Quantitative Analysis of Differential Expression of HOX Genes in Multiple Cancers

Abstract
:1. Introduction
2. Results
2.1. Gene Expression Data Obtained from TCGA and GTEx Using Xena are Comparable
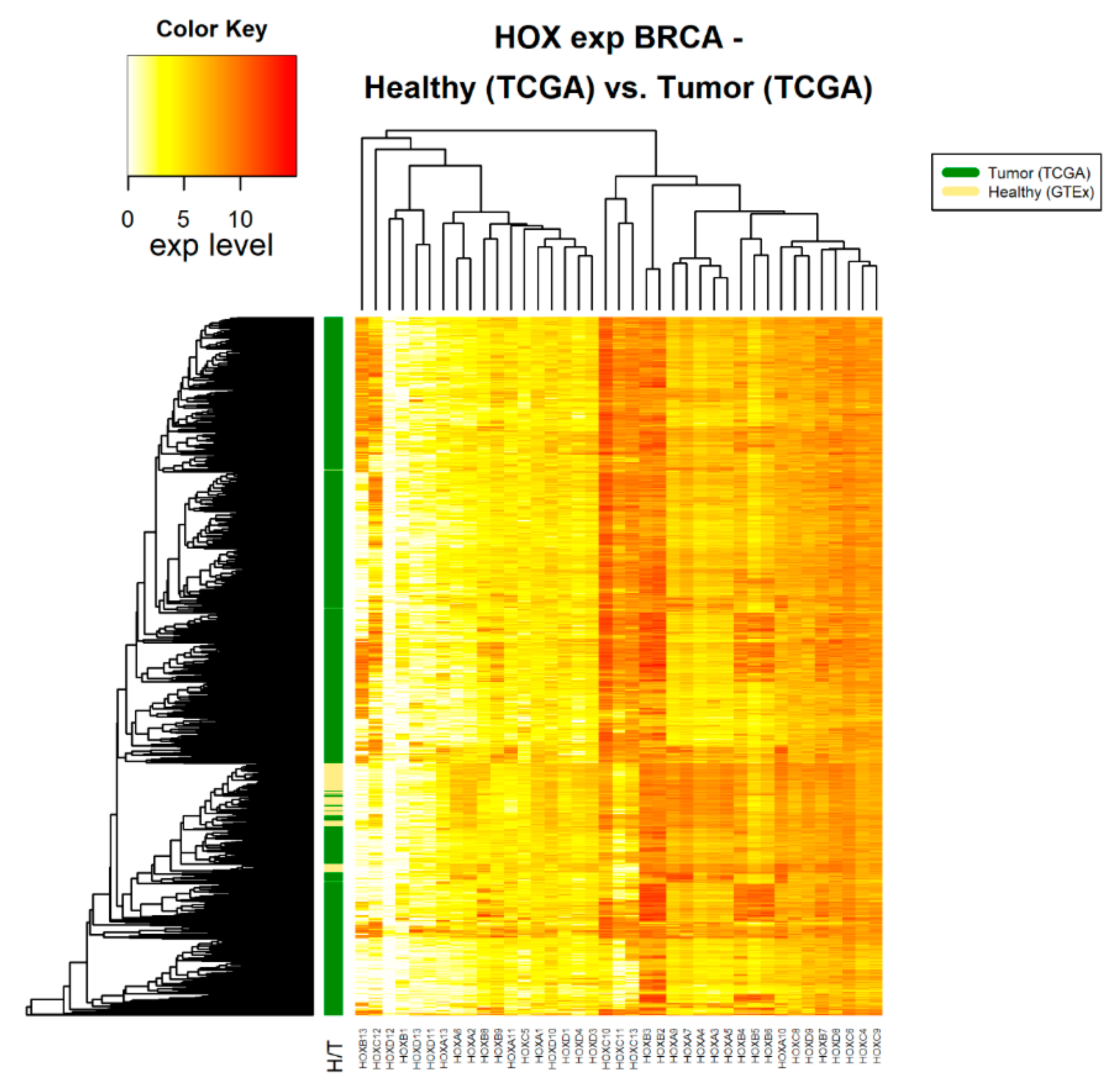
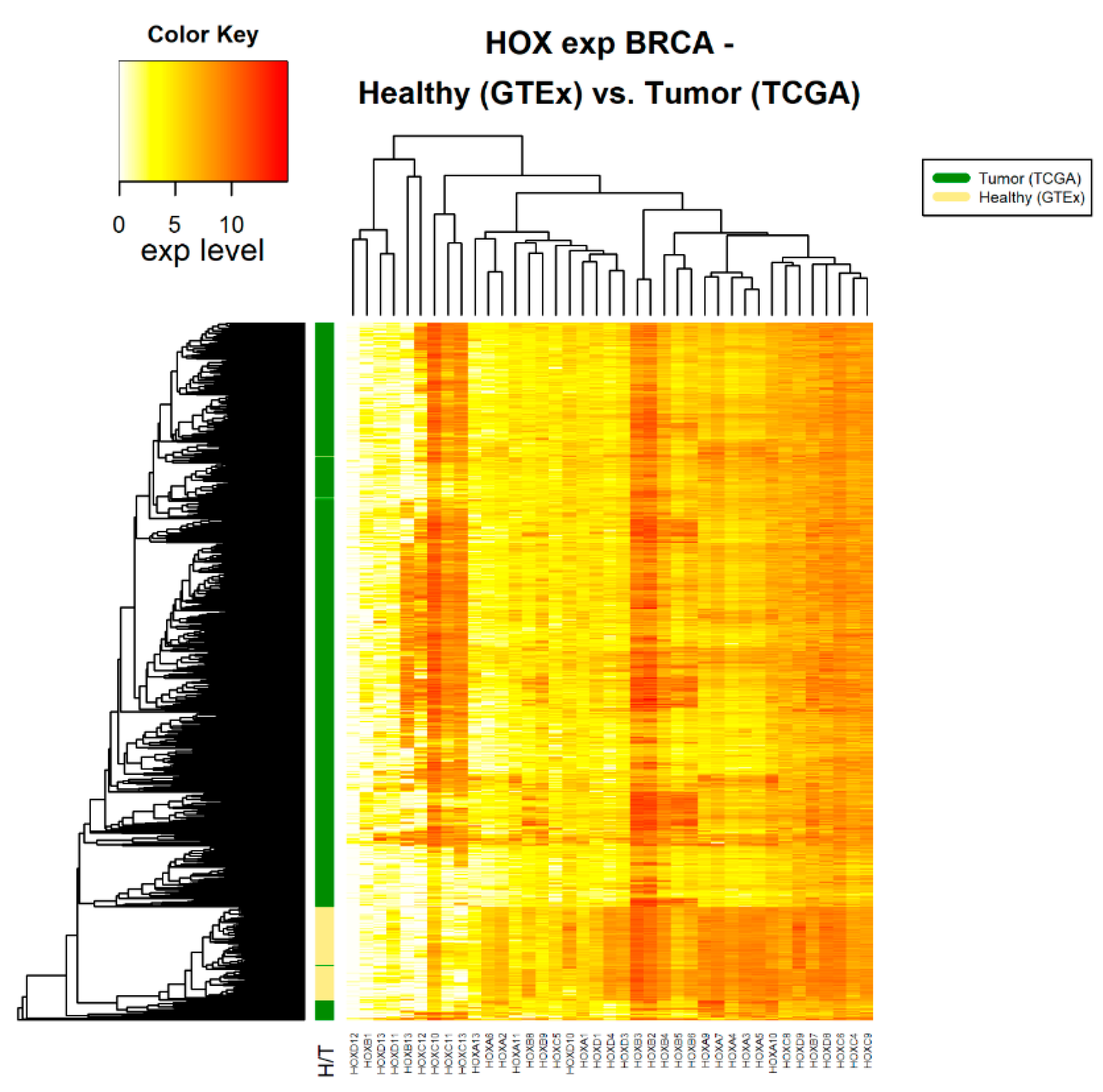
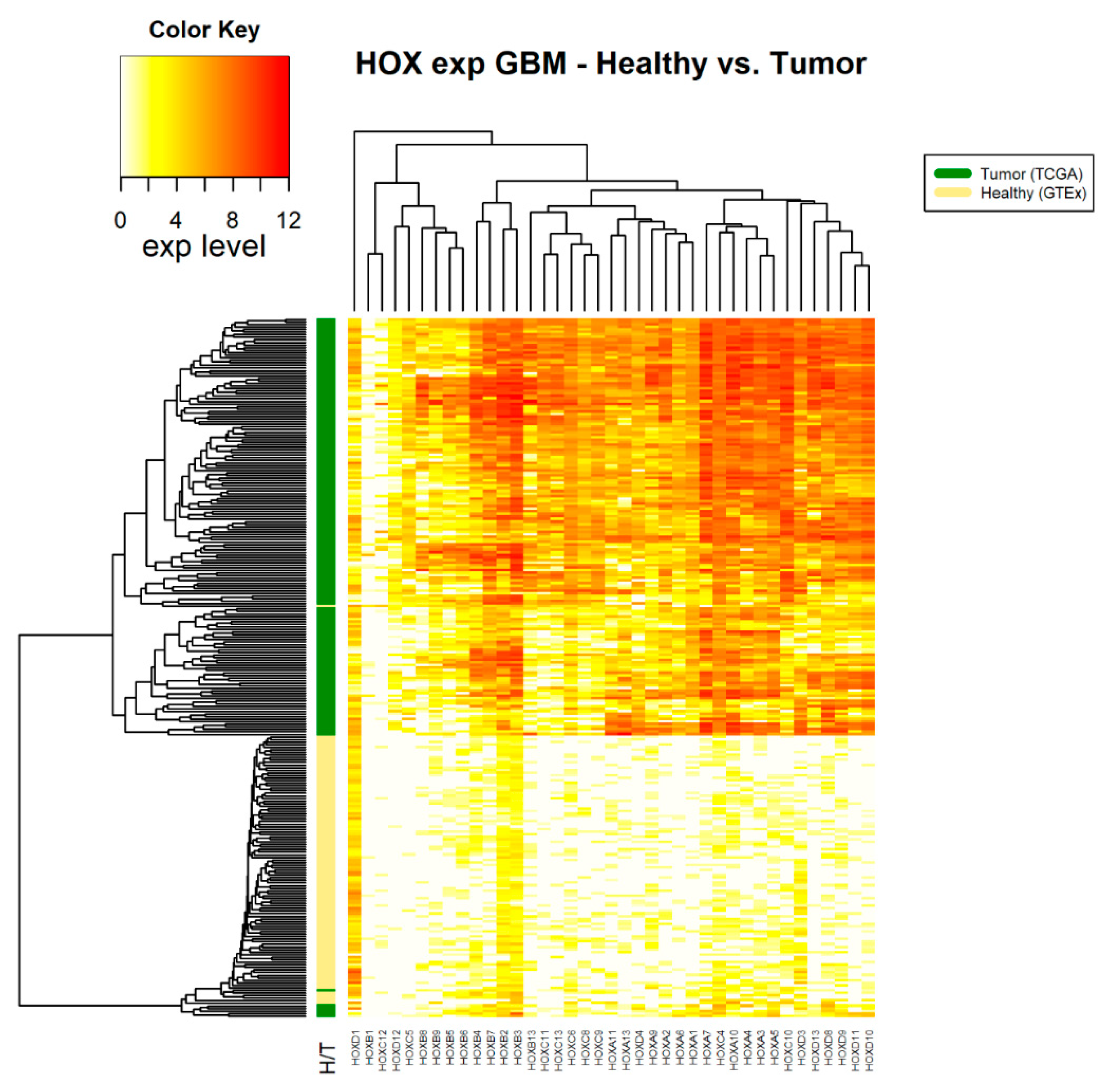
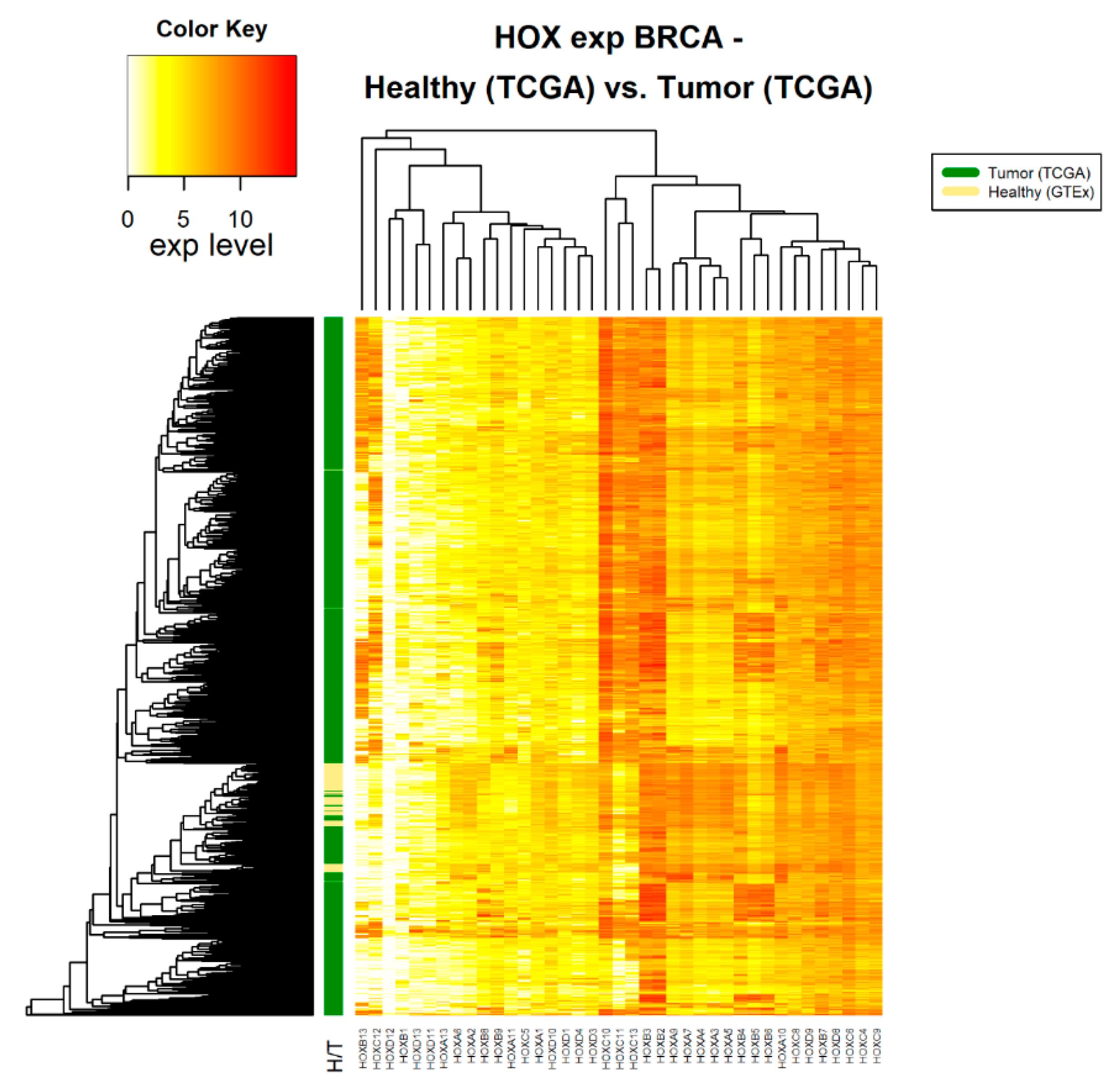
2.2. Differential Expression of HOX Gene Clusters Can Discriminate Between Tumor Samples and Healthy Samples
2.3. Differential Expression of HOX Genes in 14 Different Cancer Types
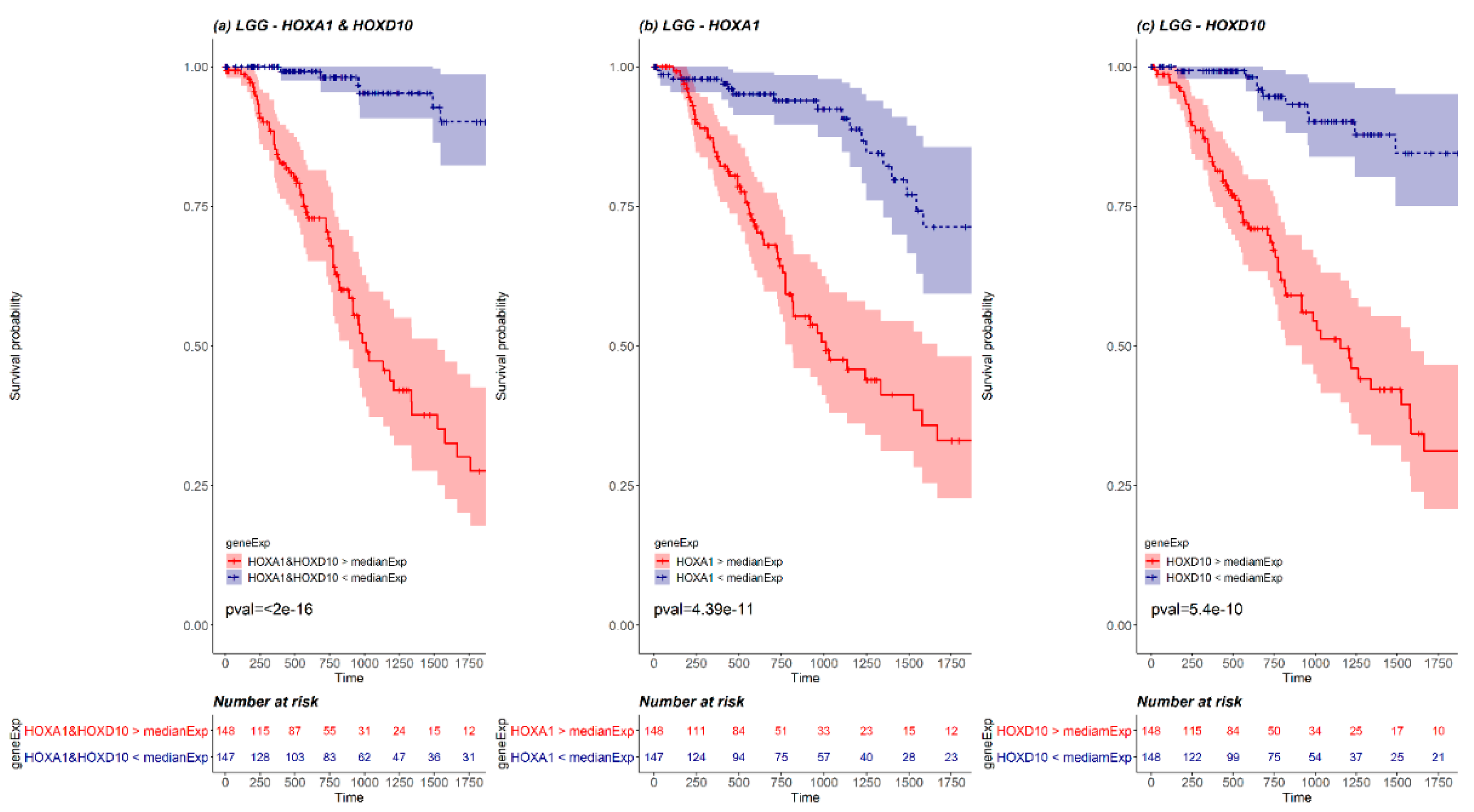
2.4. Kaplan-Meier Survival Analysis
2.5. HOX Gene Pairs with Correlated Differential Expression
2.6. Comparison of HOX Genes Expression with Expression of House Keeping Genes
2.7. Comparison of HOX Genes Expression with Expression of Transcription Factors Enocoding Genes in Brain Healthy and Tumor Tissue
2.8. Comparison of HOX Genes with Known Prognostic Markers
2.9. HOX Expression in BRCA HER2-Positive, BRCA HER2-Negative, and BRCA Triple-Negative Samples
3. Discussion
4. Materials and Methods
4.1. Data Downloads
4.2. Association of Samples to Cancer Types
4.3. Heatmaps and Data Processing
4.4. Comparison of Expression of Randomly Selected Genes Between Cancer Tissue and Healthy Tissue
4.5. Calculating Euclidean Distance Between Healthy and Tumor Samples
4.6. Verifying the Observation that Posterior HOX Genes are Expressed at Low Levels in Multiple Healthy Tissues
4.7. Kaplan-Meier Survival Analysis Based on Expression of HOX Genes
4.8. KM Survival Analysis Based on Expression of HOX Gene Pairs
4.9. Comparison of Expression of Randomly Selected Sets of 39 House Keeping Genes and HOX Genes
4.10. Analysis of HOX Genes Expression in BRCA HER2-Positive, BRCA HER2-Negative and BRCA Triple-Negative Samples
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Software Source Code
Abbreviations
| Acronyms | Definition |
| ACC | Adrenocortical carcinoma |
| BRCA | Breast invasive carcinoma |
| COAD | Colon adenocarcinoma |
| ESCA | Esophageal carcinoma |
| GBM | Glioblastoma multiforme |
| GTEx | Genotype-Tissue Expression |
| HER2 | Human Epidermal Growth Factor Receptor 2 |
| HK | Housekeeping |
| HOX | Homeobox |
| KM | Kaplan-Meier |
| LAML | Acute Myeloid Leukemia |
| LGG | Brain lower grade glioma |
| LIHC | Liver hepatocellular carcinoma |
| LUAD | Lung adenocarcinoma |
| LUSC | Lung squamous cell carcinoma |
| PAAD | Pancreatic adenocarcinoma |
| PCPG | Pheochromocytoma and paraganglioma |
| PRAD | Prostate adenocarcinoma |
| RSEM | RNA-seq by Expectation Maximization |
| STAD | Stomach adenocarcinoma |
| TALE | Three Amino Acids Loop Extension |
| TCGA | The Cancer Genome Atla |
| THCA | Thyroid carcinoma |
| TF | Transcription Factor |
References
- Bhatlekar, S.; Fields, J.Z.; Boman, B.M. HOX genes and their role in the development of human cancers. J. Mol. Med. (Berl.) 2014, 92, 811–823. [Google Scholar] [CrossRef]
- Deschamps, J.; Duboule, D. Embryonic timing, axial stem cells, chromatin dynamics, and the Hox clock. Genes Dev. 2017, 31, 1406–1416. [Google Scholar] [CrossRef] [Green Version]
- Shah, N.; Sukumar, S. The Hox genes and their roles in oncogenesis. Nat. Rev. Cancer 2010, 10, 361–371. [Google Scholar] [CrossRef]
- Bhatlekar, S.; Fields, J.Z.; Boman, B.M. Role of HOX Genes in Stem Cell Differentiation and Cancer. Stem. Cells Int. 2018, 2018, 3569493. [Google Scholar] [CrossRef] [Green Version]
- Kamkar, F.; Xaymardan, M.; Asli, N.S. Hox-Mediated Spatial and Temporal Coding of Stem Cells in Homeostasis and Neoplasia. Stem. Cells Dev. 2016, 25, 1282–1289. [Google Scholar] [CrossRef]
- Seifert, A.; Werheid, D.F.; Knapp, S.M.; Tobiasch, E. Role of Hox genes in stem cell differentiation. World J. Stem. Cells 2015, 7, 583–595. [Google Scholar] [CrossRef]
- Iimura, T.; Pourquie, O. Hox genes in time and space during vertebrate body formation. Dev. Growth Differ. 2007, 49, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Huang, Q.; Wei, G.H. The Role of HOX Transcription Factors in Cancer Predisposition and Progression. Cancers 2019, 11, 528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Zhang, X.; Liu, Q.; Yin, H.; Diao, Y.; Zhang, Z.; Wang, Y.; Gao, Y.; Ren, X.; Li, J.; et al. Emerging role of HOX genes and their related long noncoding RNAs in lung cancer. Crit. Rev. Oncol. Hematol. 2019, 139, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Primon, M.; Hunter, K.D.; Pandha, H.S.; Morgan, R. Kinase Regulation of HOX Transcription Factors. Cancers 2019, 11, 508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.; Zyoud, A.; Allegrucci, C. A Case of Identity: HOX Genes in Normal and Cancer Stem Cells. Cancers 2019, 11, 512. [Google Scholar] [CrossRef] [Green Version]
- Carithers, L.J.; Ardlie, K.; Barcus, M.; Branton, P.A.; Britton, A.; Buia, S.A.; Compton, C.C.; DeLuca, D.S.; Peter-Demchok, J.; Gelfand, E.T.; et al. A Novel Approach to High-Quality Postmortem Tissue Procurement: The GTEx Project. Biopreserv. Biobank 2015, 13, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Barger, C.J.; Branick, C.; Chee, L.; Karpf, A.R. Pan-Cancer Analyses Reveal Genomic Features of FOXM1 Overexpression in Cancer. Cancers 2019, 11, 251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frost, F.G.; Cherukuri, P.F.; Milanovich, S.; Boerkoel, C.F. Pan-cancer RNA-seq data stratifies tumours by some hallmarks of cancer. J. Cell Mol. Med. 2020, 24, 418–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Takahashi, Y.; Hamada, J.; Murakawa, K.; Takada, M.; Tada, M.; Nogami, I.; Hayashi, N.; Nakamori, S.; Monden, M.; Miyamoto, M.; et al. Expression profiles of 39 HOX genes in normal human adult organs and anaplastic thyroid cancer cell lines by quantitative real-time RT-PCR system. Exp. Cell Res. 2004, 293, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Akoglu, H. User’s guide to correlation coefficients. Turk. J. Emerg. Med. 2018, 18, 91–93. [Google Scholar] [CrossRef] [PubMed]
- di Pietro, M.; Lao-Sirieix, P.; Boyle, S.; Cassidy, A.; Castillo, D.; Saadi, A.; Eskeland, R.; Fitzgerald, R.C. Evidence for a functional role of epigenetically regulated midcluster HOXB genes in the development of Barrett esophagus. Proc. Natl. Acad. Sci. USA 2012, 109, 9077–9082. [Google Scholar] [CrossRef] [Green Version]
- Eisenberg, E.; Levanon, E.Y. Human housekeeping genes, revisited. Trends Genet. TIG 2013, 29, 569–574. [Google Scholar] [CrossRef]
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The Human Transcription Factors. Cell 2018, 175, 598–599. Available online: http://humantfs.ccbr.utoronto.ca/ (accessed on 26 May 2020). [CrossRef] [Green Version]
- Assmar, M.; Yeganeh, S.; Mansourghanaei, F.; Amirmozafari, N. Combined Evaluation of AFP, CA15-3, CA125, CA19-9, and CEA Tumor Markers in Patients with Hepatitis B and C. Iran. J. Public Health 2016, 45, 1645–1651. [Google Scholar] [PubMed]
- Battaglin, F.; Naseem, M.; Puccini, A.; Lenz, H.J. Molecular biomarkers in gastro-esophageal cancer: Recent developments, current trends and future directions. Cancer Cell Int. 2018, 18, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Q.; Li, Q.; Wang, M.; Hu, J.; Dai, J.; Niu, L.; Yuan, G.; Pan, Y. Elevated CD44 expression predicts poor prognosis in patients with low-grade glioma. Oncol. Lett. 2019, 18, 3698–3704. [Google Scholar] [CrossRef]
- Handschuh, L. Not Only Mutations Matter: Molecular Picture of Acute Myeloid Leukemia Emerging from Transcriptome Studies. J. Oncol. 2019, 2019, 7239206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, F.L.; Yu, S.J. Esophageal cancer: Risk factors, genetic association, and treatment. Asian J. Surg. 2018, 41, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Loosen, S.H.; Neumann, U.P.; Trautwein, C.; Roderburg, C.; Luedde, T. Current and future biomarkers for pancreatic adenocarcinoma. Tumour Biol. J. Int. Soc. Oncodev. Med. 2017, 39, 1010428317692231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Man, J.; Zhang, X.; Dong, H.; Li, S.; Yu, X.; Meng, L.; Gu, X.; Yan, H.; Cui, J.; Lai, Y. Screening and identification of key biomarkers in lung squamous cell carcinoma by bioinformatics analysis. Oncol. Lett. 2019, 18, 5185–5196. [Google Scholar] [CrossRef] [PubMed]
- Menyhart, O.; Nagy, A.; Gyorffy, B. Determining consistent prognostic biomarkers of overall survival and vascular invasion in hepatocellular carcinoma. R. Soc. Open Sci. 2018, 5, 181006. [Google Scholar] [CrossRef] [Green Version]
- Nwosu, Z.C.; Megger, D.A.; Hammad, S.; Sitek, B.; Roessler, S.; Ebert, M.P.; Meyer, C.; Dooley, S. Identification of the Consistently Altered Metabolic Targets in Human Hepatocellular Carcinoma. Cell. Mol. Gastroenterol. Hepatol. 2017, 4, 303–323. [Google Scholar] [CrossRef] [Green Version]
- Sasmita, A.O.; Wong, Y.P.; Ling, A.P.K. Biomarkers and therapeutic advances in glioblastoma multiforme. Asia-Pac. J. Clin. Oncol. 2018, 14, 40–51. [Google Scholar] [CrossRef] [Green Version]
- Virgilio, E.; Proietti, A.; D’Urso, R.; Cardelli, P.; Giarnieri, E.; Montagnini, M.; Giovagnoli, M.R.; Mercantini, P.; Balducci, G.; Cavallini, M. Measuring Intragastric Tumor Markers in Gastric Cancer Patients: A Systematic Literature Review on Significance and Reliability. Anticancer Res. 2017, 37, 2817–2821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Zheng, Z.; Guan, J.; Qi, D.; Zhou, S.; Shen, X.; Wang, F.; Wenkert, D.; Kirmani, B.; Solouki, T.; et al. Identification of a panel of genes as a prognostic biomarker for glioblastoma. EBioMedicine 2018, 37, 68–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Y.; Bleicher, F.; Merabet, S. A systematic survey of HOX and TALE expression profiling in human cancers. Int. J. Dev. Biol. 2018, 62, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Fattah, R.; Xiao, A.; Bomgardner, D.; Pease, C.S.; Lopes, M.B.; Hussaini, I.M. Differential expression of HOX genes in neoplastic and non-neoplastic human astrocytes. J. Pathol. 2006, 209, 15–24. [Google Scholar] [CrossRef]
- Costa, B.M.; Smith, J.S.; Chen, Y.; Chen, J.; Phillips, H.S.; Aldape, K.D.; Zardo, G.; Nigro, J.; James, C.D.; Fridlyand, J.; et al. Reversing HOXA9 oncogene activation by PI3K inhibition: Epigenetic mechanism and prognostic significance in human glioblastoma. Cancer Res. 2010, 70, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Gallo, M.; Ho, J.; Coutinho, F.J.; Vanner, R.; Lee, L.; Head, R.; Ling, E.K.; Clarke, I.D.; Dirks, P.B. A tumorigenic MLL-homeobox network in human glioblastoma stem cells. Cancer Res. 2013, 73, 417–427. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.D.; Zhang, S.; Zhang, Y.H.; Pan, X.; Feng, K.; Chen, L.; Huang, T.; Kong, X. Identification of the Gene Expression Rules That Define the Subtypes in Glioma. J. Clin. Med. 2018, 7, 350. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Jutooru, I.; Chadalapaka, G.; Corton, J.C.; Safe, S. The long non-coding RNA HOTTIP enhances pancreatic cancer cell proliferation, survival and migration. Oncotarget 2015, 6, 10840–10852. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; He, R.; Zhang, R.; Gan, B.; Zhang, Y.; Chen, G.; Hu, X. The expression of HOXA13 in lung adenocarcinoma and its clinical significance: A study based on The Cancer Genome Atlas, Oncomine and reverse transcription-quantitative polymerase chain reaction. Oncol. Lett. 2018, 15, 8556–8572. [Google Scholar] [CrossRef]
- Dong, C.Y.; Cui, J.; Li, D.H.; Li, Q.; Hong, X.Y. HOXA10AS: A novel oncogenic long noncoding RNA in glioma. Oncol. Rep. 2018, 40, 2573–2583. [Google Scholar] [CrossRef]
- Drabkin, H.A.; Parsy, C.; Ferguson, K.; Guilhot, F.; Lacotte, L.; Roy, L.; Zeng, C.; Baron, A.; Hunger, S.P.; Varella-Garcia, M.; et al. Quantitative HOX expression in chromosomally defined subsets of acute myelogenous leukemia. Leukemia 2002, 16, 186–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, R.; Han, L.; Wang, Q.; Wei, J.; Chen, L.; Zhang, J.; Kang, C.; Wang, L. HOXA13 is a potential GBM diagnostic marker and promotes glioma invasion by activating the Wnt and TGF-beta pathways. Oncotarget 2015, 6, 27778–27793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eoh, K.J.; Kim, H.J.; Lee, J.Y.; Nam, E.J.; Kim, S.; Kim, S.W.; Kim, Y.T. Upregulation of homeobox gene is correlated with poor survival outcomes in cervical cancer. Oncotarget 2017, 8, 84396–84402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischbach, N.A.; Rozenfeld, S.; Shen, W.; Fong, S.; Chrobak, D.; Ginzinger, D.; Kogan, S.C.; Radhakrishnan, A.; Le Beau, M.M.; Largman, C.; et al. HOXB6 overexpression in murine bone marrow immortalizes a myelomonocytic precursor in vitro and causes hematopoietic stem cell expansion and acute myeloid leukemia in vivo. Blood 2005, 105, 1456–1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giampaolo, A.; Felli, N.; Diverio, D.; Morsilli, O.; Samoggia, P.; Breccia, M.; Lo Coco, F.; Peschle, C.; Testa, U. Expression pattern of HOXB6 homeobox gene in myelomonocytic differentiation and acute myeloid leukemia. Leukemia 2002, 16, 1293–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Peng, Y.; Gao, D.; Zhang, M.; Yang, W.; Linghu, E.; Herman, J.G.; Fuks, F.; Dong, G.; Guo, M. Silencing HOXD10 by promoter region hypermethylation activates ERK signaling in hepatocellular carcinoma. Clin. Epigenetics 2017, 9, 116. [Google Scholar] [CrossRef] [Green Version]
- Huo, X.Y.; Zhang, X.Y.; Yuan, F.; Zhao, X.Y.; You, B.A. HOXB7 promotes proliferation and metastasis of glioma by regulating the Wnt/beta-catenin pathway. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 2476–2485. [Google Scholar] [CrossRef]
- Kang, J.U. Characterization of amplification patterns and target genes on the short arm of chromosome 7 in early-stage lung adenocarcinoma. Mol. Med. Rep. 2013, 8, 1373–1378. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; McCrudden, C.M.; Yuen, H.F.; Xi, X.; Lyu, P.; Chan, K.W.; Zhang, S.D.; Kwok, H.F. CD133 in brain tumor: The prognostic factor. Oncotarget 2017, 8, 11144–11159. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Zhao, X.; Zhou, Y.; Liu, Y.; Zhou, Q.; Ye, H.; Wang, Y.; Zeng, J.; Song, Y.; Gao, W.; et al. The long non-coding RNA HOTTIP promotes progression and gemcitabine resistance by regulating HOXA13 in pancreatic cancer. J. Transl. Med. 2015, 13, 84. [Google Scholar] [CrossRef] [Green Version]
- Long, J.; Zhang, L.; Wan, X.; Lin, J.; Bai, Y.; Xu, W.; Xiong, J.; Zhao, H. A four-gene-based prognostic model predicts overall survival in patients with hepatocellular carcinoma. J. Cell Mol. Med. 2018, 22, 5928–5938. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Jin, K.; Cruz, L.A.; Park, S.; Sadik, H.; Cho, S.; Goswami, C.P.; Nakshatri, H.; Gupta, R.; Chang, H.Y.; et al. HOXB13 mediates tamoxifen resistance and invasiveness in human breast cancer by suppressing ERalpha and inducing IL-6 expression. Cancer Res. 2013, 73, 5449–5458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starkova, J.; Zamostna, B.; Mejstrikova, E.; Krejci, R.; Drabkin, H.A.; Trka, J. HOX gene expression in phenotypic and genotypic subgroups and low HOXA gene expression as an adverse prognostic factor in pediatric ALL. Pediatr. Blood Cancer 2010, 55, 1072–1082. [Google Scholar] [CrossRef]
- Sui, B.Q.; Zhang, C.D.; Liu, J.C.; Wang, L.; Dai, D.Q. HOXB13 expression and promoter methylation as a candidate biomarker in gastric cancer. Oncol. Lett. 2018, 15, 8833–8840. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.L.; Ding, B.X.; Hua, Y.; Chen, H.; Wu, T.; Chen, Z.Q.; Yuan, C.H. HOXC10 Promotes the Metastasis of Human Lung Adenocarcinoma and Indicates Poor Survival Outcome. Front. Physiol. 2017, 8, 557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, J.; Wang, P.; Li, S.; Song, J.; He, H.; Wang, Y.; Liu, Z.; Wang, F.; Bai, H.; Fang, W.; et al. HOXB13 networking with ABCG1/EZH2/Slug mediates metastasis and confers resistance to cisplatin in lung adenocarcinoma patients. Theranostics 2019, 9, 2084–2099. [Google Scholar] [CrossRef]
- Zhan, J.; Wang, P.; Niu, M.; Wang, Y.; Zhu, X.; Guo, Y.; Zhang, H. High expression of transcriptional factor HoxB9 predicts poor prognosis in patients with lung adenocarcinoma. Histopathology 2015, 66, 955–965. [Google Scholar] [CrossRef]
- Lv, X.; Li, L.; Lv, L.; Qu, X.; Jin, S.; Li, K.; Deng, X.; Cheng, L.; He, H.; Dong, L. HOXD9 promotes epithelial-mesenchymal transition and cancer metastasis by ZEB1 regulation in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2015, 34, 133. [Google Scholar] [CrossRef] [Green Version]
- Goldman, M.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. The UCSC Xena platform for public and private cancer genomics data visualization and interpretation. bioRxiv 2019, 326470. Available online: https://xena.ucsc.edu/ (accessed on 26 May 2020).
- Vivian, J.; Rao, A.A.; Nothaft, F.A.; Ketchum, C.; Armstrong, J.; Novak, A.; Pfeil, J.; Narkizian, J.; Deran, A.D.; Musselman-Brown, A.; et al. Toil enables reproducible, open source, big biomedical data analyses. Nat. Biotechnol. 2017, 35, 314–316. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Guo, Y.; Sheng, Q.; Shyr, Y. Advanced heat map and clustering analysis using heatmap3. BioMed Res. Int. 2014, 2014, 986048. [Google Scholar] [CrossRef] [Green Version]
- van Rossum, G. Python Tutorial, Technical Report CS-R9526; Centrum voor Wiskunde en Informatica (CWI): Amsterdam, The Netherlands, 1995. [Google Scholar]
- GTEx Consortium. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organ/Cancer Tissue Type | GTEX | TCGA | Total |
|---|---|---|---|
| Adrenal_Gland/PCPG | 128 | 185 | 313 |
| Brain/LGG | 105 | 527 | 632 |
| /GBM | 173 | 173 | |
| Breast/BRCA | 179 | 1253 | 1432 |
| Colon/COAD | 308 | 455 | 763 |
| Esophagus/ESCA | 273 | 195 | 468 |
| Leukemia/LAML | 337 | 173 | 510 |
| Liver/LIHC | 110 | 422 | 532 |
| Lung/LUAD | 288 | 598 | 886 |
| /LUSC | 551 | 551 | |
| Pancreas/PAAD | 167 | 183 | 350 |
| Prostate/PRAD | 100 | 556 | 656 |
| Stomach/STAD | 175 | 451 | 626 |
| Thyroid/THCA | 280 | 571 | 851 |
| Total | 2450 | 6293 | 8743 |
| Gene Name | Cancer Type | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| BRCA | ESCA | GBM | LAML | LGG | LIHC | LUSC | PAAD | STAD | |
| HOXA5 | 7.0 | ||||||||
| HOXA7 | 3.6 * | ||||||||
| HOXA9 | 3.7 | ||||||||
| HOXA10 | 6.2 | 3.4 | 4.3 | ||||||
| HOXB4 | 4.3 | ||||||||
| HOXB8 | 3.0 | ||||||||
| HOXB9 | 4.6 | 6.2 | |||||||
| HOXB13 | 6.8 | ||||||||
| HOXC4 | 5.5 | 3.5 * | |||||||
| HOXC6 | 5.9 | ||||||||
| HOXC8 | 3.2 | 3.5 | 4.6 | ||||||
| HOXC9 | 3.4 | ||||||||
| HOXC10 | 4.7 | 5.1 | 7.5 | ||||||
| HOXC11 | 3.7 | ||||||||
| HOXC13 | 4.3 | 5.1 | |||||||
| HOXD3 | 4.3 | ||||||||
| HOXD9 | 3.0 | ||||||||
| HOXD10 | 3.6 | ||||||||
| HOXD11 | 5.2 | ||||||||
| Organ/Cancer Type | HOX Genes with a Significant KM Correlation |
|---|---|
| Brain-LGG | HOXA1, HOXA4, HOXA7, HOXA11 |
| HOXB13 | |
| HOXC4 | |
| HOXD3, HOXD4, HOXD8, HOXD9, HOXD10 | |
| Brain-GBM | HOXB2, HOXB9 |
| Leukemia-LAML | HOXA7 |
| Cancer Type\Gene Name | HOX Pairs | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| HOXA5 | HOXA9 | HOXB3 | HOXB5 | HOXB6 | HOXB7 | HOXC8 | HOXC9 | HOXC10 | HOXD9 | Total | |
| ESCA | |||||||||||
| HOXA10 | ✓ | ✓ | 3 | ||||||||
| HOXB5 | ✓ | ✓ | 2 | ||||||||
| HOXB6 | ✓ | 1 | |||||||||
| HOXC10 | ✓ | 1 | |||||||||
| GBM | |||||||||||
| HOXA3 | ✓ | 1 | |||||||||
| HOXA10 | ✓ | 1 | |||||||||
| HOXB2 | ✓ | 1 | |||||||||
| HOXB4 | ✓ | 1 | |||||||||
| HOXB5 | ✓ | 1 | |||||||||
| LAML | |||||||||||
| HOXA3 | ✓ | 1 | |||||||||
| HOXA10 | ✓ | 1 | |||||||||
| LIHC | |||||||||||
| HOXD3 | ✓ | 1 | |||||||||
| HOXD8 | ✓ | 1 | |||||||||
| LUSC | |||||||||||
| HOXC8 | ✓ | 1 | |||||||||
| STAD | |||||||||||
| HOXC6 | ✓ | ✓ | 2 | ||||||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adato, O.; Orenstein, Y.; Kopolovic, J.; Juven-Gershon, T.; Unger, R. Quantitative Analysis of Differential Expression of HOX Genes in Multiple Cancers. Cancers 2020, 12, 1572. https://doi.org/10.3390/cancers12061572
Adato O, Orenstein Y, Kopolovic J, Juven-Gershon T, Unger R. Quantitative Analysis of Differential Expression of HOX Genes in Multiple Cancers. Cancers. 2020; 12(6):1572. https://doi.org/10.3390/cancers12061572
Chicago/Turabian StyleAdato, Orit, Yaron Orenstein, Juri Kopolovic, Tamar Juven-Gershon, and Ron Unger. 2020. "Quantitative Analysis of Differential Expression of HOX Genes in Multiple Cancers" Cancers 12, no. 6: 1572. https://doi.org/10.3390/cancers12061572
APA StyleAdato, O., Orenstein, Y., Kopolovic, J., Juven-Gershon, T., & Unger, R. (2020). Quantitative Analysis of Differential Expression of HOX Genes in Multiple Cancers. Cancers, 12(6), 1572. https://doi.org/10.3390/cancers12061572