TP53-Deficient Angiosarcoma Expression Profiling in Rat Model


 , ,
, ,
Abstract
:
1. Introduction
2. Results
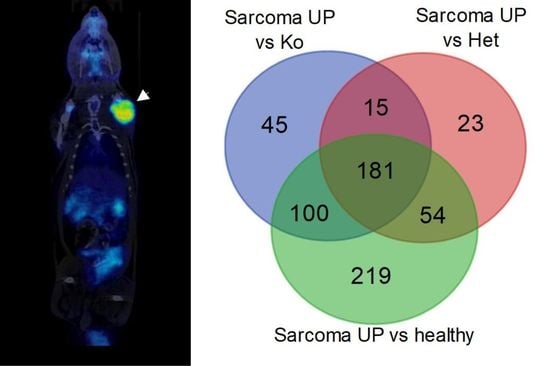
2.1. Tumors Localize in the Head and Neck, Extremities and Abdomen and Are Metabolically Active
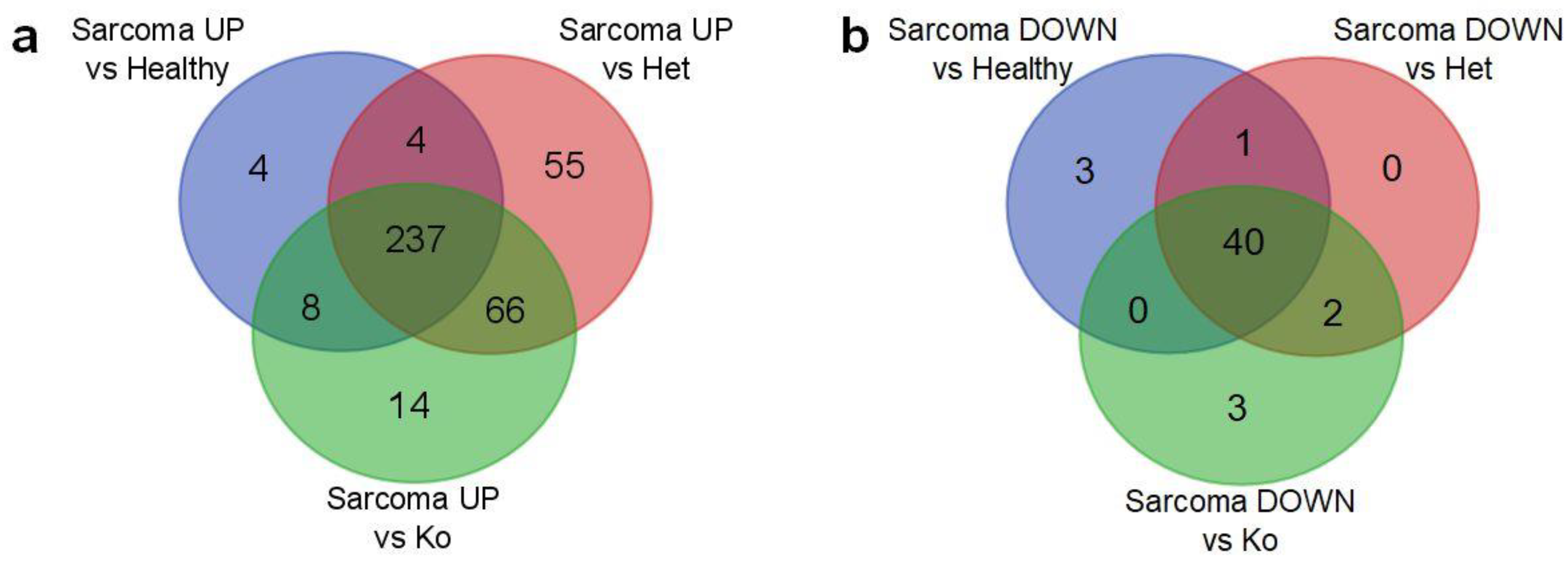
2.2. Angiosarcoma Gene Expression Differ from Normal and Non-Sarcoma Tp53 Knockout Tissues
2.3. Angiosarcoma Presents Deregulated Metabolism
2.4. Activity of p53 Signaling Pathway Is Dysregulated in Tp53 Knockout Tissues
3. Discussion
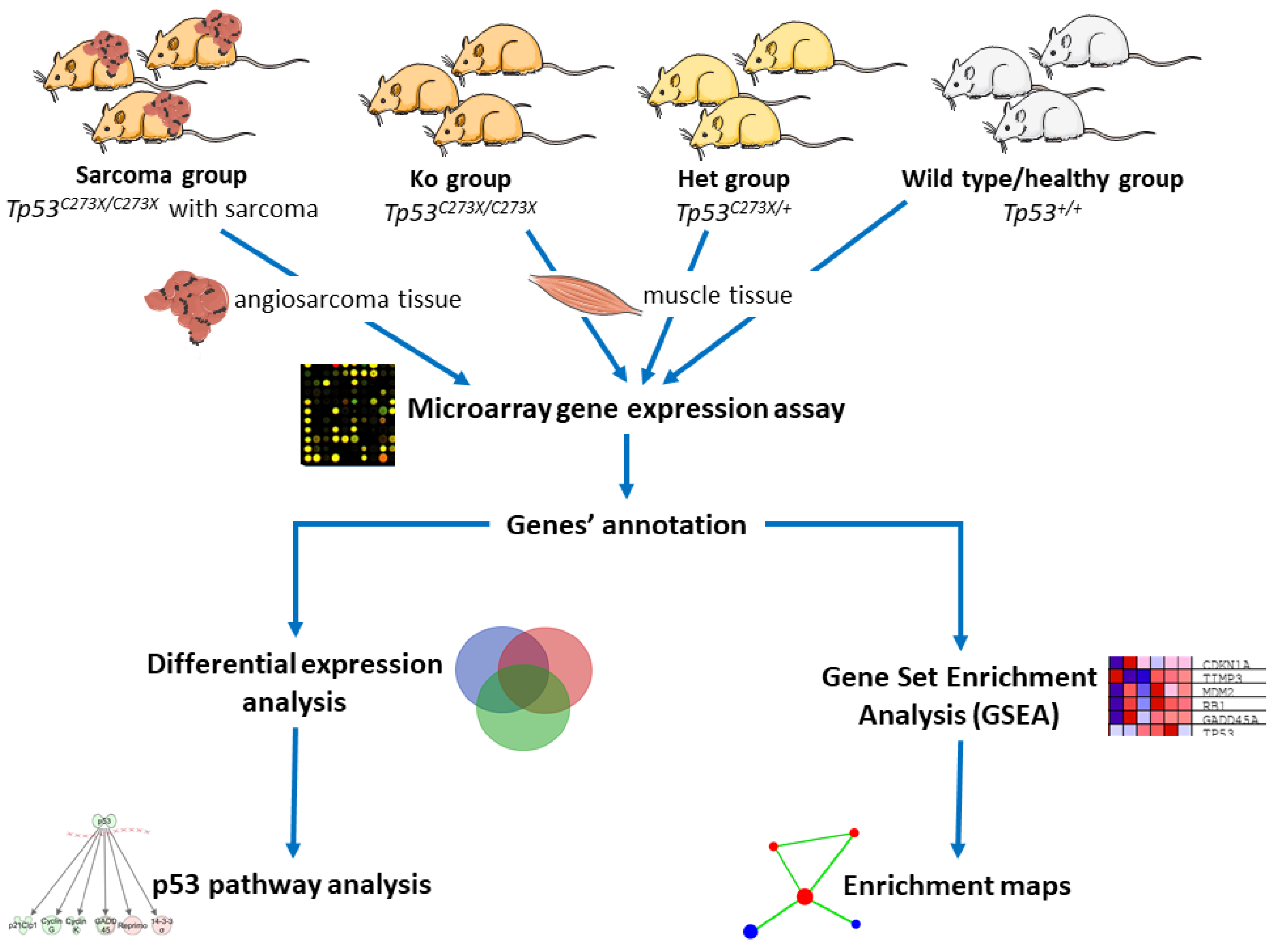
4. Materials and Methods
4.1. Animals
4.2. Magnetic Resonance Imaging
4.3. [18F]FDG PET/CT
4.4. RNA Isolation and Microarray Analysis
4.5. Data Analysis
4.6. CMap Drug Discovery
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yang, J.; Ren, Z.; Du, X.; Hao, M.; Zhou, W. The role of mesenchymal stem/progenitor cells in sarcoma: Update and dispute. Stem Cell Investig. 2014, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- Badalamenti, G.; Messina, C.; De Luca, I.; Musso, E.; Casarin, A.; Incorvaia, L. Soft tissue sarcomas in the precision medicine era: New advances in clinical practice and future perspectives. Radiol. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Casali, P.G.; Abecassis, N.; Aro, H.T.; Bauer, S.; Biagini, R.; Bielack, S.; Bonvalot, S.; Boukovinas, I.; Bovee, J.; Brodowicz, T.; et al. Soft tissue and visceral sarcomas: ESMO-EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv268–iv269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang, N.T.; Acevedo, L.A.; Mann, M.J.; Tolani, B. A review of soft-tissue sarcomas: Translation of biological advances into treatment measures. Cancer Manag. Res. 2018, 10, 1089–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, P.; Seebacher, N.A.; Hornicek, F.; Guo, Z.; Duan, Z. Advances in sarcoma gene mutations and therapeutic targets. Cancer Treat. Rev. 2018, 62, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Kato, S.M.; Kurzrock, R. Molecular Tumor Boards: Realizing Precision Oncology Therapy. Clin. Pharmacol. Ther. 2018, 103, 206–209. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.A.; Hart, M.L.; Busi, S.; Parker, T.; Goerndt, A.; Jones, K.; Amos-Landgraf, J.M.; Bryda, E.C. Fischer-344 Tp53-knockout rats exhibit a high rate of bone and brain neoplasia with frequent metastasis. Dis. Model. Mech. 2016, 9, 1139–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonizzi, G.; Cicalese, A.; Insinga, A.; Pelicci, P.G. The emerging role of p53 in stem cells. Trends Mol. Med. 2012, 18, 6–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Barys, L.; O’Reilly, T.; Young, S.; Gorbatcheva, B.; Monahan, J.; Zumstein-Mecker, S.; Choong, P.F.; Dickinson, I.; Crowe, P.; et al. Comprehensive mapping of p53 pathway alterations reveals an apparent role for both SNP309 and MDM2 amplification in sarcomagenesis. Clin. Cancer Res. 2011, 17, 416–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, G.; Ballinger, M.L.; Wong, S.; Hewitt, C.; James, P.; Young, M.A.; Cipponi, A.; Pang, T.; Goode, D.L.; Dobrovic, A.; et al. High frequency of germline TP53 mutations in a prospective adult-onset sarcoma cohort. PLoS ONE 2013, 8, e69026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consortium, A.P.G. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulligan, L.M.; Matlashewski, G.J.; Scrable, H.J.; Cavenee, W.K. Mechanisms of p53 loss in human sarcomas. Proc. Natl. Acad. Sci. USA 1990, 87, 5863–5867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Boxtel, R.; Kuiper, R.V.; Toonen, P.W.; van Heesch, S.; Hermsen, R.; de Bruin, A.; Cuppen, E. Homozygous and heterozygous p53 knockout rats develop metastasizing sarcomas with high frequency. Am. J. Pathol. 2011, 179, 1616–1622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermsen, R.; Toonen, P.; Kuijk, E.; Youssef, S.A.; Kuiper, R.; van Heesch, S.; de Bruin, A.; Cuppen, E.; Simonis, M. Lack of major genome instability in tumors of p53 null rats. PLoS ONE 2015, 10, e0122066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatachalam, S.; Tyner, S.D.; Pickering, C.R.; Boley, S.; Recio, L.; French, J.E.; Donehower, L.A. Is p53 haploinsufficient for tumor suppression? Implications for the p53+/- mouse model in carcinogenicity testing. Toxicol. Pathol. 2001, 29, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Knudson, A.G., Jr. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, M.; Shirole, N.; Tian, R.; Pal, D.; Sordella, R. The Evolution of TP53 Mutations: From Loss-of-Function to Separation-of-Function Mutants. J. Cancer Biol. Res. 2016, 4, 1091. [Google Scholar] [PubMed]
- Gonin-Laurent, N.; Gibaud, A.; Huygue, M.; Lefevre, S.H.; Le Bras, M.; Chauveinc, L.; Sastre-Garau, X.; Doz, F.; Lumbroso, L.; Chevillard, S.; et al. Specific TP53 mutation pattern in radiation-induced sarcomas. Carcinogenesis 2006, 27, 1266–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomarteli, K.; Fleckenstein, J.; Kirschner, S.; Bobu, V.; Brockmann, M.A.; Henzler, T.; Meyer, M.; Riffel, P.; Schönberg, S.O.; Veldwijk, M.R.; et al. Radiation-induced malignancies after intensity-modulated versus conventional mediastinal radiotherapy in a small animal model. Sci. Rep. 2019, 9, 15489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strzemecki, D.; Guzowska, M.; Grieb, P. Survival rates of homozygotic Tp53 knockout rats as a tool for preclinical assessment of cancer prevention and treatment. Cell. Mol. Biol. Lett. 2017, 22, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- SMART Servier Medical ART. Available online: https://smart.servier.com/ (accessed on 1 April 2020).
- Choi, J.; Curtis, S.J.; Roy, D.M.; Flesken-Nikitin, A.; Nikitin, A.Y. Local mesenchymal stem/progenitor cells are a preferential target for initiation of adult soft tissue sarcomas associated with p53 and Rb deficiency. Am. J. Pathol. 2010, 177, 2645–2658. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, C.; Gass, N.; Weber-Fahr, W.; Sartorius, A. Advantages and challenges of small animal magnetic resonance imaging as a translational tool. Neuropsychobiology 2014, 69, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Tong, C.; Li, P.; Wu, N.L.; Yan, Y.; Ying, Q.L. Production of p53 gene knockout rats by homologous recombination in embryonic stem cells. Nature 2010, 467, 211–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.X.; Wu, H.P.; Ashton, C.; Tong, C.; Ying, Q.L. Rats deficient for p53 are susceptible to spontaneous and carcinogen-induced tumorigenesis. Carcinogenesis 2012, 33, 2001–2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCoy, A.; Besch-Williford, C.L.; Franklin, C.L.; Weinstein, E.J.; Cui, X. Creation and preliminary characterization of a Tp53 knockout rat. Dis. Model. Mech. 2013, 6, 269–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, G.; Ashton, C.; Kumbhani, D.S.; Ying, Q.L. Genetic manipulations in the rat: Progress and prospects. Curr. Opin. Nephrol. Hypertens. 2011, 20, 391–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guijarro, M.V.; Ghivizzani, S.C.; Gibbs, C.P. Animal models in osteosarcoma. Front. Oncol. 2014, 4, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campanile, C.; Arlt, M.J.; Kramer, S.D.; Honer, M.; Gvozdenovic, A.; Brennecke, P.; Fischer, C.R.; Sabile, A.A.; Muller, A.; Ametamey, S.M.; et al. Characterization of different osteosarcoma phenotypes by PET imaging in preclinical animal models. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2013, 54, 1362–1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, J.; Hiniker, S.M.; Lucas, D.R.; Griffith, K.A.; McHugh, J.B.; Meirovitz, A.; Thomas, D.G.; Chugh, R.; Herman, J.M. Sporadic versus Radiation-Associated Angiosarcoma: A Comparative Clinicopathologic and Molecular Analysis of 48 Cases. Sarcoma 2013, 2013, 798403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandt, L.P.; Albers, J.; Hejhal, T.; Pfundstein, S.; Goncalves, A.F.; Catalano, A.; Wild, P.J.; Frew, I.J. Mouse genetic background influences whether Hras(G12V) expression plus Cdkn2a knockdown causes angiosarcoma or undifferentiated pleomorphic sarcoma. Oncotarget 2018, 9, 19753–19766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodd, R.D.; Mito, J.K.; Kirsch, D.G. Animal models of soft-tissue sarcoma. Dis. Model. Mech. 2010, 3, 557–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrand-Rosenberg, S. Animal models of tumor immunity, immunotherapy and cancer vaccines. Curr. Opin. Immunol. 2004, 16, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Olson, B.; Li, Y.; Lin, Y.; Liu, E.T.; Patnaik, A. Mouse Models for Cancer Immunotherapy Research. Cancer Discov. 2018, 8, 1358–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reich, M.; Liefeld, T.; Gould, J.; Lerner, J.; Tamayo, P.; Mesirov, J.P. GenePattern 2.0. Nat. Genet. 2006, 38, 500–501. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Mi, H.; Muruganujan, A.; Thomas, P.D. PANTHER in 2013: Modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013, 41, D377–D386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Antonescu, C. Malignant vascular tumors—An update. Mod. Pathol. 2014, 27 (Suppl. S1), S30–S38. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Wang, J.; He, C.; Fang, M. Angiosarcoma: A review of diagnosis and current treatment. Am. J. Cancer Res. 2019, 9, 2303–2313. [Google Scholar] [PubMed]
- Kurisetty, V.; Bryan, B.A. Aberrations in Angiogenic Signaling and MYC Amplifications are Distinguishing Features of Angiosarcoma. Angiol. Open Access 2013, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171, 1437–1452. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes Upregulated In: | |||||
|---|---|---|---|---|---|
| Healthy | Het | Ko | Sarcoma | ||
| Genes Downregulated In: | Healthy | 0 | 0 | 554 | |
| Het | 0 | 0 | 273 | ||
| Ko | 0 | 0 | 341 | ||
| Sarcoma | 691 | 223 | 358 | ||
| Pathways Upregulated In: | |||||
|---|---|---|---|---|---|
| Healthy | Het | Ko | Sarcoma | ||
| Pathways Downregulated in: | Healthy | 1 | 1 | 253 | |
| Het | 61 | 0 | 362 | ||
| Ko | 5 | 0 | 325 | ||
| Sarcoma | 44 | 43 | 45 | ||
| Compounds Groups | Enrichment Scores |
|---|---|
| topoisomerase inhibitor | −97.84 |
| ellipticine | −98.37 |
| amsacrine | −98.28 |
| mitoxantrone | −97.89 |
| amonafide | −97.85 |
| topotecan | −97.72 |
| teniposide | −96.72 |
| SN-38 | −96.44 |
| irinotecan | −96.11 |
| camptothecin | −95.88 |
| doxorubicin | −95.42 |
| pidorubicine | −95.28 |
| daunorubicin | −95.14 |
| pirarubicin | −94.78 |
| HDAC inhibitor | −95.95 |
| panobinostat | −98.45 |
| scriptaid | −98.31 |
| THM-I-94 | −97.57 |
| vorinostat | −96.79 |
| belinostat | −96.72 |
| apicidin | −95.00 |
| trichostatin-a | −94.85 |
| ISOX | −94.79 |
| HC-toxin | −94.26 |
| dacinostat | −92.65 |
| givinostat | −91.73 |
| CDK inhibitor | −94.38 |
| PHA-793887 | −97.96 |
| JNJ-7706621 | −97.43 |
| AT-7519 | −95.84 |
| aminopurvalanol-a | −93.70 |
| DNA synthesis inhibitor | −92.69 |
| mitomycin-c | −94.37 |
| JAK inhibitor | −92.60 |
| JAK3-inhibitor-VI | −93.52 |
| TG-101348 | −92.57 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smyczyńska, U.; Strzemecki, D.; Czarnecka, A.M.; Fendler, W.; Fiedorowicz, M.; Wełniak-Kamińska, M.; Guzowska, M.; Synoradzki, K.; Cheda, Ł.; Rogulski, Z.; et al. TP53-Deficient Angiosarcoma Expression Profiling in Rat Model. Cancers 2020, 12, 1525. https://doi.org/10.3390/cancers12061525
Smyczyńska U, Strzemecki D, Czarnecka AM, Fendler W, Fiedorowicz M, Wełniak-Kamińska M, Guzowska M, Synoradzki K, Cheda Ł, Rogulski Z, et al. TP53-Deficient Angiosarcoma Expression Profiling in Rat Model. Cancers. 2020; 12(6):1525. https://doi.org/10.3390/cancers12061525
Chicago/Turabian StyleSmyczyńska, Urszula, Damian Strzemecki, Anna M. Czarnecka, Wojciech Fendler, Michał Fiedorowicz, Marlena Wełniak-Kamińska, Magdalena Guzowska, Kamil Synoradzki, Łukasz Cheda, Zbigniew Rogulski, and et al. 2020. "TP53-Deficient Angiosarcoma Expression Profiling in Rat Model" Cancers 12, no. 6: 1525. https://doi.org/10.3390/cancers12061525
APA StyleSmyczyńska, U., Strzemecki, D., Czarnecka, A. M., Fendler, W., Fiedorowicz, M., Wełniak-Kamińska, M., Guzowska, M., Synoradzki, K., Cheda, Ł., Rogulski, Z., & Grieb, P. (2020). TP53-Deficient Angiosarcoma Expression Profiling in Rat Model. Cancers, 12(6), 1525. https://doi.org/10.3390/cancers12061525