PDX1 DNA Methylation Distinguishes Two Subtypes of Pancreatic Neuroendocrine Neoplasms with a Different Prognosis

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Results
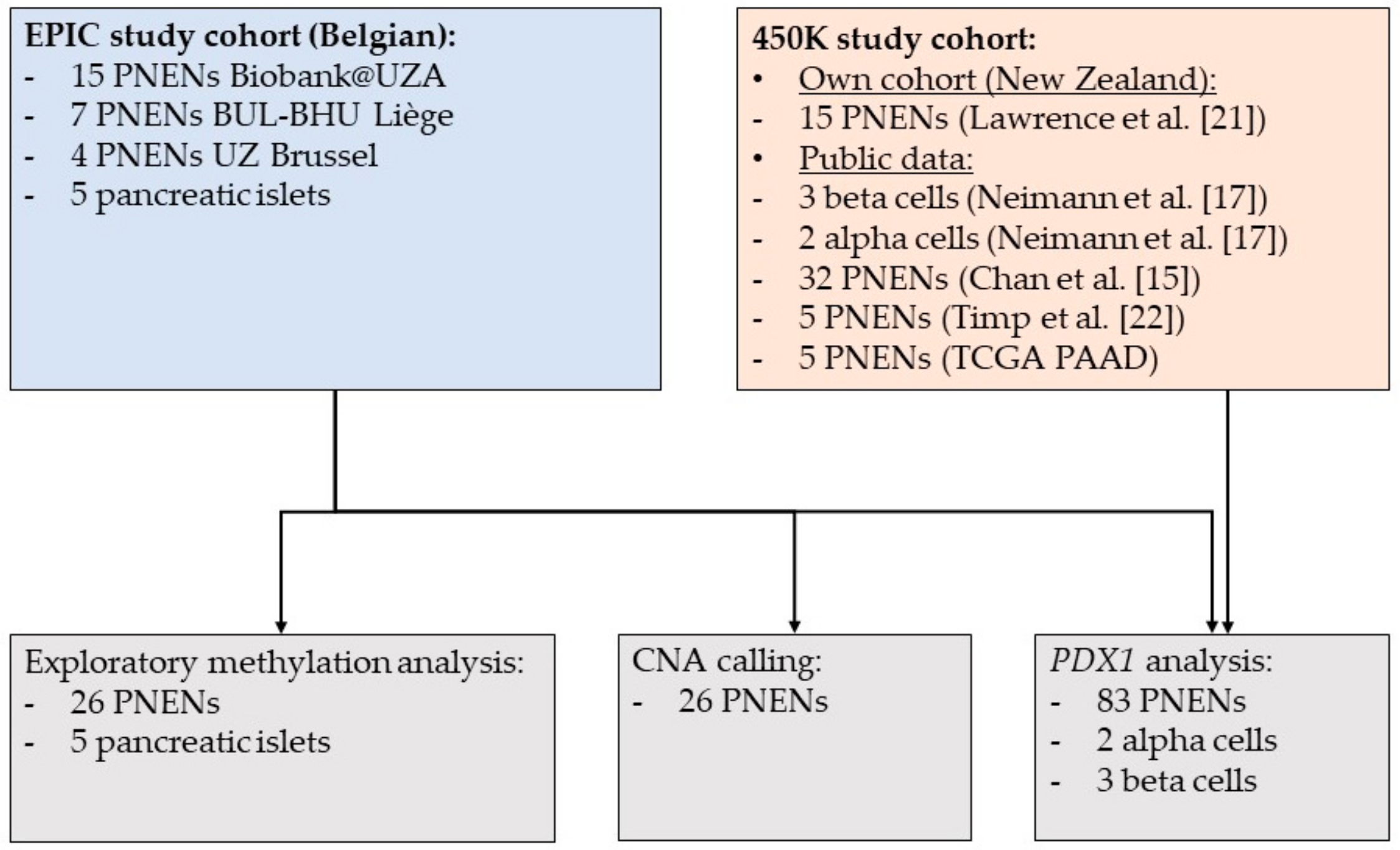
2.1. Patient Characteristics
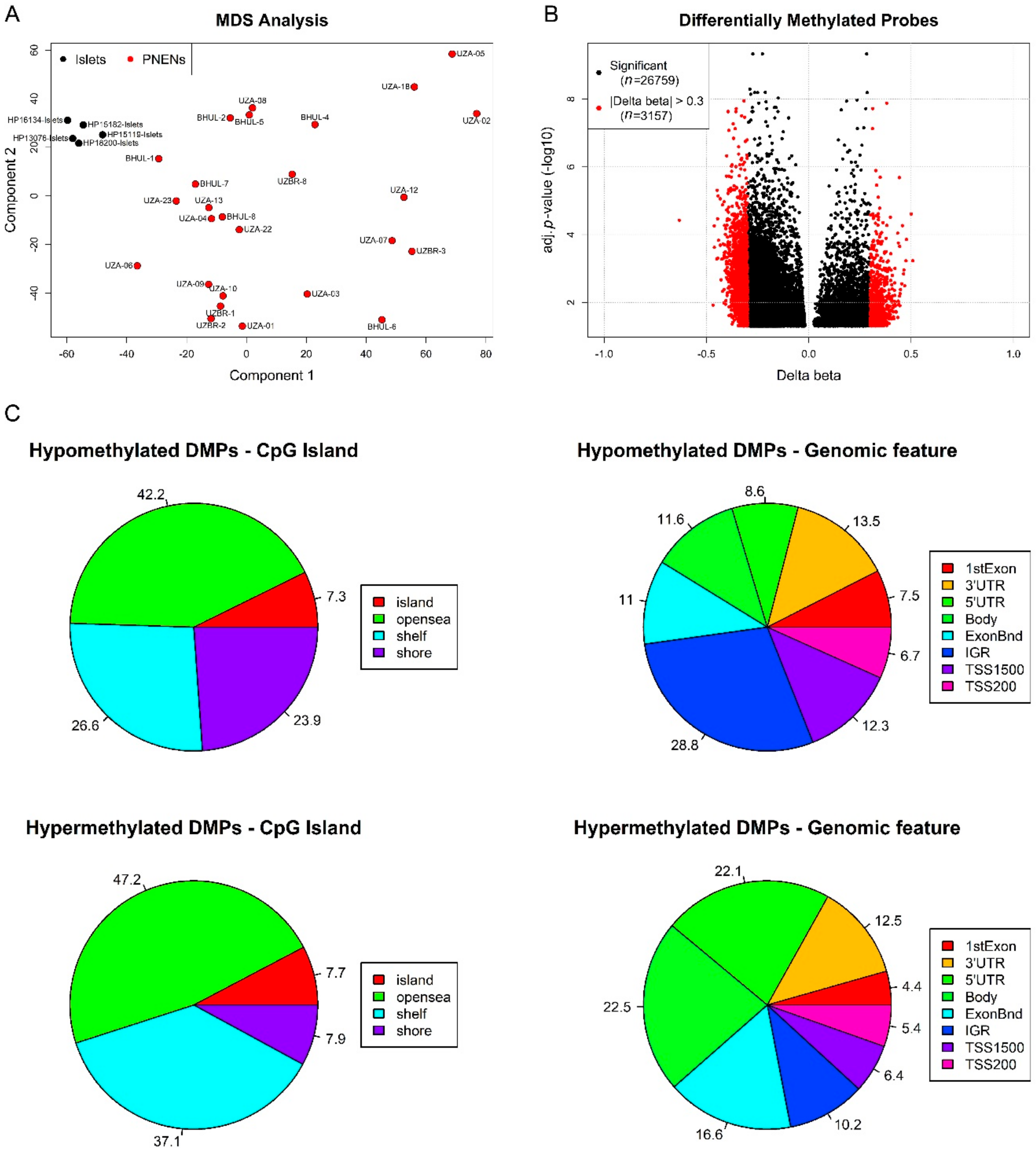
2.2. Exploratory DNA Methylation Analysis of EPIC Study Cohort
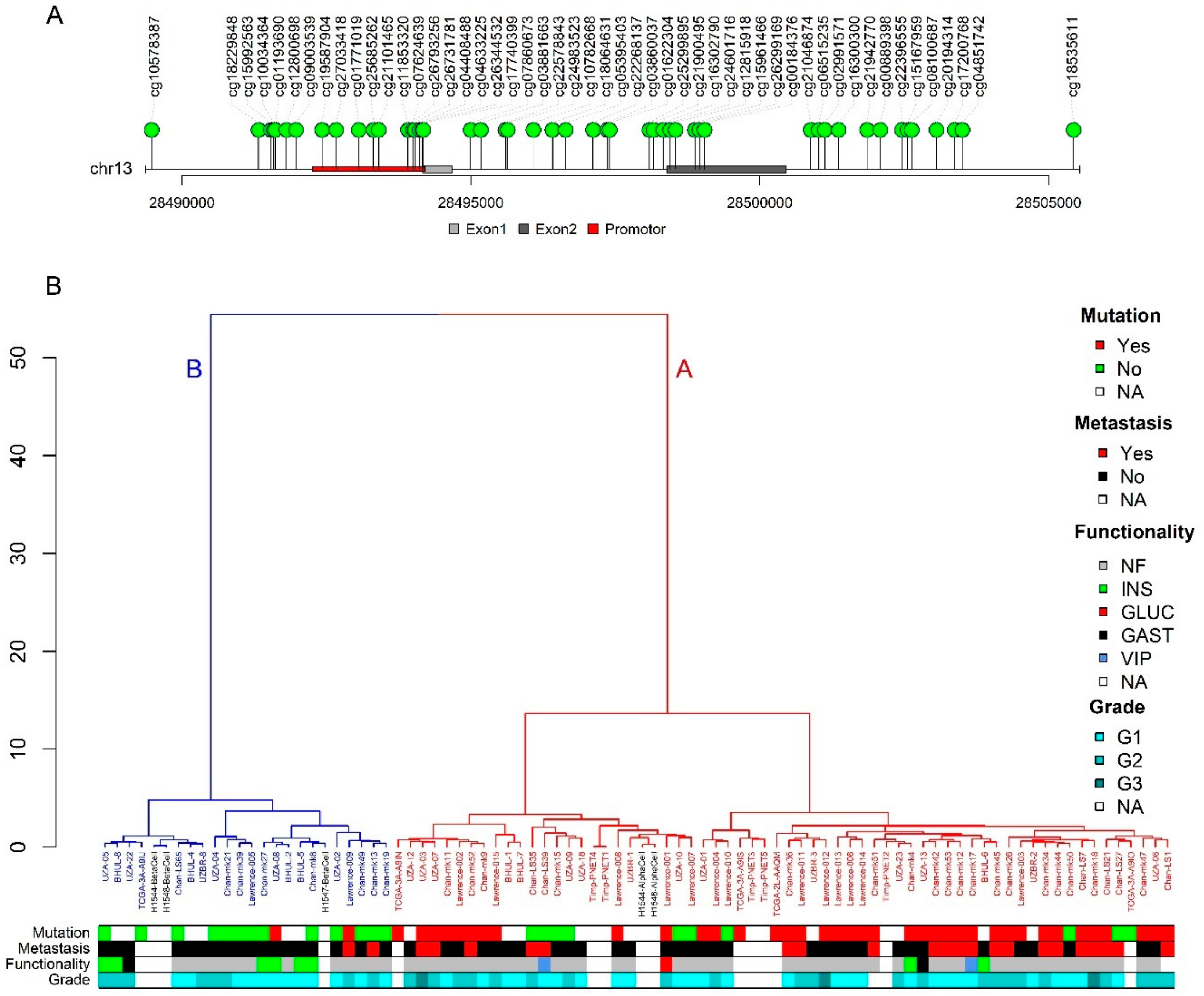
2.3. PDX1 Methylation Status Distinguishes A and B Subtype
2.4. Subtype Associates with Functionality and Metastatic Potential
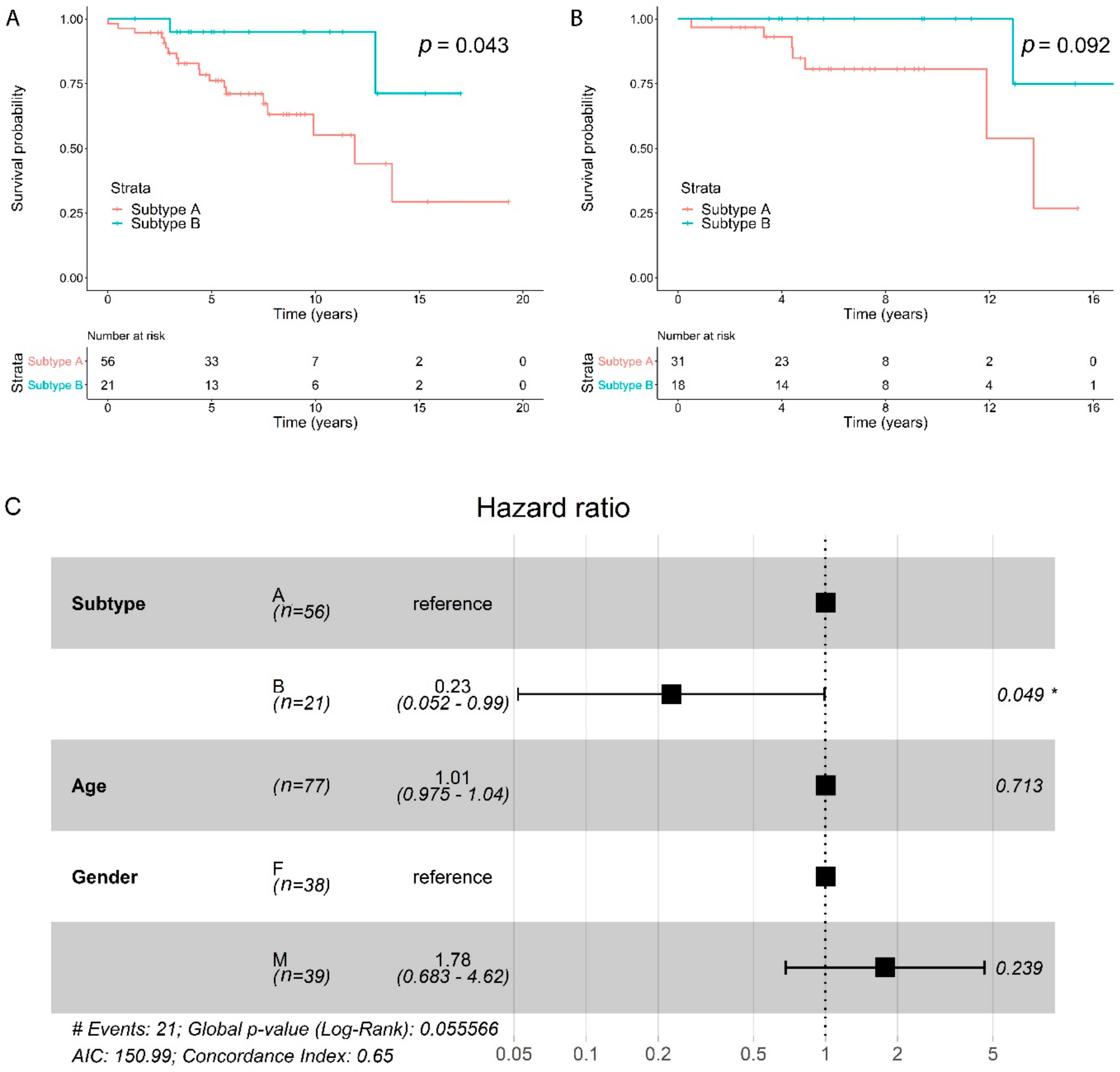
2.5. Subtypes Have a Different Prognosis
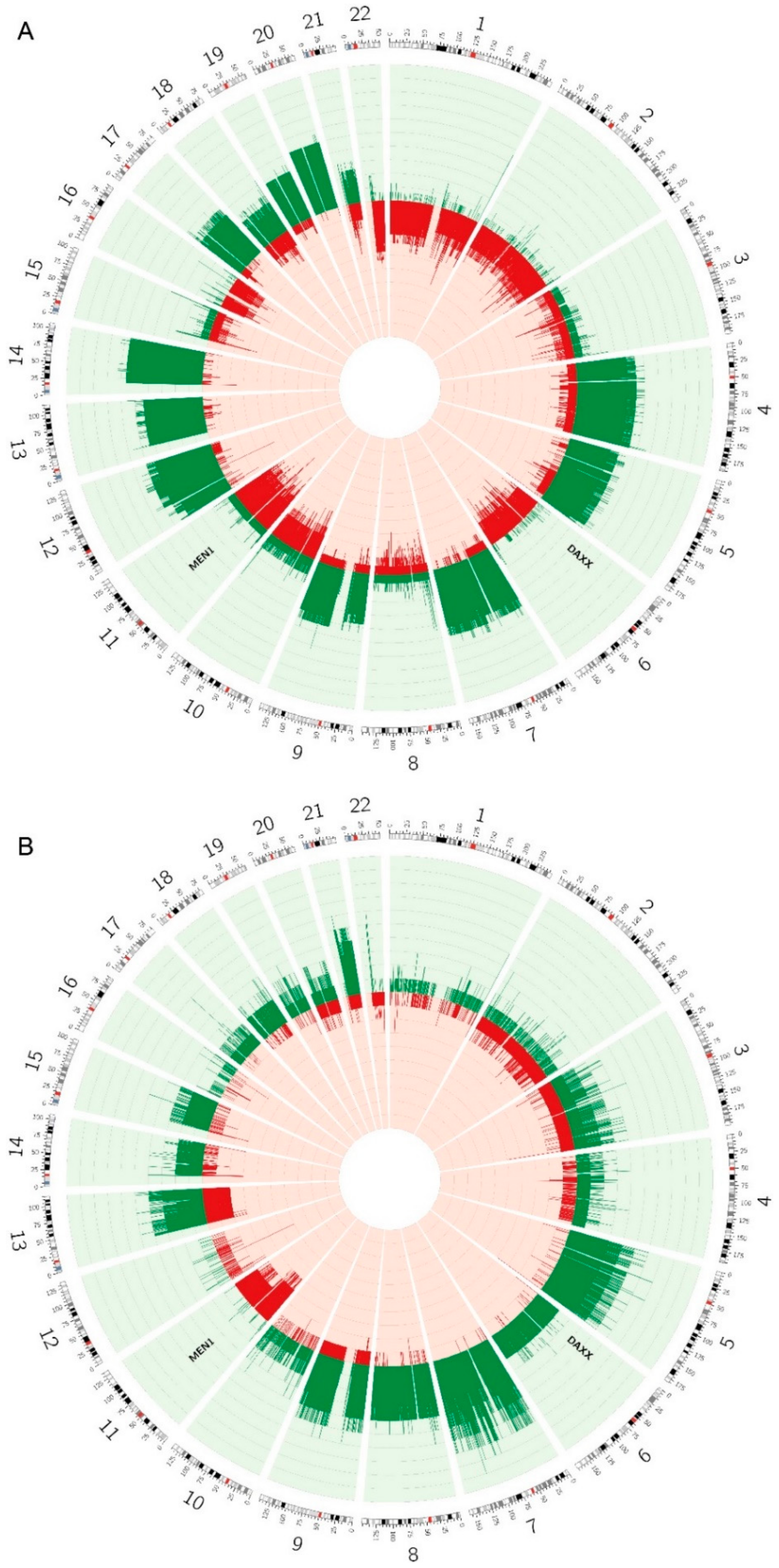
2.6. Copy Number Alterations are Different between the Two Subtypes
3. Discussion
4. Materials and Methods
4.1. Study Population
4.2. Tissue Procurement and Nucleotide Extraction
4.3. Mutation Analysis
4.4. DNA Methylation Analysis
4.5. Additional DNA Methylation Data
4.6. Exploratory DNA Methylation Data Analysis
4.7. PDX1 Data Analysis
4.8. Copy Number Alteration Calling
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- O’Shea, T.; Druce, M. When should genetic testing be performed in patients with neuroendocrine tumours? Rev. Endocr. Metab. Disord. 2017, 18, 499–515. [Google Scholar] [CrossRef] [Green Version]
- Dasari, A.; Shen, C.; Halperin, D.; Zhao, B.; Zhou, S.; Xu, Y.; Shih, T.; Yao, J.C. Trends in the Incidence, Prevalence, and Survival Outcomes in Patients With Neuroendocrine Tumors in the United States. JAMA Oncol. 2017, 3, 1335–1342. [Google Scholar] [CrossRef]
- Lemos, M.C.; Thakker, R.V. Multiple endocrine neoplasia type 1 (MEN1): Analysis of 1336 mutations reported in the first decade following identification of the gene. Hum. Mutat. 2008, 29, 22–32. [Google Scholar] [CrossRef]
- Lonser, R.R.; Glenn, G.M.; Walther, M.; Chew, E.Y.; Libutti, S.K.; Linehan, W.M.; Oldfield, E.H. von Hippel-Lindau disease. Lancet (Lond. Engl.) 2003, 361, 2059–2067. [Google Scholar] [CrossRef]
- Pellegata, N.S.; Quintanilla-Martinez, L.; Siggelkow, H.; Samson, E.; Bink, K.; Hofler, H.; Fend, F.; Graw, J.; Atkinson, M.J. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc. Natl. Acad. Sci. USA 2006, 103, 15558–15563. [Google Scholar] [CrossRef] [Green Version]
- McClatchey, A.I. Neurofibromatosis. Annu. Rev. Pathol. Mech. Dis. 2007, 2, 191–216. [Google Scholar] [CrossRef]
- Dworakowska, D.; Grossman, A.B. Are neuroendocrine tumours a feature of tuberous sclerosis? A systematic review. Endocr.-Relat. Cancer 2009, 16, 45–58. [Google Scholar] [CrossRef] [Green Version]
- Jiao, Y.; Shi, C.; Edil, B.H.; de Wilde, R.F.; Klimstra, D.S.; Maitra, A.; Schulick, R.D.; Tang, L.H.; Wolfgang, C.L.; Choti, M.A.; et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 2011, 331, 1199–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarpa, A.; Chang, D.K.; Nones, K.; Corbo, V.; Patch, A.-M.; Bailey, P.; Lawlor, R.T.; Johns, A.L.; Miller, D.K.; Mafficini, A.; et al. Whole-genome landscape of pancreatic neuroendocrine tumours. Nature 2017, 543, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Mafficini, A.; Scarpa, A. Genetics and Epigenetics of Gastroenteropancreatic Neuroendocrine Neoplasms. Endocr. Rev. 2019. [Google Scholar] [CrossRef] [PubMed]
- Boons, G.; Vandamme, T.; Peeters, M.; Van Camp, G.; Op de Beeck, K. Clinical applications of (epi)genetics in gastroenteropancreatic neuroendocrine neoplasms: Moving towards liquid biopsies. Rev. Endocr. Metab. Disord. 2019, 20, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, A.; Mukherjee, S.; Lack, J.; Gara, S.K.; Wang, S.; Quezado, M.M.; Keutgen, X.M.; Wu, X.; Cam, M.; Kumar, S.; et al. Distinct genome-wide methylation patterns in sporadic and hereditary nonfunctioning pancreatic neuroendocrine tumors. Cancer 2019, 125, 1247–1257. [Google Scholar] [CrossRef] [PubMed]
- Zerbi, A.; Falconi, M.; Rindi, G.; Delle Fave, G.; Tomassetti, P.; Pasquali, C.; Capitanio, V.; Boninsegna, L.; Di Carlo, V. Clinicopathological features of pancreatic endocrine tumors: A prospective multicenter study in Italy of 297 sporadic cases. Am. J. Gastroenterol. 2010, 105, 1421–1429. [Google Scholar] [CrossRef] [PubMed]
- Schimmack, S.; Svejda, B.; Lawrence, B.; Kidd, M.; Modlin, I.M. The diversity and commonalities of gastroenteropancreatic neuroendocrine tumors. Langenbeck’s Arch. Surg. 2011, 396, 273–298. [Google Scholar] [CrossRef]
- Chan, C.S.; Laddha, S.V.; Lewis, P.W.; Koletsky, M.S.; Robzyk, K.; Da Silva, E.; Torres, P.J.; Untch, B.R.; Li, J.; Bose, P.; et al. ATRX, DAXX or MEN1 mutant pancreatic neuroendocrine tumors are a distinct alpha-cell signature subgroup. Nat. Commun. 2018, 9, 4158. [Google Scholar] [CrossRef] [Green Version]
- Cejas, P.; Drier, Y.; Dreijerink, K.M.A.; Brosens, L.A.A.; Deshpande, V.; Epstein, C.B.; Conemans, E.B.; Morsink, F.H.M.; Graham, M.K.; Valk, G.D.; et al. Enhancer signatures stratify and predict outcomes of non-functional pancreatic neuroendocrine tumors. Nat. Med. 2019, 25, 1260–1265. [Google Scholar] [CrossRef]
- Neiman, D.; Moss, J.; Hecht, M.; Magenheim, J.; Piyanzin, S.; Shapiro, A.M.J.; de Koning, E.J.P.; Razin, A.; Cedar, H.; Shemer, R.; et al. Islet cells share promoter hypomethylation independently of expression, but exhibit cell-type-specific methylation in enhancers. Proc. Natl. Acad. Sci. USA 2017, 114, 13525–13530. [Google Scholar] [CrossRef] [Green Version]
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013, 153, 307–319. [Google Scholar] [CrossRef] [Green Version]
- Goethals, S.; De Wilde, A.; Lesage, K.; Smits, E.; Pauwels, P.; Peeters, M. Tumorbank@uza: A Collection of Tissue, Fluid Samples and Associated Data of Oncology Patients for the Use in Translational Research. Open J. Biores. 2018, 5, 4. [Google Scholar] [CrossRef] [Green Version]
- Vande Loock, K.; Van der Stock, E.; Debucquoy, A.; Emmerechts, K.; Van Damme, N.; Marbaix, E. The Belgian Virtual Tumorbank: A Tool for Translational Cancer Research. Front. Med. 2019, 6, 120. [Google Scholar] [CrossRef]
- Lawrence, B.; Blenkiron, C.; Parker, K.; Tsai, P.; Fitzgerald, S.; Shields, P.; Robb, T.; Yeong, M.L.; Kramer, N.; James, S.; et al. Recurrent loss of heterozygosity correlates with clinical outcome in pancreatic neuroendocrine cancer. NPJ Genom. Med. 2018, 3, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timp, W.; Bravo, H.C.; McDonald, O.G.; Goggins, M.; Umbricht, C.; Zeiger, M.; Feinberg, A.P.; Irizarry, R.A. Large hypomethylated blocks as a universal defining epigenetic alteration in human solid tumors. Genome Med. 2014, 6, 61. [Google Scholar] [CrossRef] [PubMed]
- Morris, T.J.; Butcher, L.M.; Feber, A.; Teschendorff, A.E.; Chakravarthy, A.R.; Wojdacz, T.K.; Beck, S. ChAMP: 450k Chip Analysis Methylation Pipeline. Bioinformatics (Oxf. Engl.) 2014, 30, 428–430. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Kuan, P.F. methylGSA: A Bioconductor package and Shiny app for DNA methylation data length bias adjustment in gene set testing. Bioinformatics (Oxf. Engl.) 2018, 35, 1958–1959. [Google Scholar] [CrossRef]
- McLean, C.Y.; Bristor, D.; Hiller, M.; Clarke, S.L.; Schaar, B.T.; Lowe, C.B.; Wenger, A.M.; Bejerano, G. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010, 28, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Hovestadt, V.; Zapatka, M. Conumee: Enhanced Copy-Number Variation Analysis Using Illumina DNA Methylation Arrays; R package version 1.18.0; Bioconductor Open Source Software for Bioinformatics. Available online: http://bioconductor.org/packages/conumee/ (accessed on 14 May 2019).
- Boons, G.; Vandamme, T.; Peeters, M.; Beyens, M.; Driessen, A.; Janssens, K.; Zwaenepoel, K.; Roeyen, G.; Van Camp, G.; Op de Beeck, K. Cell-Free DNA From Metastatic Pancreatic Neuroendocrine Tumor Patients Contains Tumor-Specific Mutations and Copy Number Variations. Front. Oncol. 2018, 8, 467. [Google Scholar] [CrossRef] [Green Version]
- Pipinikas, C.P.; Dibra, H.; Karpathakis, A.; Feber, A.; Novelli, M.; Oukrif, D.; Fusai, G.; Valente, R.; Caplin, M.; Meyer, T.; et al. Epigenetic dysregulation and poorer prognosis in DAXX-deficient pancreatic neuroendocrine tumours. Endocr.-Relat. Cancer 2015, 22, L13–L18. [Google Scholar] [CrossRef] [Green Version]
- Ehrlich, M. DNA hypomethylation in cancer cells. Epigenomics 2009, 1, 239–259. [Google Scholar] [CrossRef] [Green Version]
- Naumov, V.A.; Generozov, E.V.; Zaharjevskaya, N.B.; Matushkina, D.S.; Larin, A.K.; Chernyshov, S.V.; Alekseev, M.V.; Shelygin, Y.A.; Govorun, V.M. Genome-scale analysis of DNA methylation in colorectal cancer using Infinium HumanMethylation450 BeadChips. Epigenetics 2013, 8, 921–934. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Han, H.; De Carvalho, D.D.; Lay, F.D.; Jones, P.A.; Liang, G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell 2014, 26, 577–590. [Google Scholar] [CrossRef]
- Vandamme, T.; Beyens, M.; de Beeck, K.O.; Dogan, F.; van Koetsveld, P.M.; Pauwels, P.; Mortier, G.; Vangestel, C.; de Herder, W.; Van Camp, G.; et al. Long-term acquired everolimus resistance in pancreatic neuroendocrine tumours can be overcome with novel PI3K-AKT-mTOR inhibitors. Br. J. Cancer 2016, 114, 650–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandamme, T.; Beyens, M.; Boons, G.; Schepers, A.; Kamp, K.; Biermann, K.; Pauwels, P.; De Herder, W.W.; Hofland, L.J.; Peeters, M.; et al. Hotspot DAXX, PTCH2 and CYFIP2 mutations in pancreatic neuroendocrine neoplasms. Endocr.-Relat. Cancer 2019, 26, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Ma, L.; Teruya-Feldstein, J.; Rojo, F.; Salmena, L.; Alimonti, A.; Egia, A.; Sasaki, A.T.; Thomas, G.; Kozma, S.C.; et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J. Clin. Investig. 2008, 118, 3065–3074. [Google Scholar] [CrossRef] [PubMed]
- Zitzmann, K.; Rüden, J.V.; Brand, S.; Göke, B.; Lichtl, J.; Spöttl, G.; Auernhammer, C.J. Compensatory activation of Akt in response to mTOR and Raf inhibitors—A rationale for dual-targeted therapy approaches in neuroendocrine tumor disease. Cancer Lett. 2010, 295, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Valentino, J.D.; Li, J.; Zaytseva, Y.Y.; Mustain, W.C.; Elliott, V.A.; Kim, J.T.; Harris, J.W.; Campbell, K.; Weiss, H.; Wang, C.; et al. Cotargeting the PI3K and RAS pathways for the treatment of neuroendocrine tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 1212–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyens, M.; Vandamme, T.; Peeters, M.; Camp, G.V.; Beeck, K.O.d. Resistance to targeted treatment of gastroenteropancreatic neuroendocrine tumors. Endocr.-Relat. Cancer 2019, 26, R109. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.S.; Xu, H.X.; Wang, W.Q.; Li, S.; Li, H.; Li, T.J.; Zhang, W.H.; Liu, L.; Yu, X.J. Tumor-infiltrating platelets predict postoperative recurrence and survival in resectable pancreatic neuroendocrine tumor. World J. Gastroenterol. 2019, 25, 6248–6257. [Google Scholar] [CrossRef]
- Maßberg, D.; Hatt, H. Human Olfactory Receptors: Novel Cellular Functions Outside of the Nose. Physiol. Rev. 2018, 98, 1739–1763. [Google Scholar] [CrossRef]
- Cui, T.; Tsolakis, A.V.; Li, S.C.; Cunningham, J.L.; Lind, T.; Oberg, K.; Giandomenico, V. Olfactory receptor 51E1 protein as a potential novel tissue biomarker for small intestine neuroendocrine carcinomas. Eur. J. Endocrinol. 2013, 168, 253–261. [Google Scholar] [CrossRef] [Green Version]
- Cives, M.; Pelle, E.; Quaresmini, D.; Rizzo, F.M.; Tucci, M.; Silvestris, F. The Tumor Microenvironment in Neuroendocrine Tumors: Biology and Therapeutic Implications. Neuroendocrinology 2019, 109, 83–99. [Google Scholar] [CrossRef]
- Cuny, T.; de Herder, W.; Barlier, A.; Hofland, L.J. Role of the tumor microenvironment in digestive neuroendocrine tumors. Endocr.-Relat. Cancer 2018, 25, R519–R544. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, D.; Kojima, M.; Suzuki, T.; Sugimoto, M.; Kobayashi, S.; Takahashi, S.; Konishi, M.; Gotohda, N.; Ikeda, M.; Nakatsura, T.; et al. Profiling the Tumour Immune Microenvironment in Pancreatic Neuroendocrine Neoplasms with Multispectral Imaging Indicates Distinct Subpopulation Characteristics Concordant with WHO 2017 Classification. Sci. Rep. 2018, 8, 13166. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, M.J.; Subramaniam, P.S.; Tang, L.H.; Grunn, A.; Aburi, M.; Rieckhof, G.; Komissarova, E.V.; Hagan, E.A.; Bodei, L.; Clemons, P.A.; et al. A precision oncology approach to the pharmacological targeting of mechanistic dependencies in neuroendocrine tumors. Nat. Genet. 2018, 50, 979–989. [Google Scholar] [CrossRef] [PubMed]
- Maggio, I.; Manuzzi, L.; Lamberti, G.; Ricci, A.D.; Tober, N.; Campana, D. Landscape and Future Perspectives of Immunotherapy in Neuroendocrine Neoplasia. Cancers 2020, 12. [Google Scholar] [CrossRef] [Green Version]
- Strosberg, J.R.; Mizuno, N.; Doi, T.; Grande, E.; Delord, J.-P.; Shapira-Frommer, R.; Bergsland, E.K.; Shah, M.H.; Fakih, M.; Takahashi, S.; et al. Efficacy and Safety of Pembrolizumab in Previously Treated Advanced Neuroendocrine Tumors: Results From the Phase 2 KEYNOTE-158 Study. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 2018, 362. [Google Scholar] [CrossRef] [Green Version]
- Câmara-de-Souza, A.B.; Toyoshima, M.T.K.; Giannella, M.L.; Freire, D.S.; Camacho, C.P.; Lourenço, D.M.; Rocha, M.S.; Bacchella, T.; Jureidini, R.; Machado, M.C.C.; et al. Insulinoma: A retrospective study analyzing the differences between benign and malignant tumors. Pancreatology 2018, 18, 298–303. [Google Scholar] [CrossRef]
- Cao, Y.; Gao, Z.; Li, L.; Jiang, X.; Shan, A.; Cai, J.; Peng, Y.; Li, Y.; Jiang, X.; Huang, X.; et al. Whole exome sequencing of insulinoma reveals recurrent T372R mutations in YY1. Nat. Commun. 2013, 4, 2810. [Google Scholar] [CrossRef] [Green Version]
- Hackeng, W.M.; Schelhaas, W.; Morsink, F.H.M.; Heidsma, C.M.; van Eeden, S.; Valk, G.D.; Vriens, M.R.; Heaphy, C.M.; Nieveen van Dijkum, E.J.M.; Offerhaus, G.J.A.; et al. Alternative Lengthening of Telomeres and Differential Expression of Endocrine Transcription Factors Distinguish Metastatic and Non-metastatic Insulinomas. Endocr. Pathol. 2020. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Herrera, P.L.; Carreira, C.; Bonnavion, R.; Seigne, C.; Calender, A.; Bertolino, P.; Zhang, C.X. α Cell–Specific Men1 Ablation Triggers the Transdifferentiation of Glucagon-Expressing Cells and Insulinoma Development. Gastroenterology 2010, 138, 1954–1965.e1958. [Google Scholar] [CrossRef]
- Arslan, M.S.; Ozbek, M.; Karakose, M.; Tutal, E.; Ucan, B.; Yilmazer, D.; Dilli, A.; Gultekin, S.S.; Cakal, E.; Delibasi, T. Transformation of nonfunctioning pancreatic tumor into malignant insulinoma after 3 years: An uncommon clinical course of insulinoma. Arch. Endocrinol. Metab. 2015, 59, 270–272. [Google Scholar] [CrossRef] [Green Version]
- Nagano, Y.; Kim, D.H.; Zhang, L.; White, J.A.; Yao, J.C.; Hamilton, S.R.; Rashid, A. Allelic alterations in pancreatic endocrine tumors identified by genome-wide single nucleotide polymorphism analysis. Endocr.-Relat. Cancer 2007, 14, 483–492. [Google Scholar] [CrossRef]
- Zikusoka, M.N.; Kidd, M.; Eick, G.; Latich, I.; Modlin, I.M. The molecular genetics of gastroenteropancreatic neuroendocrine tumors. Cancer Interdisc. Int. J. Am. Cancer Soc. 2005, 104, 2292–2309. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2018, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, T.M.; Shan, Y.S.; Chu, P.Y.; Jiang, S.S.; Hung, W.C.; Chen, Y.L.; Tu, H.C.; Lin, H.Y.; Tsai, H.J.; Chen, L.T. The regulatory role of aberrant Phosphatase and Tensin Homologue and Liver Kinase B1 on AKT/mTOR/c-Myc axis in pancreatic neuroendocrine tumors. Oncotarget 2017, 8, 98068–98083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd RV, O.R.; Klöppel, G.; Rosai, J. WHO Classification of Tumours of Endocrine Organs, 4th ed.; IARC Press: Lyon, France, 2017. [Google Scholar]
- Salgado, R.; Denkert, C.; Demaria, S.; Sirtaine, N.; Klauschen, F.; Pruneri, G.; Wienert, S.; Van den Eynden, G.; Baehner, F.L.; Penault-Llorca, F.; et al. The evaluation of tumor-infiltrating lymphocytes (TILs) in breast cancer: Recommendations by an International TILs Working Group 2014. Ann. Oncol. Off. J. Eur. Soc. Med Oncol. 2015, 26, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics (Oxf. Engl.) 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Laird, P.W.; Shen, H. Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Res 2017, 45, e22. [Google Scholar] [CrossRef]
- Teschendorff, A.E.; Marabita, F.; Lechner, M.; Bartlett, T.; Tegner, J.; Gomez-Cabrero, D.; Beck, S. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics (Oxf. Engl.) 2013, 29, 189–196. [Google Scholar] [CrossRef] [Green Version]
- Smyth, G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3. [Google Scholar] [CrossRef]
- Jaffe, A.E.; Murakami, P.; Lee, H.; Leek, J.T.; Fallin, M.D.; Feinberg, A.P.; Irizarry, R.A. Bump hunting to identify differentially methylated regions in epigenetic epidemiology studies. Int J Epidemiol. 2012, 41. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, R.; Shimodaira, H. Pvclust: An R package for assessing the uncertainty in hierarchical clustering. Bioinformatics (Oxf. Engl.) 2006, 22, 1540–1542. [Google Scholar] [CrossRef] [PubMed]
- Galili, T. Dendextend: An R package for visualizing, adjusting and comparing trees of hierarchical clustering. Bioinformatics (Oxf. Engl.) 2015, 31, 3718–3720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krzywinski, M.I.; Schein, J.E.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinicopathological Parameter | Subtype A | Subtype B | p-Value |
|---|---|---|---|
| Gender | 0.20 | ||
| F | 25 | 13 | |
| M | 32 | 8 | |
| Mutation MEN1/ATRX/DAXX | 7.6E-06 | ||
| No | 10 | 13 | |
| Yes | 38 | 2 | |
| WHO Grade | 0.66 | ||
| G1 | 27 | 12 | |
| G2 | 22 | 8 | |
| G3 | 3 | 0 | |
| Functionality | 0.011 | ||
| Gastrinoma | 1 | 1 | |
| Glucagonoma | 1 | 0 | |
| Insulinoma | 2 | 6 | |
| Non-functional | 46 | 13 | |
| VIPoma | 2 | 0 | |
| LVI | 0.059 | ||
| No | 14 | 8 | |
| Yes | 31 | 5 | |
| Distant metastasis | 0.022 | ||
| No | 32 | 18 | |
| Yes | 21 | 2 | |
| Recurrence | 5.9E-04 | ||
| No | 14 | 16 | |
| Yes | 13 | 0 | |
| Age | 57.1 (±12.1) | 53.1 (±15.0) | 0.29 |
| Tumor size | 4.4 (±2.6) | 3.77 (±3.2) | 0.18 |
| Patient | Gene | Mutation | Allelic Ratio WES | Sanger Validated? |
|---|---|---|---|---|
| UZA-01 | MEN1 | chr11:64572212 CG > C (frameshift) | 100% | Yes |
| UZA-01 | MEN1 | chr11:64571924 G > T (stopgain) | 11% | No |
| UZA-03 | DAXX | chr6:33288573 G > A (stopgain) | 60% | Yes |
| UZA-06 | DAXX | chr6:33289250 G > A (missense) | 8% | No |
| UZA-07 | MEN1 | chr11:64575561 C > A (missense) | 53% | Yes |
| UZA-07 | DAXX | chr6:33287917 C > A (stopgain) | 48% | Yes |
| UZA-08 | ATRX | chrX:76938655 T > C (missense) | 5% | No |
| Lawrence-001 | MEN1 | chr11:64577293 T/TC (frameshift) | / | Yes |
| Lawrence-002 | MEN1 | chr11:64575561 C/CA (frameshift) | / | Yes |
| Lawrence-003 | MEN1 | chr11:64575530 C/T (missense) | / | Yes |
| Lawrence-004 | MEN1 | chr11:64573723 TGTCCGCCCAGGC/T (in-frame deletion) | / | Yes |
| Lawrence-004 | ATRX | chrX:76944420 TCTAGGAGAAAGGA/T (splicing) | / | Yes |
| Lawrence-006 | MEN1 | chr11:64572613 G/GCAA (in-frame insertion) | / | Yes |
| Lawrence-008 | MEN1 | chr11:64573744 CT/C (frameshift) | / | Yes |
| Lawrence-009 | ATRX | chrX:76813035 C/A (stopgain) | / | Yes |
| Lawrence-011 | MEN1 | chr11:64572078 63Nucleotides/G (frameshift) | / | No |
| Lawrence-011 | DAXX | chr6:33287491 33Nucleotides/C (frameshift) | / | Yes |
| Lawrence-012 | MEN1 | chr11:64577329 TAGAC/T (frameshift) | / | Yes |
| Lawrence-012 | ATRX | chrX:76778831 CTACAAT/C (in-frame deletion) | / | Yes |
| Lawrence-013 | MEN1 | chr11:64572643 G/A (stopgain) | / | Yes |
| Lawrence-014 | MEN1 | chr11:64575550 C/A (missense) | / | Yes |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boons, G.; Vandamme, T.; Ibrahim, J.; Roeyen, G.; Driessen, A.; Peeters, D.; Lawrence, B.; Print, C.; Peeters, M.; Van Camp, G.; et al. PDX1 DNA Methylation Distinguishes Two Subtypes of Pancreatic Neuroendocrine Neoplasms with a Different Prognosis. Cancers 2020, 12, 1461. https://doi.org/10.3390/cancers12061461
Boons G, Vandamme T, Ibrahim J, Roeyen G, Driessen A, Peeters D, Lawrence B, Print C, Peeters M, Van Camp G, et al. PDX1 DNA Methylation Distinguishes Two Subtypes of Pancreatic Neuroendocrine Neoplasms with a Different Prognosis. Cancers. 2020; 12(6):1461. https://doi.org/10.3390/cancers12061461
Chicago/Turabian StyleBoons, Gitta, Timon Vandamme, Joe Ibrahim, Geert Roeyen, Ann Driessen, Dieter Peeters, Ben Lawrence, Cristin Print, Marc Peeters, Guy Van Camp, and et al. 2020. "PDX1 DNA Methylation Distinguishes Two Subtypes of Pancreatic Neuroendocrine Neoplasms with a Different Prognosis" Cancers 12, no. 6: 1461. https://doi.org/10.3390/cancers12061461
APA StyleBoons, G., Vandamme, T., Ibrahim, J., Roeyen, G., Driessen, A., Peeters, D., Lawrence, B., Print, C., Peeters, M., Van Camp, G., & Op de Beeck, K. (2020). PDX1 DNA Methylation Distinguishes Two Subtypes of Pancreatic Neuroendocrine Neoplasms with a Different Prognosis. Cancers, 12(6), 1461. https://doi.org/10.3390/cancers12061461