1. Introduction
Prostate cancer (PCa) is the most commonly diagnosed cancer in men in developed countries and the second most common cancer worldwide [
1]. Several studies have reported that PCa pathobiology is influenced by obesity, specifically, more aggressive carcinoma, poorer treatment outcomes, and increased cancer-specific mortality in obese men [
2,
3,
4,
5,
6,
7,
8,
9]. A range of systemic mechanisms has been proposed to underpin this association, including altered adipokine and inflammatory profiles, hyperinsulinemia, and dyslipidemia [
10,
11,
12].
Significant attention has focused on the role that the local adipose bed that surrounds the prostate, called periprostatic adipose tissue (PPAT), plays in influencing PCa biology. For example, higher grade or more aggressive PCa has been linked to greater PPAT thickness (for review, see [
13]), adipose infiltrating into the tumor [
14], and invasion of PCa into PPAT (defined as an extracapsular extension) [
15,
16,
17]. Additionally, a reciprocal interaction between adipocytes and PCa cells has been reported, both in terms of PPAT stimulating PCa aggressiveness but also PCa cells altering adipocyte biology [
18,
19,
20,
21,
22]. Direct tumor–adipocyte interaction does occur in vivo in multiple situations. Specifically, the vasculature that services the prostate crosses PPAT, thereby facilitating local tissue crosstalk, at the anterior surface of the prostate, where there is direct contact with PPAT [
23], as well as during extracapsular extension of the tumor. As such, it is conceivable that the poorer PCa outcomes observed in obese populations are, in part, due to a paracrine relationship between PPAT and PCa that may be altered in an obese setting. However, there are no existing reports on the PPAT lipid phenotype, including lipolysis, from men with localized PCa, and whether metabolic rewiring of PPAT associates with patients’ metabolic traits and indices of PCa aggressiveness.
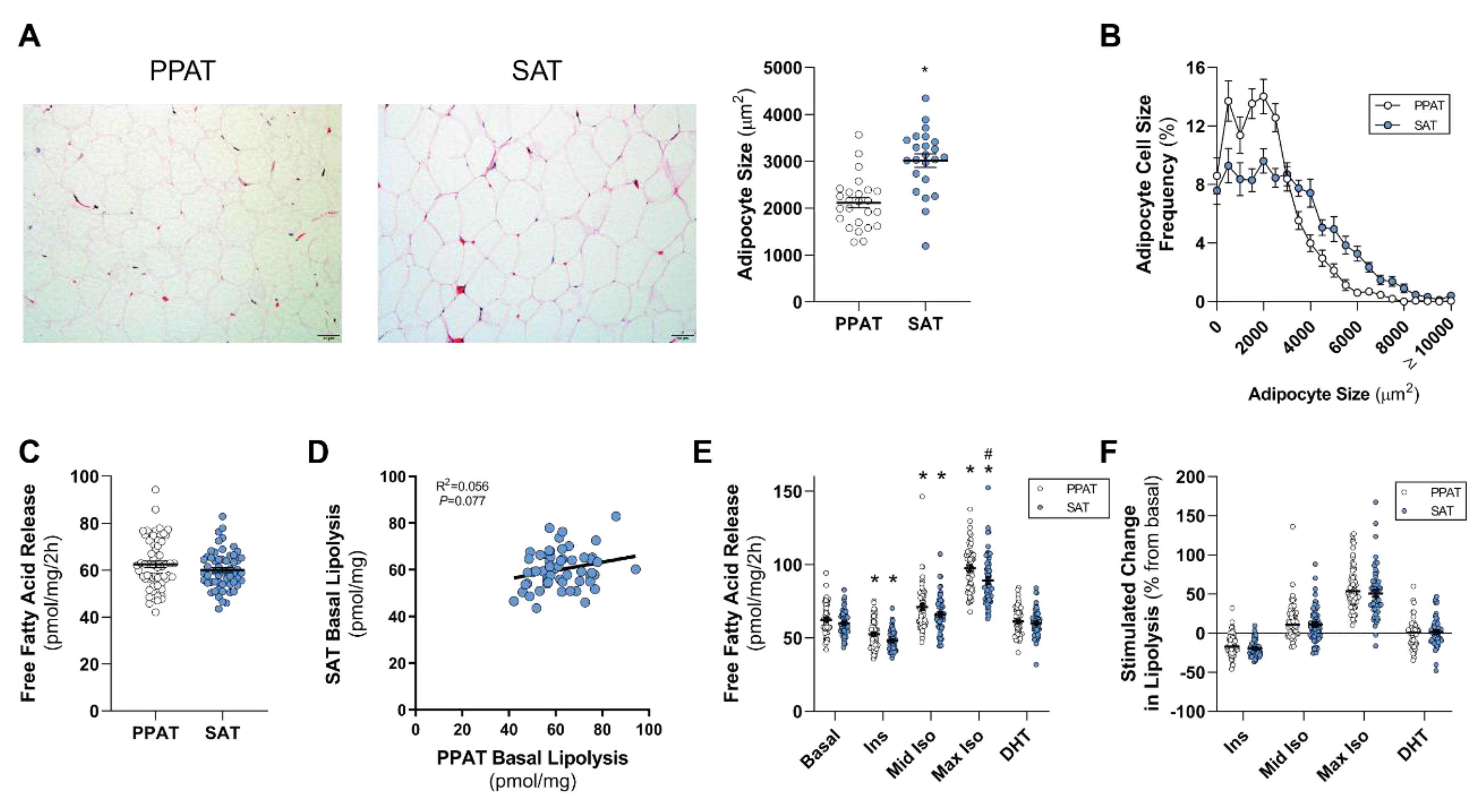
In this study, we assess the lipolytic profile and adipocyte size of PPAT and compare it to that of SAT in men undergoing radical prostatectomy for localized PCa. Further, we explore the relationship between adipose lipid biology and metabolic profiles and PCa clinical features. We hypothesize that lipid-attributes of PPAT differ from SAT, and that PPAT lipolysis is increased in men with more aggressive PCa and in obese men.
3. Discussion
Several studies have reported that the clinical outcomes of PCa are influenced by obesity, with obese patients more likely to have more aggressive carcinoma, poor treatment outcomes, and increased cancer-specific mortality [
7,
8]. A hallmark feature of obesity is adipose tissue expansion and altered adipocyte fatty acid-triacylglycerol metabolism [
37], and when combined with the reports that the amount of PPAT correlates with higher grade or aggressiveness of disease (for review, see [
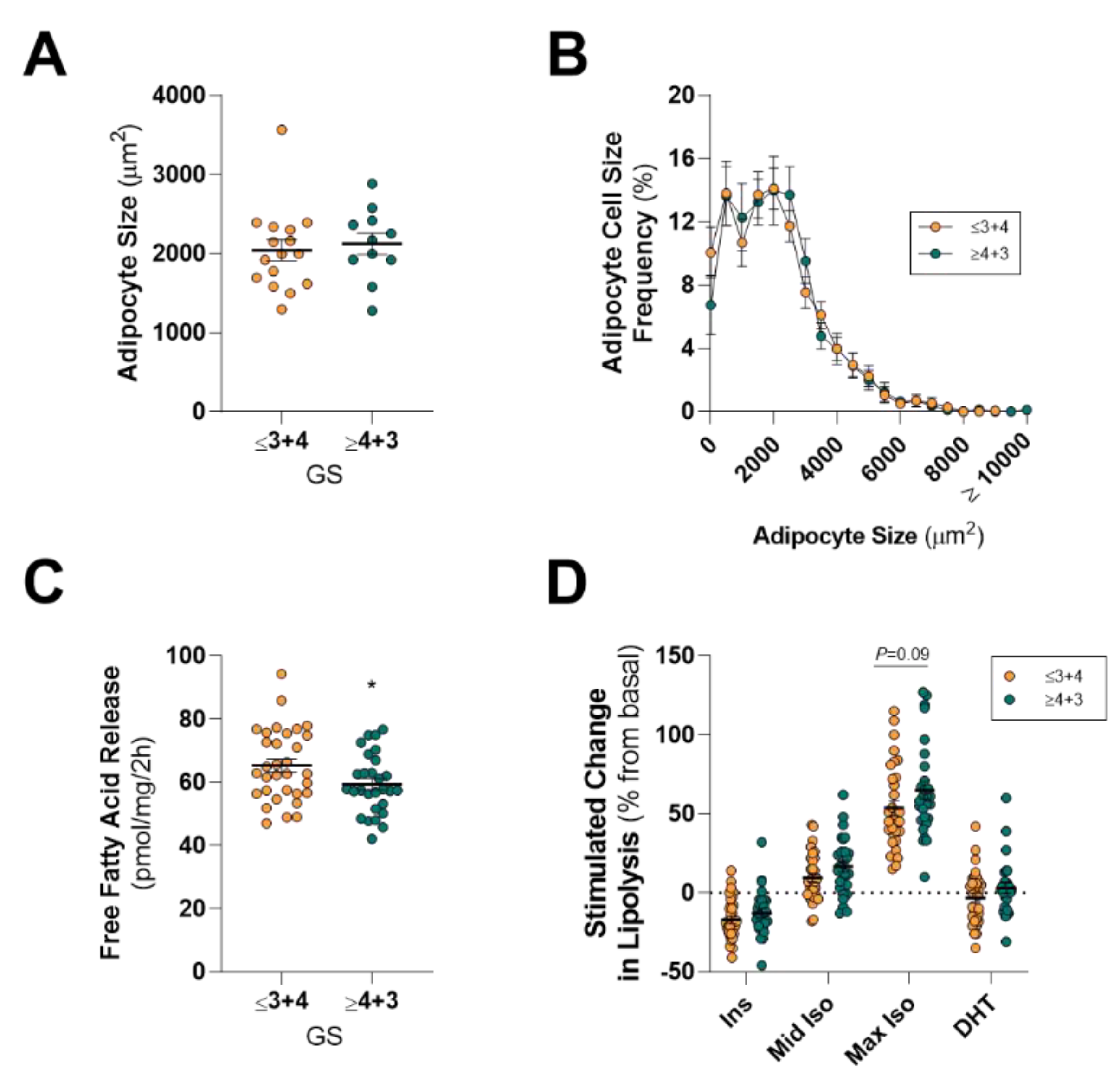
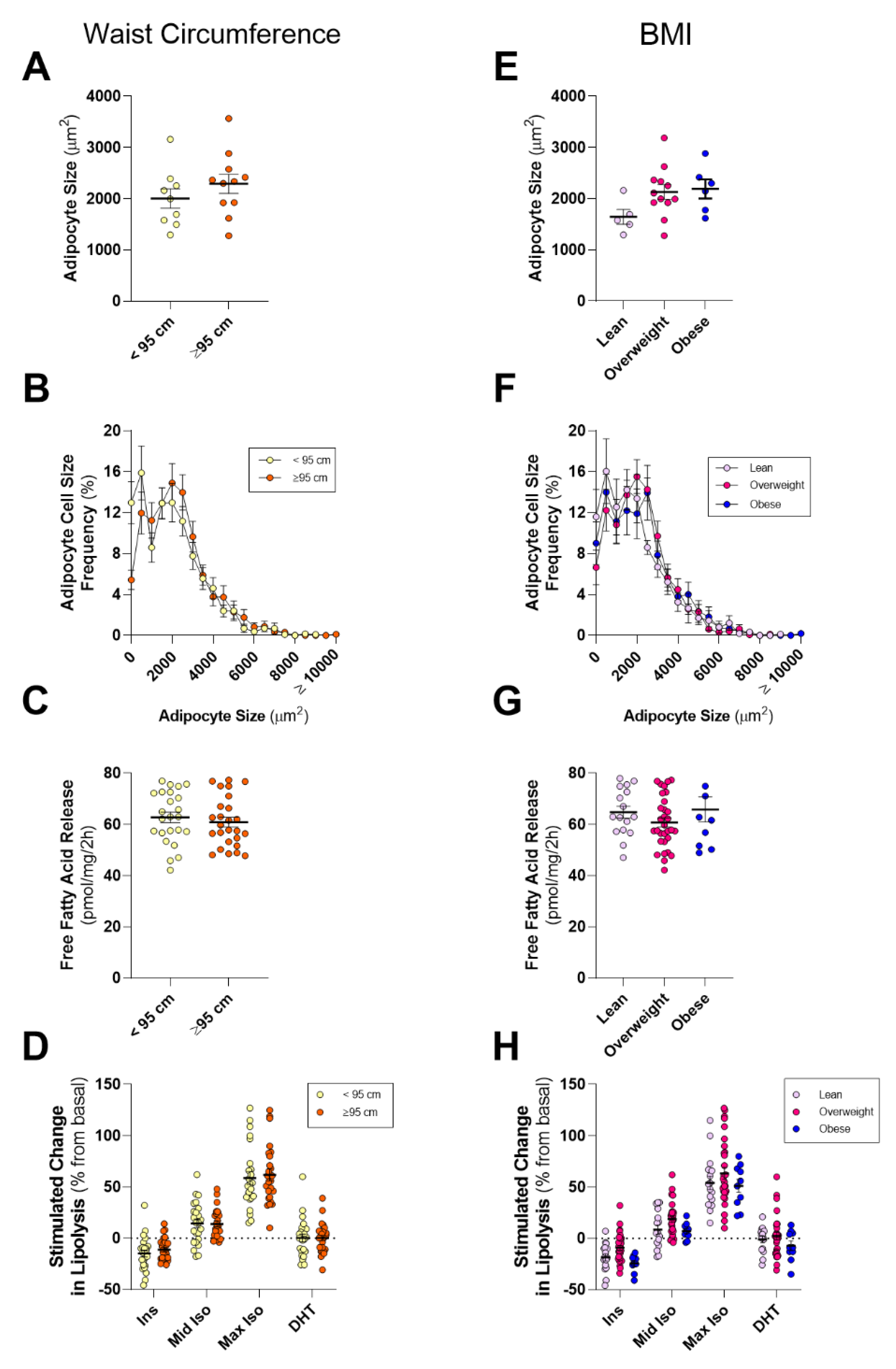
13]), it is an attractive hypothesis that an adipose–PCa axis underpins these clinical observations in obese patients. Here, we show that PPAT adipocytes are smaller than those adipocytes from the same person’s SAT samples and, despite subtle differences in the fatty acid species profile secreted, there was no difference between PPAT and SAT explants in the amount of fatty acid release on a per-milligram basis. PPAT lipolysis responded as expected to insulin (anti-lipolytic) and isoproterenol (pro-lipolytic); however, PPAT was more sensitive to 1 µM isoproterenol stimulation than SAT explants. Counter to our hypothesis, the basal rate of fatty acid release from PPAT explants was greater in men with less aggressive PCa, yet we observed no changes in the abundance of fatty acid species secreted by PPAT explants nor the rate of hormone-stimulated lipolysis or adipocyte size between men with more or less aggressive disease. Further, there was no difference in the rate of fatty acid release from PPAT explants collected from low or high WC patients. These observations, in an adequately sized cohort of 60 patients, demonstrate that there is no remodeling of PPAT lipolysis or adipocyte size that associates with PCa aggressiveness or metabolic features (i.e., WC, BMI) in men undergoing radical prostatectomy.
There is a growing appreciation that the “host” plays a major role in supporting the establishment and progression of cancer [
38,
39,
40]. A key example is the influence that obesity has on cancer risk and progression. Whereas obesity, as determined by BMI, is not associated with PCa risk [
3,
41], it is associated with altered PCa progression [
2,
4]. Specifically, several studies have reported a correlation between BMI and PCa aggressiveness [
2,
3,
4,
5,
6], while other studies have not [
42,
43]. We observed no differences in the mean BMI between high-grade PCa disease (GS ≤ 3 + 4) compared to men with low-grade disease (GS ≥ 4 + 3;
Table 3); however, our study was not designed to test this, and so we are underpowered for this analysis. In any event, BMI is a flawed measure of adiposity/obesity whereas WC, as a measure of abdominal obesity, has been shown to be a better predictor of obesity-associated all-cause mortality in populations over 50 yrs. [
33,
44]. Additionally, men with a larger WC (i.e., greater central obesity) have elevated risk of high-grade PCa and PCa mortality [
5,
6]. In this study of 60 men with an average age of 62 years old, WC was greater in men with high-grade PCa disease (GS ≥ 3 + 4) compared to men with low-grade disease (GS ≤ 4 + 3;
Table 3), which is consistent with similar studies of men in this age group with PCa [
43,
45]. Despite this increase in WC, there was no difference in the levels of circulating factors, including insulin, glucose and lipids, which have been proposed to be key mediators of the effect of obesity on PCa progression (see review in [
12]). As such, the role that circulating factors play in the link between obesity and altered PCa progress remains to be elucidated.
Due to its anatomical relationship with the prostate and its suggested role in PCa, PPAT has received significant attention in recent years (for reviews, see [
13,
46]). To date, there has been limited characterization of PPAT biology, with interleukin-6, pro-MMP-9, and other proteins reported to be secreted from PPAT [
21,
47,
48]. We report here that PPAT has similar attributes to other visceral adipose tissue beds. Specifically, PPAT has smaller adipocytes compare to SAT, which is in line with published observations that SAT adipocytes are larger than omental or mesentery visceral adipose beds [
49]. We did not observe any difference in the basal rate of fatty acid release from PPAT compared to SAT, whereas others have reported that omental visceral adipose tissue has higher basal lipolytic rates compared to SAT [
50,
51]. Further, PPAT was less responsive to the anti-lipolytic effect of insulin compared to SAT, which is consistent with published observations made in other visceral adipose beds [
52,
53]. Whilst we did not see depot-specific differences in the bulk release of fatty acids from PPAT and SAT, we did observe subtle changes in the profile of fatty acid species released from each depot. Specifically, we observed less PUFA species released from PPAT compared to SAT, including 18:3 n-6, 20:3 n-6, 22:5 n-3, and 22:6 n-3, and increased release of saturated species, such as 16:0 and 20:0. Other studies have assessed the intracellular fatty acid profile of PPAT and SAT from men with PCa [
30,
31,
54] but have not performed a direct comparison between PPAT and SAT. It is important to note that the plasma free fatty acid profiles do not correlate well with the fatty acid composition of adipose tissue [
55]. This supports our observations that show that saturated fatty acids are the predominant species secreted by PPAT and SAT, whereas others have shown that monounsaturated fatty acids are the major species of intracellular lipids in PPAT, SAT, and visceral adipose tissue [
30,
31,
54,
55,
56]. Overall, our results demonstrate that PPAT biology differs from SAT biology and that these differences are similar to reported differences between SAT and other visceral adipose depots [
49,
50,
51]. However, we were not able to collect visceral adipose tissue, and so we are limited to the comparison of PPAT biology to SAT. A visceral adipose tissue control would help elucidate differences between the PPAT and visceral adipose tissue biology to inform on whether the presence of local tumors influences PPAT and not overall visceral adipose tissue biology.
In this study, we observed lower levels of PUFAs (18:3 n-6, 20:3 n-6, 20:4 n-6, 22:5 n-3, 22:6 n-3) being secreted from PPAT explants compared with those secreted from SAT. The reduction in the levels of secreted fatty acid species may be simply due to less of those species being stored within adipocytes of adipose tissue. To date, intracellular PPAT and SAT fatty acid species has not been defined. That said, one study did report that the levels of PUFA 20:4 n-6 extracted from PPAT (i.e., intracellular) obtained from men with PCa were lower than from the control group (benign prostatic hyperplasia) [
54]. We did not observe any differences in the levels of PUFAs released from PPAT from men with less or more aggressive disease, but we did not have access to cancer-free men. Interestingly, another study [
31] reported that PUFA species (20:3n-6, 20:4n-6, 22:5n-6, 22:5n-3, 22:6n-3) were less abundant within the malignant prostate gland compared to a benign prostatic hyperplasia control group. Based upon these observations (see
Table S1), one could speculate that the lower levels of PUFA in PPAT, in PPAT in men with PCa, and the malignant prostate gland itself are due to these PUFAs being released and converted into prostaglandins, leukotrienes, and related compounds, which was also suggested by Careaga and colleagues [
54]. It is well known that prostaglandins, leukotrienes, and related compounds derived from n-6 PUFAs are proinflammatory, whereas n-3 PUFAs are antiinflammatory [
57] and can reduce telomere shortening, which is related to aging and chronic diseases [
58]. A recent review by Frietas and Campos [
59] suggested that there is preclinical evidence that n-3 PUFAs and their metabolites might modulate pivotal pathways underlying complications secondary to cancer. Furthermore, a review of human interventional and observational studies reported that higher n-3 PUFA intakes were associated with decreased PCa mortality, but more research is warranted [
60].
Several studies have shown that PPAT-derived signals, including proteins and lipids, can exacerbate PCa progression in vitro [
21,
48,
61]. Complimenting this evidence of paracrine interaction are a number of reports that more aggressive PCa disease is associated with infiltrative adipose tissue into the prostate capsule [
14] and greater PPAT thickness, independent of BMI status [
42,
62]. A key question we set out to answer is whether aspects of ex vivo PPAT lipid biology differ between patients with less or more aggressive PCa. Our rationale was that more aggressive disease (i.e., GS ≥ 4 + 3, extracapsular extension) is associated with increased basal PPAT lipolysis, and thereby smaller adipocytes, due to tumor-derived signaling “rewiring” PPAT [
20]. Surprisingly, we show that men diagnosed with less aggressive (GS ≤ 3 + 4) PCa had a rate of basal fatty acid release from PPAT explants at a per-unit level that was 9% greater (~6 pmol/mg/2 h) than men diagnosed with a more aggressive cancer (
Figure 2). However, we did not see any change in the rate of basal fatty acid release from PPAT explants from patients with extracapsular extension (
Figure S2) or positive lymph nodes. This is important since extracapsular extension is defined as the invasion of PCa into PPAT, resulting in the juxtaposition of PCa cells with cells of the adipose tissue, and patients with this pathological feature have poorer prognostic outcomes [
15,
16,
17,
63,
64,
65,
66]. Whilst we report a subtle difference in basal fatty acid release between less and more aggressive disease, there was no difference in the hormone-stimulated PPAT lipolysis. The net effect is that there was no difference in mean adipocyte size or the distribution of adipocyte size in PPAT from men with low- and high-grade PCa. It is important to highlight that adipocyte size is influenced by a range of factors, including extracellular FA availability, pro- and antilipolytic stimulation, and differentiation status [
67,
68,
69]. Since we observed only a minor difference in the total amount of fatty acid released from PPAT during a two-hour incubation, we hypothesized that there might be differences in the composition of fatty acid species released. This is because a study reported that PPAT lipids (i.e., intracellular lipids) were enriched with monounsaturated fatty acids in men with more aggressive disease [
30]. However, another study reported that there were only minor differences in the fatty acid composition of PPAT from patients undergoing radical prostatectomy for localized prostate tumors and patients undergoing adenomectomy for benign prostatic hyperplasia [
30,
54]. We saw no differences in the profile of fatty acid species released from PPAT from men with low- or high-grade PCa undergoing radical prostatectomy surgery. As such, we conclude that there is no “rewiring” of PPAT lipolysis that associates with high-grade disease. However, this does not preclude the potential for differences in fatty acid release in vivo where the PPAT will be under hormonal stimulation, which fluctuates dependent upon physiological responses to the environment [
70,
71], as well as tumor-derived secreted factors that likely influence PPAT lipid biology [
19].
Like many other cancer types (see reviews in [
72,
73]), PCa biology is influenced by obesity [
13]. PCa that exists in an obese host is associated with more aggressive disease and increased cancer-specific mortality [
2,
3,
4,
5,
6,
7,
8]. Obesity is defined by expansion and alterations in the biology of adipose tissue, including infiltration and activation of immune cells, larger adipocytes, and altered adipocytokine and adipokine release, commonly referred to as adipose tissue dysfunction. This dysfunction in adipose biology has been proposed to be a key driver of altered cancer progression, including PCa (see reviews [
46,
74]). As such, we hypothesized that PPAT lipid biology would be altered in obese men, as defined by a large WC or BMI. Counter to our hypothesis, we observed no differences in adipocyte size, basal- or hormone-stimulated lipolysis, or in fatty acid species abundance when dividing the cohort by WC or BMI. We did observe that men with more aggressive localized disease (GS ≥ 4 + 3) showed a trend to have a greater mean WC compared to men with less aggressive disease (GS ≤ 3 + 4;
p = 0.054), but there was no difference in mean BMI. Whilst we did not see changes in PPAT lipid biology in lean or obese men, this does not negate the role of other aspects of PPAT biology that may influence PCa aggressiveness. For example, the gene expression profile of PPAT from obese men favors hypercellularity and reduced immune-surveillance [
75], as well as having higher MMP9 activity [
20] and increased expression of the chemokine CXCL1 [
76]. PPAT from obese men exhibits increased angiogenic capacity, and conditioned media generated from PPAT from obese subjects promotes PC3 PCa cell line proliferation more than conditioned media from lean subjects [
35]. Likewise, PC-3 invasion was increased following co-cultivation with adipose tissue from high-fat-diet obese mice compared to cells cultured with adipose tissue from low-fat-diet lean mice [
19]. Importantly, others have shown that there was no difference in patient-derived xenografts of moderate-grade localized PCa tumorigenicity in lean and high-fat diet-induced obese severe combined immunodeficiency (SCID) mice [
77]. It is important to note that PPAT thickness, as measured by MRI or similar techniques, does not correlate with BMI [
78] and so the relationship between patient obesity, PPAT, and PCa biology remains complex and unresolved.


 ,
, 



{kind=link}
{kind=link}
{kind=link}