens, L.M.; et al. Dissecting the Stromal Signaling and Regulation of Myeloid Cells and Memory Effector T Cells in Pancreatic Cancer. Clin. Cancer Res. 2019, 25, 5351–5363. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Molecule | Source Cell | Target Cell | Effect | Reference |
|---|---|---|---|---|
| Annexin A6 | CAFs | Cancer cells | CAF derived annexin A6+ extracellular vesicles found to support pancreatic cancer aggressiveness | Leca et al., J Clin Invest, 2016 [30] |
| CXCL12 | CAFs | Cancer cells | Effector of immunosuppression by FAP+ CAFs | Feig et al., Proc Natl Acad Sci, 2013 [31] |
| Collagen | CAFs | Cancer cells | Well aligned collagen in the stroma is associated with worse prognosis | Drifka et al., Oncotarget, 2016 [32] |
| Asporin | Activated pancreatic stellate cells, CAFs | Cancer cells | Enhances EMT, promotes cancer cell invasion and metastases | Wang et al., Cancer Lett, 2017 [33] |
| TGF-B | CAFs, Cancer cells | CAFs, Cancer cells, CD8 T-cells | Increases CAF stiffness and elongation Suppresses CD8 T cell acquisition to the tumor as well as function | Stylianou et al., Biochim Biophys Acta Gen Subj, 2018 [29] Ahmadzadeh & Rosenberg, J Immunol, 2005 [34] |
| MAPK, STAT3 signaling | CAFs | Cancer cells | Paracrine CAF TGF-B promotes MAPK and STAT3 signaling, which causes EMT and enhanced PDAC proliferation | Ligorio et al., Cell, 2019 [35] |
| Il-8 | Senescent CAFs | Cancer cells | Prometastatic phenotype | Wang et al., 2017. [36] |
| Big-h3 | CAFs | Tumor CD8+ T-cells | Inhibits tumor specific CD8 T-cells and increases tumor growth | Goehrig et al., Gut, 2019 [37] |
| FAP | CAFs | Cancer cells | Cancer cell motility, invasiveness and progression. Also, tumor angiogenesis and ECM deposition. | Kawase et al., BCM Gastroenterol, 2015 [38] |
| Podoplanin | CAFs | Cancer Cells | Worse outcome | Hu et al., Cell Physiol Biochem, 2018 [39] |
| Il-6 | CAFs | Cancer cells | Survivin (apoptosis inhibitor) expression | Duluc et al., EMBO Mol Med, 2015 [40] |
| VEGF | Pancreatic stellate cells | Tumor stroma | Angiogenesis | Masamune et al., Am J Physiol Gastrointest Liver Physiol, 2008 [41] |
| SDF-1 | CAFs | Cancer cells | Tumor progression and resistance to gemcitabine | Wei et al., Cell Death Dis, 2018 [42] |
| Thymic stromal lymphopoietin (TSLP) | CAFs, simulated by cancer cell Il-1 | Immune cells | Development of Th2 immunity, worse survival | Brunetto et al., J Immunother Cancer, 2019 [43] |
| PDAC CAF Sub-Type | Characteristics | Proposed Role | Reference |
|---|---|---|---|
| iCAF (inflammatory CAF) | Il-6high, aSMAhigh, adjacent to tumor cells | Tumorigenesis and cancer progression | Ohund et al., JEM, 2017 [48] |
| myCAF (myofibroblasts CAF) | Il-6low, aSMAlow, distant from tumor cells | Ohund et al., JEM, 2017 [48] | |
| apCAFs (antigen presenting CAFs) | Express CD74 and MHC class II | Activate CD4 T-cells | Elyada et al., Cancer Discov, 2019 [64] |
| FAP+ CAFs | Express FAP | Escaping the immune system (blocking of CD8+ anti-tumor T cells) | Zhang et al., Oncotarget, 2016 [65] |
| CD10+GPR77+ CAFs | Enhances PDAC cell invasion | Su et al., Cell, 2018 [66] | |
| Podoplanin+ CAFs | Express podoplanin | Worse prognosis | Hu et al., Cell Physiol Biochem, 2018. [39] |
| aSMA+ PDAC myofibroblasts | Express aSMA | Depletion of this sub-type resulted in more aggressive tumors and decreased survival in mice | Ozdemir et al., Cancer cell, 2015 [20] |
| PDGFRa+ SAA1+ CAFs | Express PDGFRa and SAA1 | Stimulate PDAC tumor growth in mice | Djurec et al., Proc Natl Acad Sci, 2018 |
| “Sub-type A” | aSMAlow, Vimentinlow, Proliferationhigh, ECM+ | “invasive front”, poor prognosis | Neuzillet et al., J Pathol, 2018 [50] |
| “Sub-type B” | aSMAhigh, Vimentinhigh, Proliferationlow, ECM+ | Intermediate prognosis | Neuzillet et al., J Pathol, 2018 [50] |
| “Sub-type C” | ECM+, Immune++ | Good prognosis | Neuzillet et al., J Pathol, 2018 [50] |
| “Sub-type D” | aSMAhigh, Vimentinhigh, Proliferationlow, ECM+ | Poor prognosis | Neuzillet et al., J Pathol, 2018 [50] |
| Treatment | Result | Reference |
|---|---|---|
| Depletion of CAF-derived annexin (67) | Impaired tumor cell survival and migration in mouse model | Leca et al., J Clin Invest, 2016 [30] |
| Sonic hedgehog inhibitor (cyclopamine, CPA) PD1 checkpoint blockade Sonic hedgehog inhibitor (vismodegib) | Tumor infiltration by cytotoxic CD8+ T cells and prolongation of survival in murine models Decreases desmoplasia, improves effects of chemo- and cancer nano-therapies in mice | Zhou et al., Biomaterials, 2018. [55] Mpekris et al., J Control Release, 2017 [84] Hedgehog inhibitor clinical trials are ongoing |
| Depletion of Big-h3 | Reduction of pancreatic tumor growth by functionally reprogramming F4/80 macrophages in tumor environment | Goehrig et al., Gut, 2019. [37] |
| Treatment with Nutilin-3a induces p53 activation | Induces p53 activation in the stroma, reverses activation of pancreatic stellate cells, and decreases stromal fibrosis | Saison-Ridinger et al.m PLoS One, 2017 [85] |
| Anti-SPARC with Nab-paclitaxel | Positive results in clinical trials vs. human pancreas cancer | Multiple clinical reviewed in the following: Vaz et al., Pancreas, 2015 [86] |
| miRNA therapies that target ZEB and its downstream pathway | Strategy to improve outcome by inhibiting multiple gene pathways | Bronsert et al., Surgery, 2014 [67] |
| PEGPH20 (digests hyaluronic acid) | Improves delivery of immuno- and chemotherapy to the tumor | Wong et al., Curr Oncol Rep, 2017 [87] Clinical trials ongoing |
| Galunisertib (TGFB-1 inhibition) | Inhibits TGFB-1 receptor, found to show improved overall survival in combination with gemcitabine versus gemcitabine alone. | Melisi, Br J Cancer, 2018 [88] Clinical trials ongoing |
| Depletion of ANXA6 extracellular vesicles in CAFs | Impaired pancreas cancer migration and invasion. | Leca et al., J Clin Invest, 2016 [30] |
| AMD3100 (a CXCL12 chemokine receptor inhibitor) | Resulted in rapid T cell accumulation in tumor that synergized with PDL1 to destroy pancreas cancer cells | Feig et al., PNAS, 2013 [31] |
| DKK3 blocking monoclonal antibody | Inhibited pancreas cancer progression, tumor growth, and prolongs survival in mouse model | Zhou et al., ACS Nano, 2018 [89] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Norton, J.; Foster, D.; Chinta, M.; Titan, A.; Longaker, M. Pancreatic Cancer Associated Fibroblasts (CAF): Under-Explored Target for Pancreatic Cancer Treatment. Cancers 2020, 12, 1347. https://doi.org/10.3390/cancers12051347
Norton J, Foster D, Chinta M, Titan A, Longaker M. Pancreatic Cancer Associated Fibroblasts (CAF): Under-Explored Target for Pancreatic Cancer Treatment. Cancers. 2020; 12(5):1347. https://doi.org/10.3390/cancers12051347
Chicago/Turabian StyleNorton, Jeffrey, Deshka Foster, Malini Chinta, Ashley Titan, and Michael Longaker. 2020. "Pancreatic Cancer Associated Fibroblasts (CAF): Under-Explored Target for Pancreatic Cancer Treatment" Cancers 12, no. 5: 1347. https://doi.org/10.3390/cancers12051347
APA StyleNorton, J., Foster, D., Chinta, M., Titan, A., & Longaker, M. (2020). Pancreatic Cancer Associated Fibroblasts (CAF): Under-Explored Target for Pancreatic Cancer Treatment. Cancers, 12(5), 1347. https://doi.org/10.3390/cancers12051347