Omics Integration Analyses Reveal the Early Evolution of Malignancy in Breast Cancer


 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
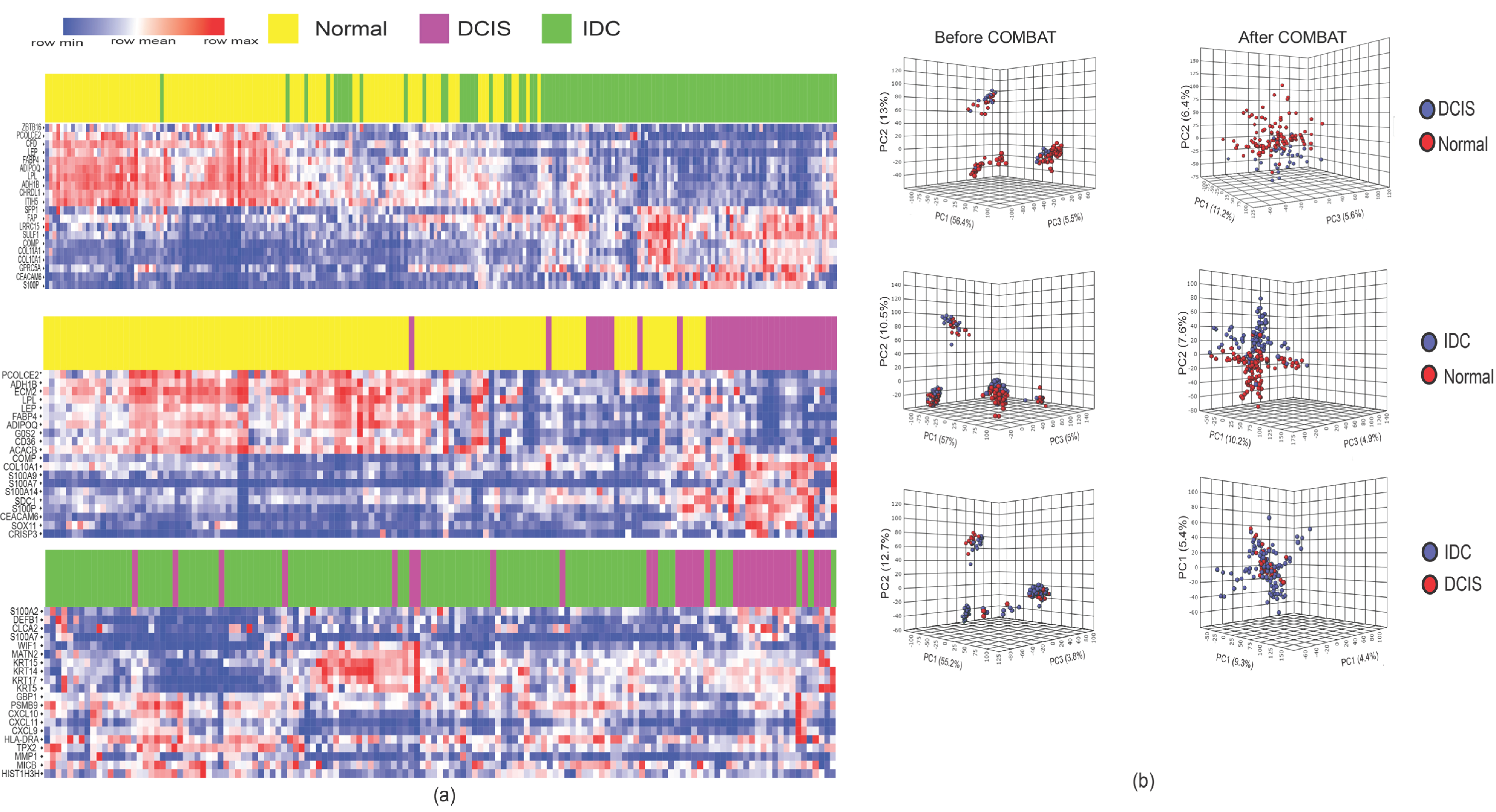
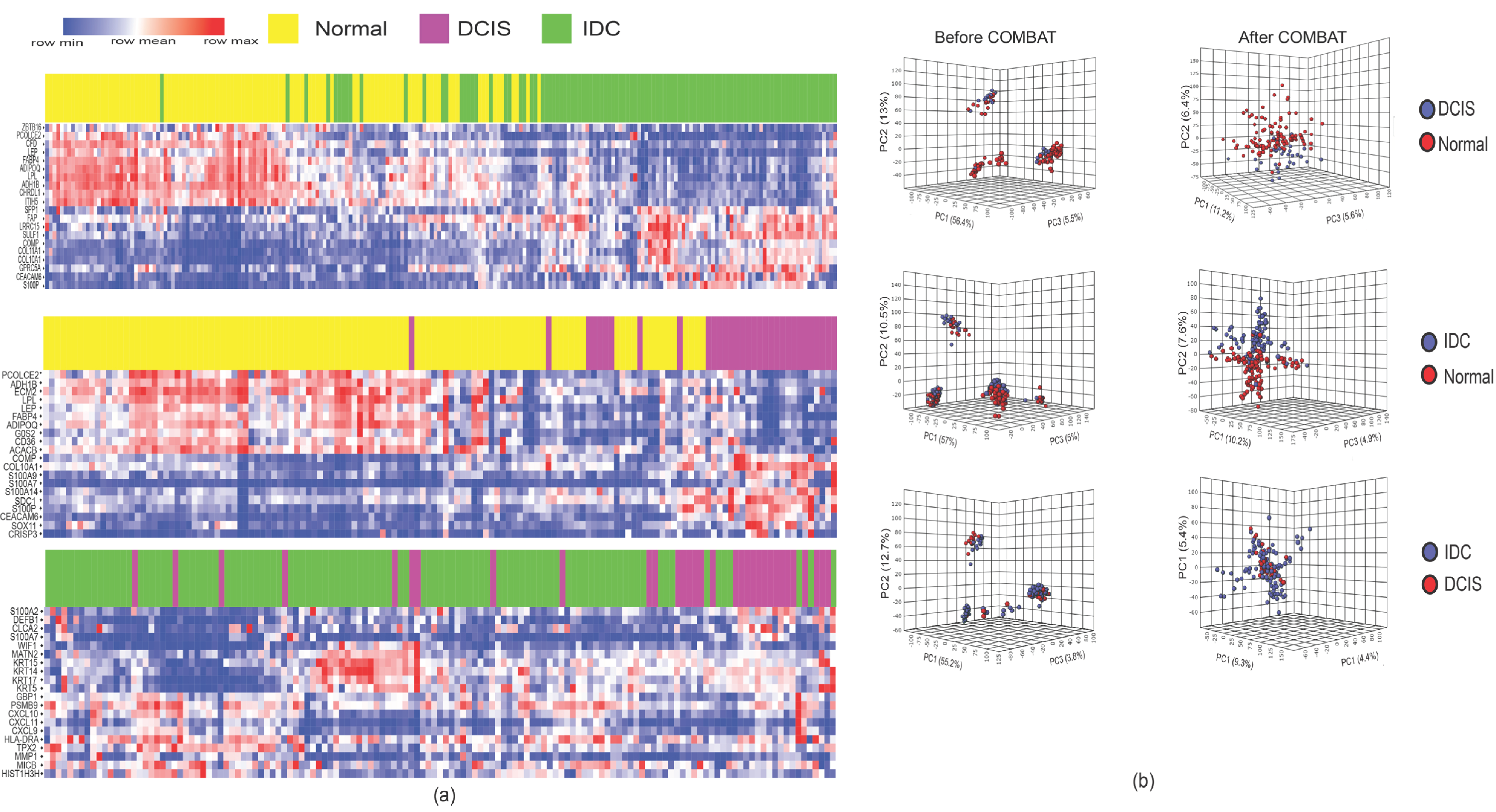
2.1. DCIS and IDC Are Indistinguishable Based on Gene Expression Profiles
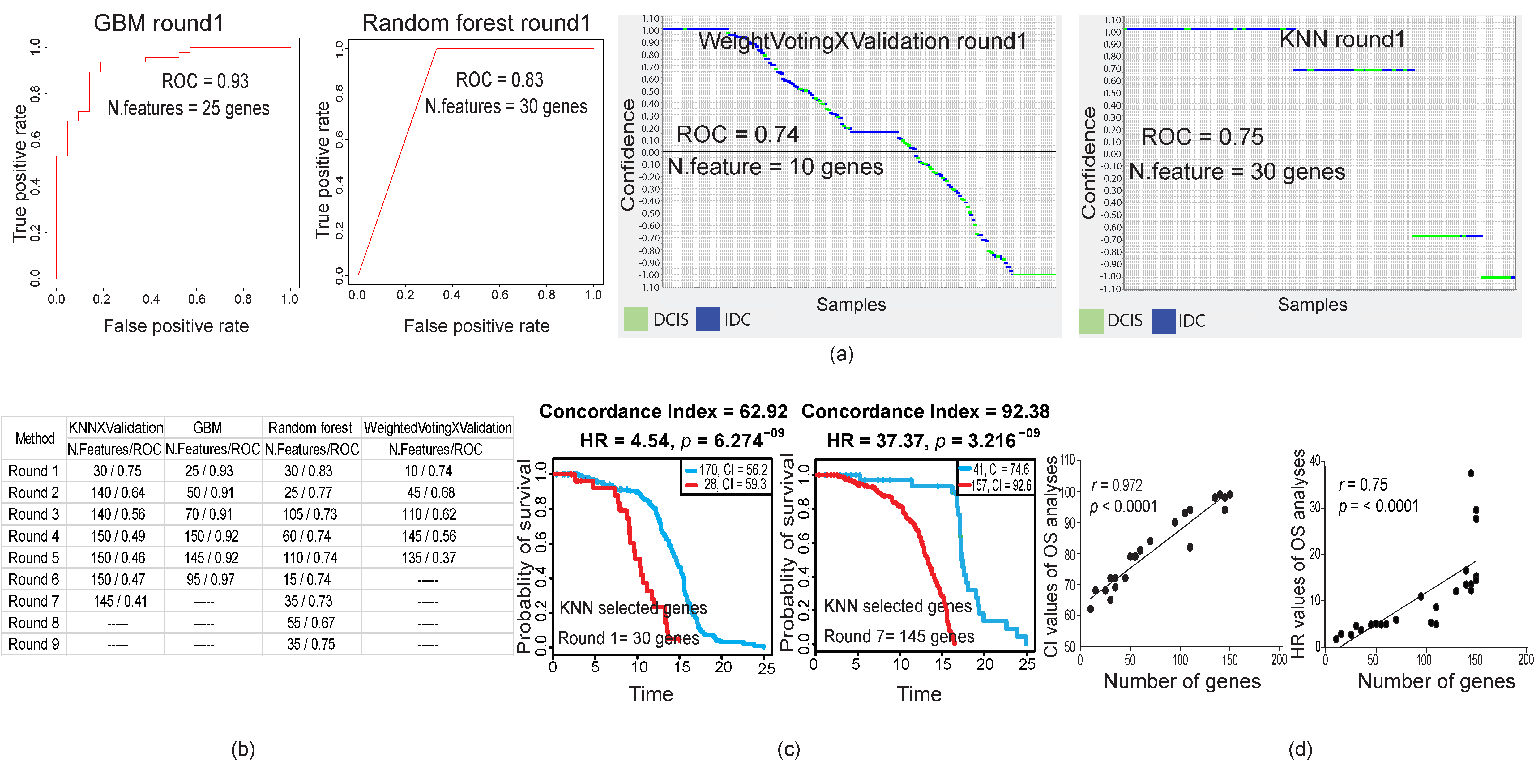
2.2. Stage-Dependent Gene Expression Analyses Did Not Generate a Malignancy-Predictive Feature
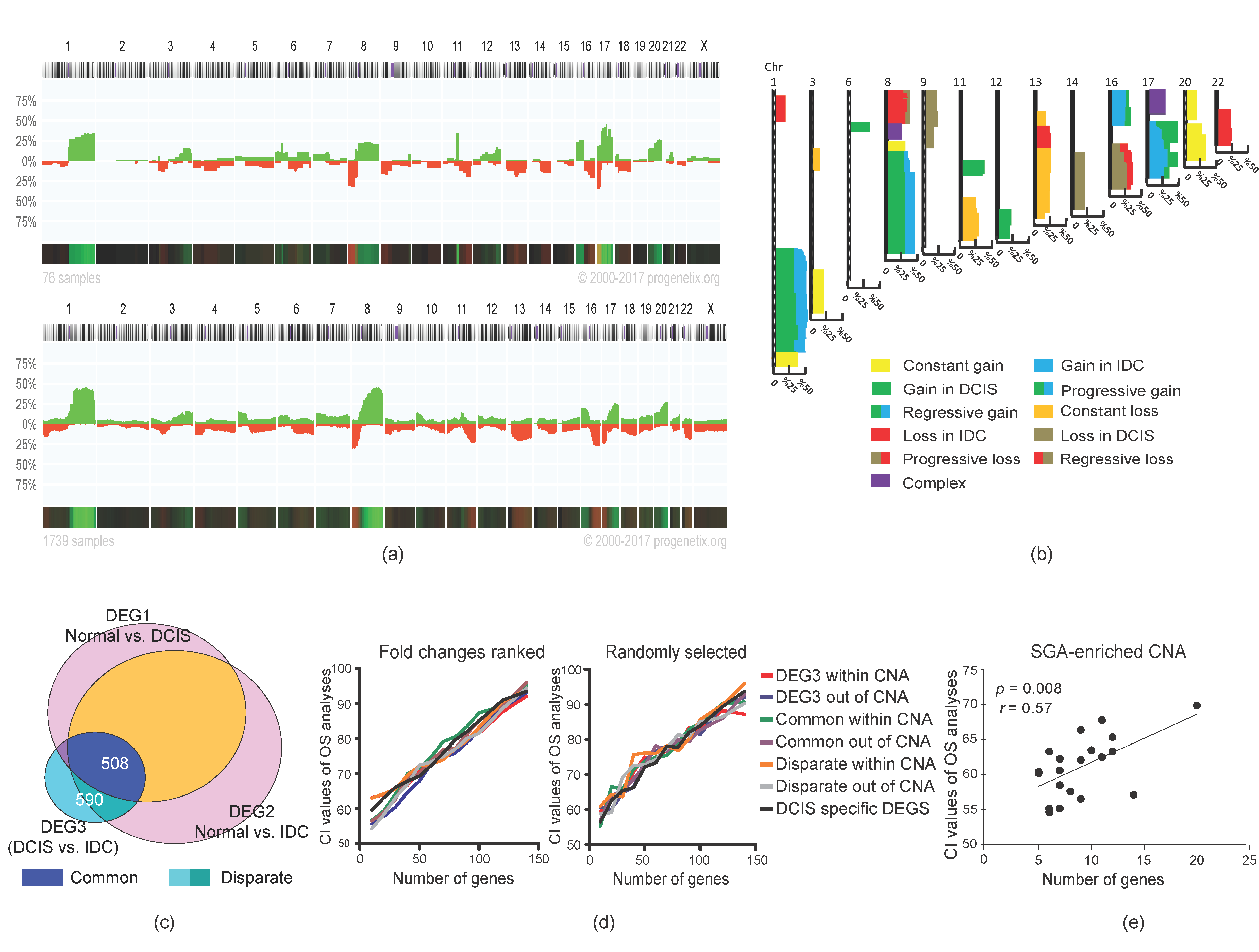
2.3. Stage-Dependent CNA and SGA Integrative Analyses Did Not Generate Malignancy-Predictive Features
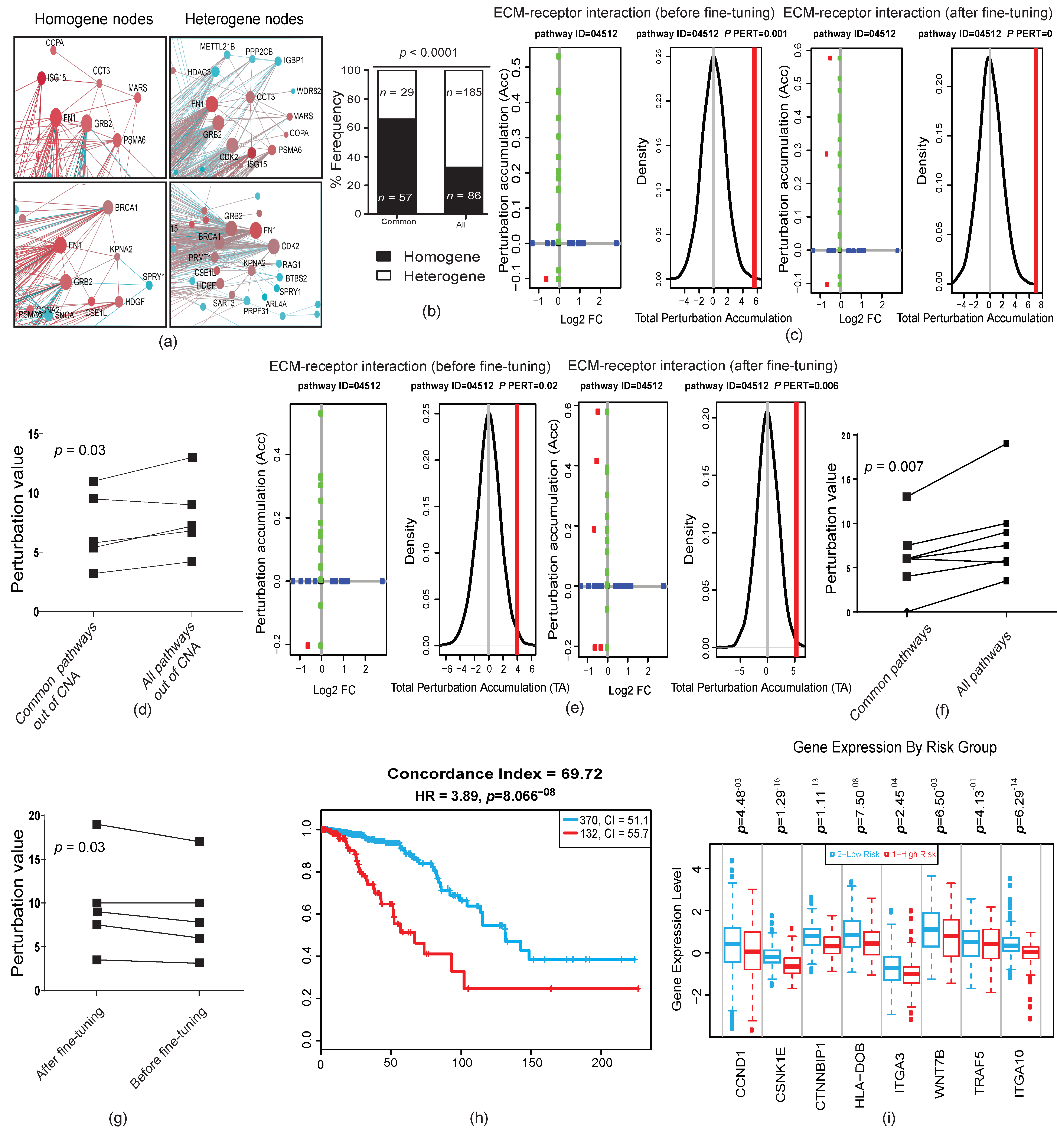
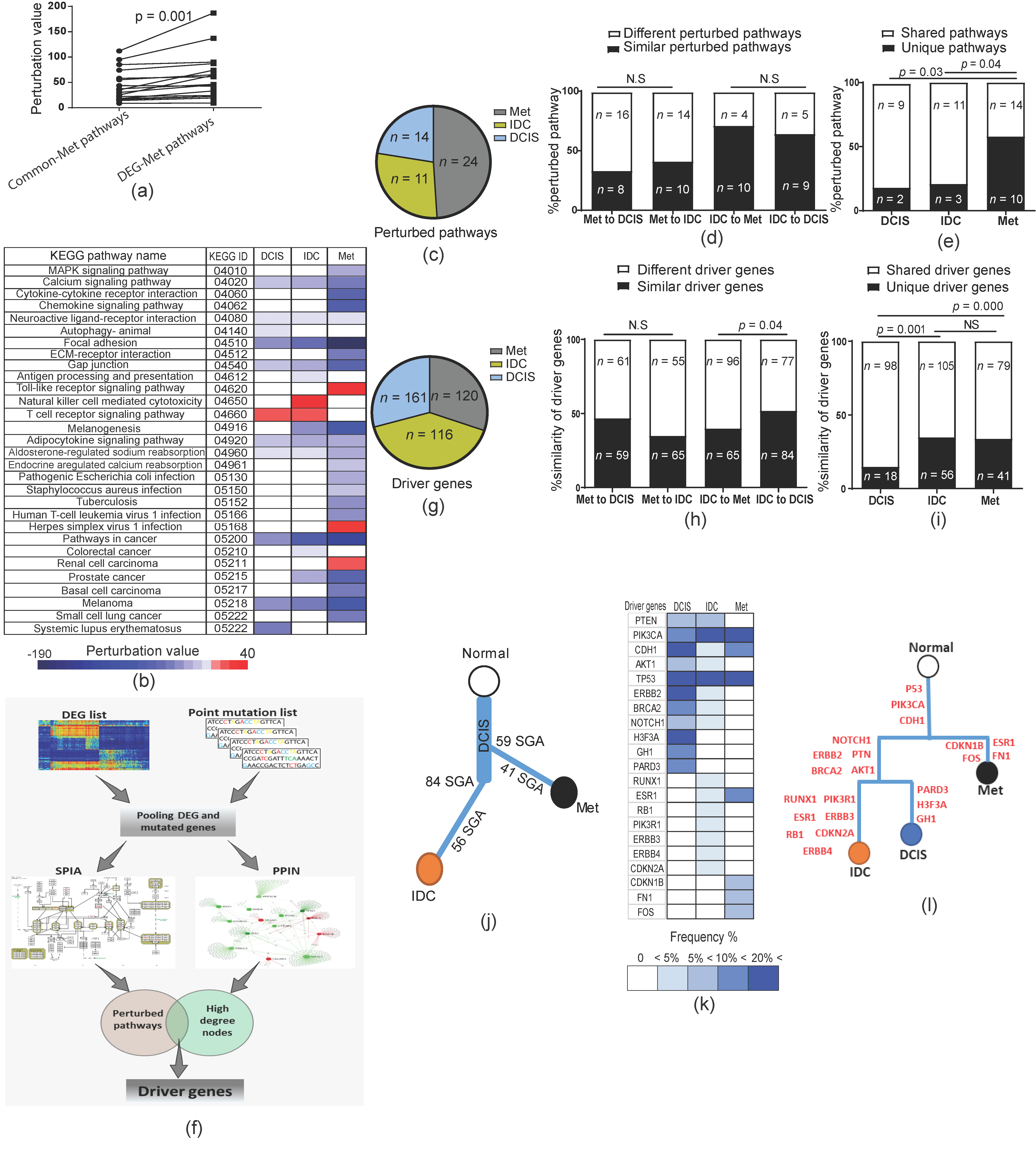
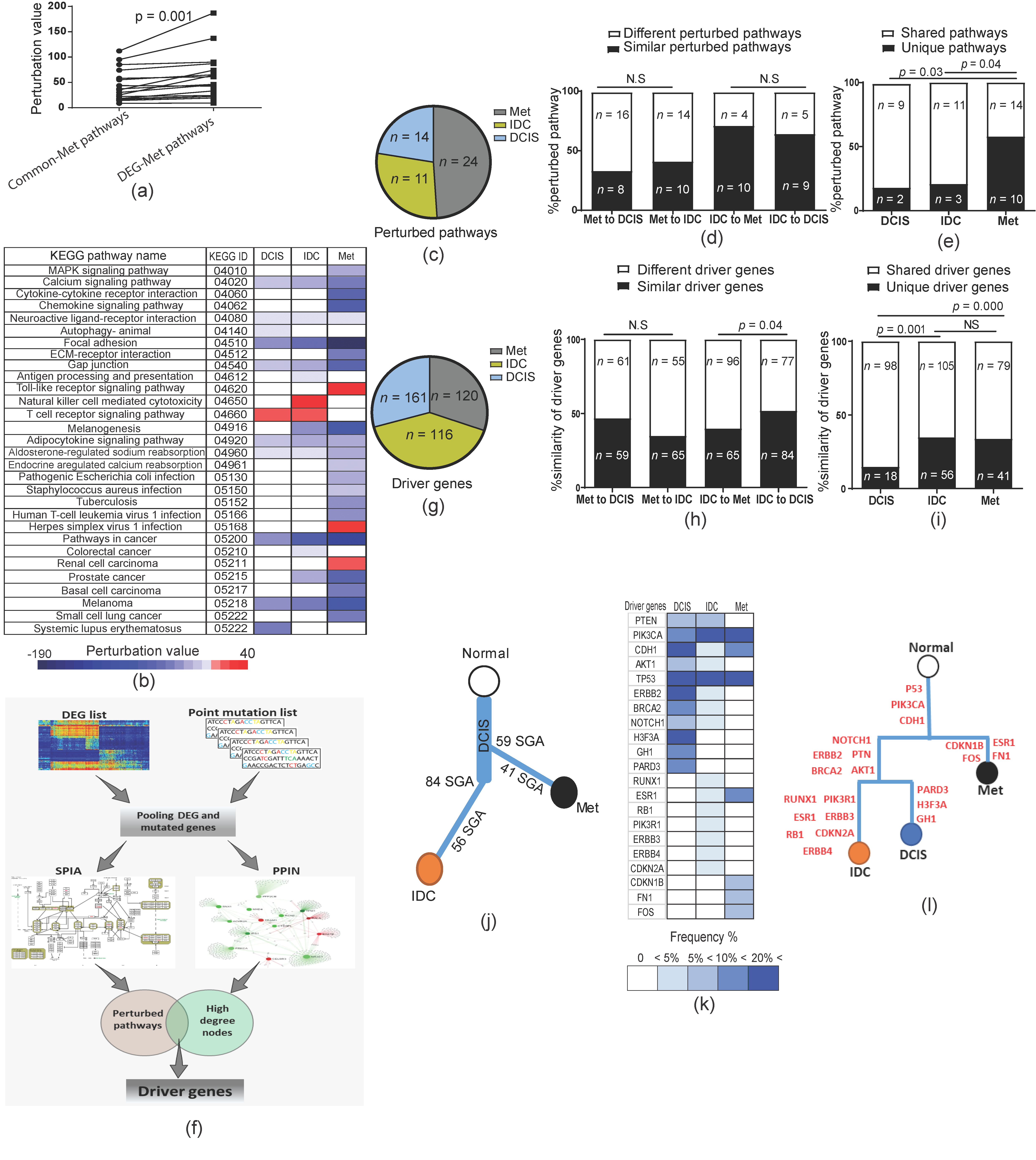
2.4. Genetic Changes in Earlier Tumor Stages Were More Functional Than in Later Stages
2.5. Forward and Backward Evolution Models Were Exploited for the Fine-Tuning of Operating Pathways during Tumor Progression
2.6. Metastases Diverge from Primary Tumor at Early Stages
3. Discussion
4. Materials and Methods
4.1. Data Collection, Preprocessing, and Identification of Differentially Expressed Genes
4.2. (Un)Supervised Clustering and Machine Learning
4.3. Survival Analysis
4.4. Integration of Copy-Number Alteration and Single Gene Alteration
4.5. Network Analysis
4.6. Enrichment Analysis
4.7. NGS Data and Point-Mutation Analysis
4.8. Identification of Driver Genes
4.9. Phylogenetic Analyses of Driver Genes
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Merlo, L.; Pepper, J.W.; Reid, B.J.; Maley, C.C. Cancer as an evolutionary and ecological process. Nat. Rev. Cancer 2006, 6, 924–935. [Google Scholar] [CrossRef] [PubMed]
- Pepper, J.W.; Findlay, C.S.; Kassen, R.; Spencer, S.; Maley, C.C. Cancer research meets evolutionary biology. Evol. Appl. 2009, 2, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Hanada, K.; Okazaki, A.; Hirano, N.; Izumi, Y.; Teraoka, Y.; Ikemoto, J.; Kanemitsu, K.; Hino, F.; Fukuda, T.; Yonehara, S. Diagnostic strategies for early pancreatic cancer. J. Gastroenterol. 2014, 50, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Woodard, G.A.; Jones, K.D.; Jablons, D.M. Lung cancer staging and prognosis. Cancer Treat. Res. 2016, 170, 47–75. [Google Scholar] [CrossRef] [PubMed]
- Kehoe, J.; Khatri, V.P. Staging and prognosis of colon cancer. Surg. Oncol. Clin. N. Am. 2006, 15, 129–146. [Google Scholar] [CrossRef]
- Cheng, L.; Montironi, R.; Bostwick, D.G.; Lopez-Beltran, A.; Berney, D.M. Staging of prostate cancer. Histopathology 2012, 60, 87–117. [Google Scholar] [CrossRef]
- Svedman, F.C.; Pillas, D.; Taylor, A.; Kaur, M.; Linder, R.; Hansson, J. Stage-specific survival and recurrence in patients with cutaneous malignant melanoma in Europe—A systematic review of the literature. Clin. Epidemiol. 2016, 8, 109–122. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.; Li, N.; Sun, Y.; Li, B.; Xu, L.; Wu, L. Clinical outcome and prognostic factors of patients with early-stage epithelial ovarian cancer. Oncotarget 2016, 8, 23862–23870. [Google Scholar] [CrossRef] [Green Version]
- Hudis, C.A. Biology before anatomy in early breast cancer—Precisely the point. N. Engl. J. Med. 2015, 373, 2079–2080. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, H.; Obradović, M.M.S.; Hoffmann, M.; Harper, K.L.; Sosa, M.S.; Werner-Klein, M.; Nanduri, S.L.K.; Werno, C.; Ehrl, C.; Maneck, M.; et al. Early dissemination seeds metastasis in breast cancer. Nature 2016, 540, 552–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.; Ding, J.; Ma, Z.; Sun, R.; Seoane, J.A.; Shaffer, J.S.; Suarez, C.J.; Berghoff, A.S.; Cremolini, C.; Falcone, A.; et al. Quantitative evidence for early metastatic seeding in colorectal cancer. Nat. Genet. 2019, 51, 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M.; Mailey, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M. Evolutionary determinants of cancer. Cancer Discov. 2015, 5, 806–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGranahan, N.; Swanton, C. Clonal heterogeneity and tumor evolution: Past, present, and the future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef]
- Jiang, Y.; Qiu, Y.; Minn, A.J.; Zhang, N.R. Assessing intratumor heterogeneity and tracking longitudinal and spatial clonal evolutionary history by next-generation sequencing. Proc. Natl. Acad. Sci. USA 2016, 113, E5528–E5537. [Google Scholar] [CrossRef] [Green Version]
- Lawson, D.A.; Kessenbrock, K.; Davis, R.T.; Pervolarakis, N.; Werb, Z. Tumour heterogeneity and metastasis at single-cell resolution. Nature 2018, 20, 1349–1360. [Google Scholar] [CrossRef]
- Lipinski, K.A.; Barber, L.; Davies, M.N.; Ashenden, M.; Sottoriva, A.; Gerlinger, M. Cancer evolution and the limits of predictability in precision cancer medicine. Trends Cancer 2016, 2, 49–63. [Google Scholar] [CrossRef] [Green Version]
- Williams, M.J.; Werner, B.; Barnes, C.P.; Graham, T.A.; Sottoriva, A. Identification of neutral tumor evolution across cancer types. Nat. Genet. 2016, 48, 238–244. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Research; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113. [Google Scholar]
- Virnig, B.A.; Tuttle, T.M.; Shamliyan, T.; Kane, R.L. Ductal carcinoma in situ of the breast: A systematic review of incidence, treatment, and outcomes. J. Natl. Cancer Inst. 2010, 102, 170–178. [Google Scholar] [CrossRef] [Green Version]
- Sakorafas, G.H.; Tsiotou, A.G. Ductal carcinoma in situ (DCIS) of the breast: Evolving perspectives. Cancer Treat Rev. 2000, 26, 103–125. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.-Z.; Huang, H.; Wu, C.H.; Jung, M.; Dritschilo, A.; Riegel, A.T.; Wellstein, A. Omics-based molecular target and biomarker identification. Adv. Struct. Saf. Stud. 2011, 719, 547–571. [Google Scholar] [CrossRef] [Green Version]
- Hasin, Y.; Seldin, M.; Lusis, A.J. Multi-omics approaches to disease. Genome Biol. 2017, 18, 83. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Zhou, F.; Ren, J.; Li, X.; Jiang, Y.; Ma, S. A Selective review of multi-level omics data integration using variable selection. High-Throughput 2019, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, I.; Verma, S.; Kumar, S.; Jere, A.; Anamika, K. Multi-omics data integration, interpretation, and its application. Bioinform. Biol. Insights 2020, 14. [Google Scholar] [CrossRef] [Green Version]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef]
- Liao, S.; Desouki, M.M.; Gaile, D.P.; Shepherd, L.; Nowak, N.J.; Conroy, J.; Barry, W.T.; Geradts, J. Differential copy number aberrations in novel candidate genes associated with progression from in situ to invasive ductal carcinoma of the breast. Genes Chromosom. Cancer 2012, 51, 1067–1078. [Google Scholar] [CrossRef] [Green Version]
- Tarca, A.L.; Draghici, S.; Khatri, P.; Hassan, S.S.; Mittal, P.; Kim, J.-S.; Kim, C.J.; Kusanovic, J.P.; Romero, R. A novel signaling pathway impact analysis. Bioinformatics 2008, 25, 75–82. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.P.; Ma, J. DawnRank: Discovering personalized driver genes in cancer. Genome Med. 2014, 6, 56. [Google Scholar] [CrossRef]
- Ling, S.; Hu, Z.; Yang, Z.; Yang, F.; Li, Y.; Lin, P.; Chen, K.; Dong, L.; Cao, L.; Tao, Y.; et al. Extremely high genetic diversity in a single tumor points to prevalence of non-Darwinian cell evolution. Proc. Natl. Acad. Sci. USA 2015, 112, E6496–E6505. [Google Scholar] [CrossRef] [Green Version]
- Pereira, B.; Chin, S.-F.; Rueda, O.M.; Vollan, H.-K.M.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.-J.; et al. The somatic mutation profiles of 2,433 breast cancers refine their genomic and transcriptomic landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, R.; Davis, A.; McDonald, T.O.; Sei, E.; Shi, X.; Wang, Y.; Tsai, P.-C.; Casasent, A.; Waters, J.; Zhang, H.; et al. Punctuated copy number evolution and clonal stasis in triple-negative breast cancer. Nat. Genet. 2016, 48, 1119–1130. [Google Scholar] [CrossRef] [PubMed]
- Visser, L.L.; Elshof, L.E.; Van De Vijver, K.; Groen, E.J.; Almekinders, M.M.; Sanders, J.; Bierman, C.; Peters, D.; Hofland, I.; Broeks, A.; et al. Discordant marker expression between invasive breast carcinoma and corresponding synchronous and preceding DCIS. Am. J. Surg. Pathol. 2019, 43, 1574–1582. [Google Scholar] [CrossRef] [PubMed]
- Miligy, I.M.; Toss, M.S.; Gorringe, K.L.; Lee, A.H.S.; Ellis, I.; Green, A.R.; Rakha, E.A. The clinical and biological significance of HER2 over-expression in breast ductal carcinoma in situ: A large study from a single institution. Br. J. Cancer 2019, 120, 1075–1082. [Google Scholar] [CrossRef]
- Dobrescu, A.; Chang, M.; Kirtani, V.; Turi, G.K.; Hennawy, R.; Hindenburg, A. Study of estrogen receptor and progesterone receptor expression in breast ductal carcinoma in situ by immunohistochemical staining in ER/PgR-negative invasive breast cancer. ISRN Oncol. 2011, 2011, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Harper, K.L.; Sosa, M.S.; Entenberg, D.; Hosseini, H.; Cheung, J.F.; Nobre, R.; Avivar-Valderas, A.; Nagi, C.; Girnius, N.; Davis, R.J.; et al. Mechanism of early dissemination and metastasis in Her2+ mammary cancer. Nature 2016, 540, 588–592. [Google Scholar] [CrossRef]
- Ma, X.-J.; Salunga, R.; Tuggle, J.T.; Gaudet, J.; Enright, E.; McQuary, P.; Payette, T.; Pistone, M.; Stecker, K.; Zhang, B.M.; et al. Gene expression profiles of human breast cancer progression. Proc. Natl. Acad. Sci. USA 2003, 100, 5974–5979. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.-M.; Zhao, B.; Bai, Y.; Iamarino, A.; Gaffney, S.G.; Schlessinger, J.; Lifton, R.P.; Rimm, D.L.; Townsend, J.P. Early and multiple origins of metastatic lineages within primary tumors. Proc. Natl. Acad. Sci. USA 2016, 113, 2140–2145. [Google Scholar] [CrossRef] [Green Version]
- Makohon-Moore, A.P.; Zhang, M.; Reiter, J.G.; Bozic, I.; Allen, B.; Kundu, D.; Chatterjee, K.; Wong, F.; Jiao, Y.; Kohutek, Z.A.; et al. Limited heterogeneity of known driver gene mutations among the metastases of individual patients with pancreatic cancer. Nat. Genet. 2017, 49, 358–366. [Google Scholar] [CrossRef]
- Casasent, A.; Schalck, A.; Gao, R.; Sei, E.; Long, A.; Pangburn, W.; Casasent, T.; Meric-Bernstam, F.; Edgerton, M.E.; Navin, N. Multiclonal invasion in breast tumors identified by topographic single cell sequencing. Cell 2018, 172, 205–217. [Google Scholar] [CrossRef] [Green Version]
- Gautier, L.; Cope, L.; Bolstad, B.M.; Irizarry, R.A. Affy—Analysis of affymetrix genechip data at the probe level. Bioinformatics 2004, 20, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Irizarry, R.; MacDonald, J.; Gentry, J. GCRMA: Background Adjustment Using Sequence Information, R package version 2.38.0. 2014. Available online: https://bioc.ism.ac.jp/packages/3.7/bioc/html/gcrma.html (accessed on 3 June 2020).
- Smyth, G.K. Limma: Linear models for microarray data. In Bioinformatics and Computational Biology Solutions Using R and Bioconductor; Springer: New York, NY, USA, 2005; pp. 397–420. [Google Scholar]
- Xia, J.; E Gill, E.; Hancock, R.E. NetworkAnalyst for statistical, visual and network-based meta-analysis of gene expression data. Nat. Protoc. 2015, 10, 823–844. [Google Scholar] [CrossRef] [PubMed]
- McCall, M.; Jaffee, H.A.; Irizarry, R.A. fRMA ST: Frozen robust multiarray analysis for Affymetrix Exon and Gene ST arrays. Bioinformatics 2012, 28, 3153–3154. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2006, 8, 118–127. [Google Scholar] [CrossRef]
- Reich, M.; Liefeld, T.; Gould, J.; Lerner, J.; Tamayo, P.; Mesirov, J.P. GenePattern 2.0. Nat. Genet. 2006, 38, 500–501. [Google Scholar] [CrossRef]
- Ridgeway, G.; Southworth, M.H.; RUnit, S. Package ‘gbm’. Viitattu 2013, 10, 40. [Google Scholar]
- Liaw, A.; Wiener, M. Classification and regression by randomforest. R News 2002, 2, 18–22. [Google Scholar]
- Korn, J.; Gould, J. KNN Documentation. Available online: https://www.genepattern.org/modules (accessed on 3 June 2020).
- Ross, K. WeightedVotingXValidation Documentation. 2005. Available online: https://www.genepattern.org/modules (accessed on 3 June 2020).
- Aguirre-Gamboa, R.; Gomez-Rueda, H.; Martinez-Ledesma, E.; Martinez-Torteya, A.; Chacolla-Huaringa, R.; Rodriguez-Barrientos, A.; Tamez-Pena, J.; Trevino, V. SurvExpress: An online biomarker validation tool and database for cancer gene expression data using survival analysis. PLoS ONE 2013, 8, e74250. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, S.K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2014, 43, 447–452. [Google Scholar] [CrossRef]
- Breuer, K.; Foroushani, A.K.; Laird, M.; Chen, C.; Sribnaia, A.; Lo, R.; Winsor, G.L.; Hancock, R.E.W.; Brinkman, F.; Lynn, D.J. InnateDB: Systems biology of innate immunity and beyond—Recent updates and continuing curation. Nucleic Acids Res. 2012, 41, D1228–D1233. [Google Scholar] [CrossRef]
- Orchard, S.; Ammari, M.; Aranda, B.; Breuza, L.; Briganti, L.; Broackes-Carter, F.; Campbell, N.; Chavali, G.; Chen, C.; Del-Toro, N.; et al. The MIntAct project—IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2013, 42, 358–363. [Google Scholar] [CrossRef] [Green Version]
- Chatr-Aryamontri, A.; Ceol, A.; Palazzi, L.M.; Nardelli, G.; Schneider, M.V.; Castagnoli, L.; Cesareni, G. MINT: The Molecular INTeraction database. Nucleic Acids Res. 2006, 35, 572–574. [Google Scholar] [CrossRef] [PubMed]
- Xenarios, I. DIP, the database of interacting proteins: A research tool for studying cellular networks of protein interactions. Nucleic Acids Res. 2002, 30, 303–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bader, G.; Donaldson, I.; Wolting, C.; Ouellette, B.F.F.; Pawson, T.; Hogue, C.W. BIND—The biomolecular interaction network database. Nucleic Acids Res. 2001, 29, 242–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatr-Aryamontri, A.; Breitkreutz, B.-J.; Oughtred, R.; Boucher, L.; Heinicke, S.; Chen, D.; Stark, C.; Breitkreutz, A.; Kolas, N.; O’Donnell, L.; et al. The BioGRID interaction database: 2015 update. Nucleic Acids Res. 2014, 43, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Skanderup, A.J.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cbioportal. Sci. Signal. 2013, 6, 1. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Skanderup, A.J.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Akavia, U.D.; Litvin, O.; Kim, J.; Sanchez-Garcia, F.; Kotliar, D.; Causton, H.C.; Pochanard, P.; Mozes, E.; Garraway, L.A.; Pe’Er, D. An integrated approach to uncover drivers of cancer. Cell 2010, 143, 1005–1017. [Google Scholar] [CrossRef] [Green Version]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarhadi, S.; Salehzadeh-Yazdi, A.; Damaghi, M.; Zarghami, N.; Wolkenhauer, O.; Hosseini, H. Omics Integration Analyses Reveal the Early Evolution of Malignancy in Breast Cancer. Cancers 2020, 12, 1460. https://doi.org/10.3390/cancers12061460
Sarhadi S, Salehzadeh-Yazdi A, Damaghi M, Zarghami N, Wolkenhauer O, Hosseini H. Omics Integration Analyses Reveal the Early Evolution of Malignancy in Breast Cancer. Cancers. 2020; 12(6):1460. https://doi.org/10.3390/cancers12061460
Chicago/Turabian StyleSarhadi, Shamim, Ali Salehzadeh-Yazdi, Mehdi Damaghi, Nosratollah Zarghami, Olaf Wolkenhauer, and Hedayatollah Hosseini. 2020. "Omics Integration Analyses Reveal the Early Evolution of Malignancy in Breast Cancer" Cancers 12, no. 6: 1460. https://doi.org/10.3390/cancers12061460
APA StyleSarhadi, S., Salehzadeh-Yazdi, A., Damaghi, M., Zarghami, N., Wolkenhauer, O., & Hosseini, H. (2020). Omics Integration Analyses Reveal the Early Evolution of Malignancy in Breast Cancer. Cancers, 12(6), 1460. https://doi.org/10.3390/cancers12061460