Beyond the Paclitaxel and Vinca Alkaloids: Next Generation of Plant-Derived Microtubule-Targeting Agents with Potential Anticancer Activity

Abstract
:1. Introduction
2. Microtubule Stabilizing Agents
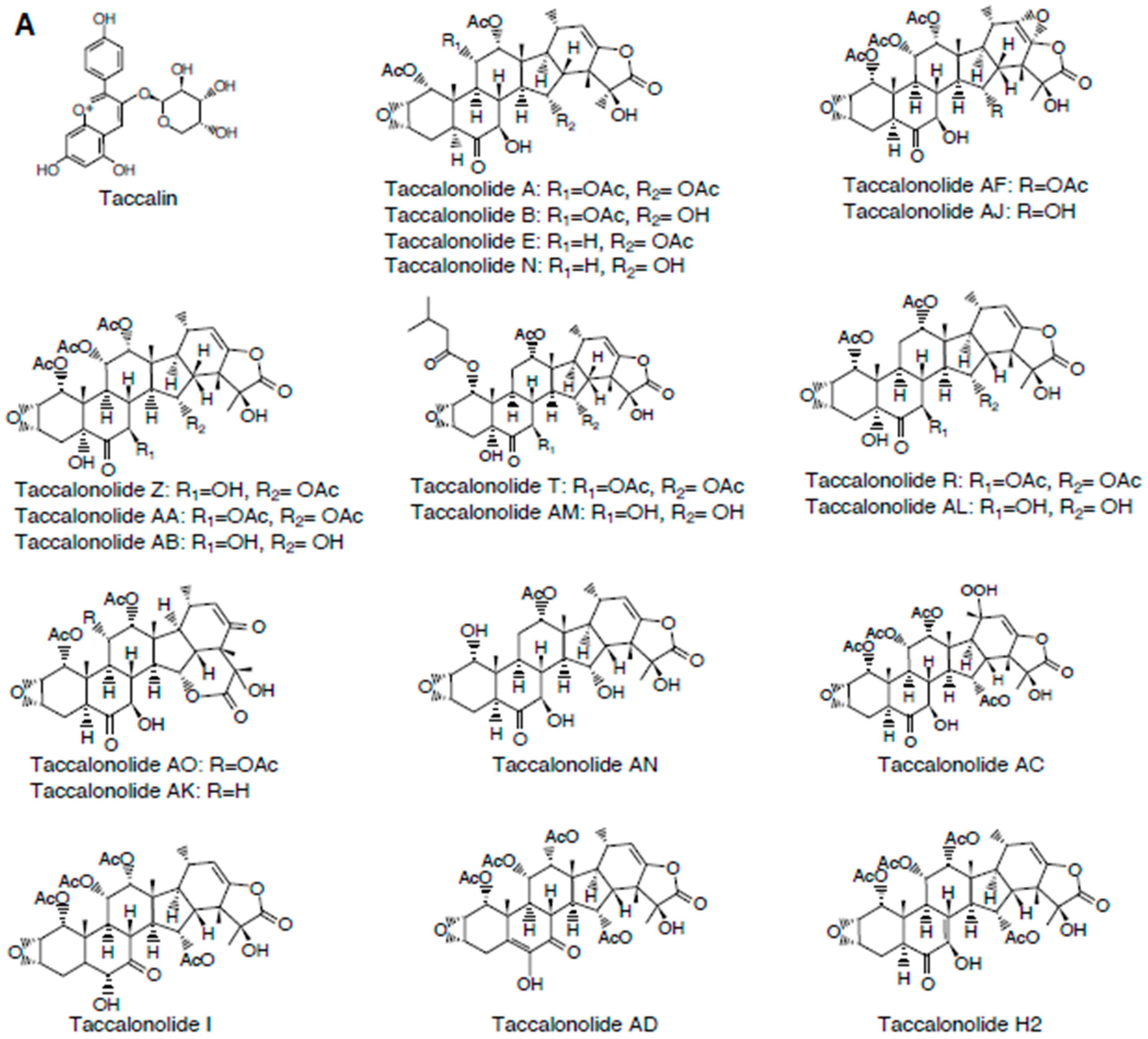
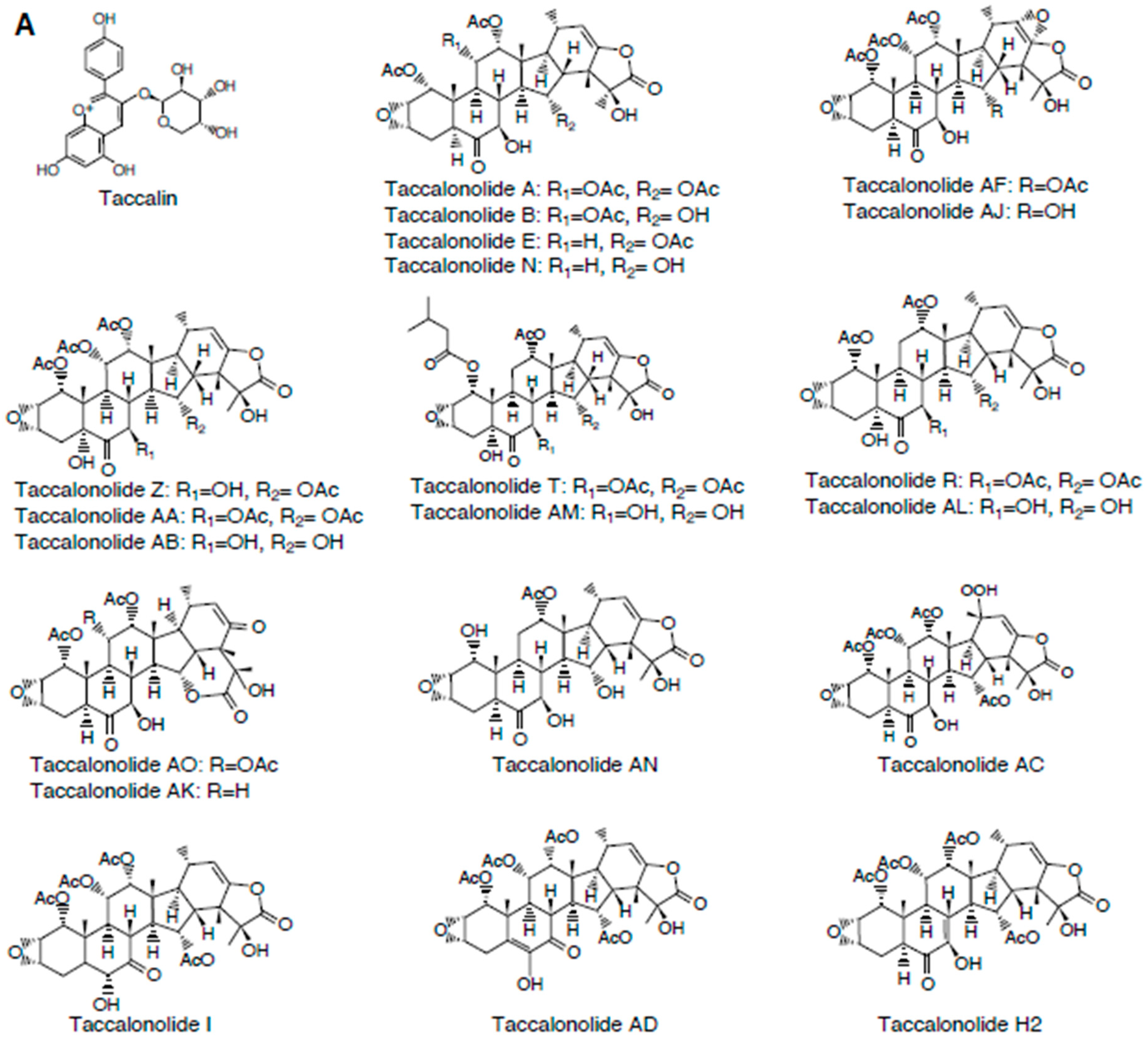
2.1. Taccalonolides
2.1.1. Mechanism of Action
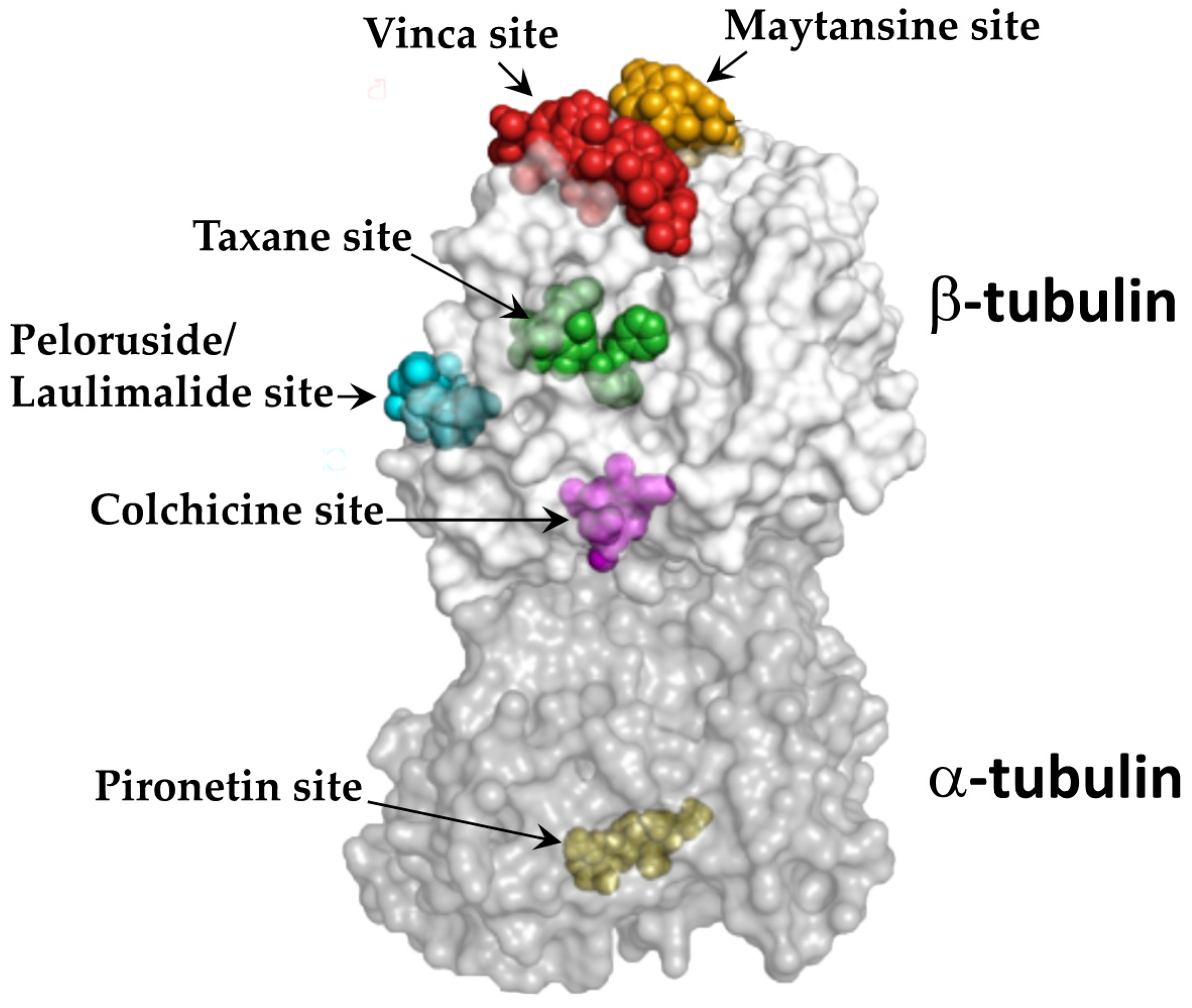
2.1.2. Tubulin Binding Sites
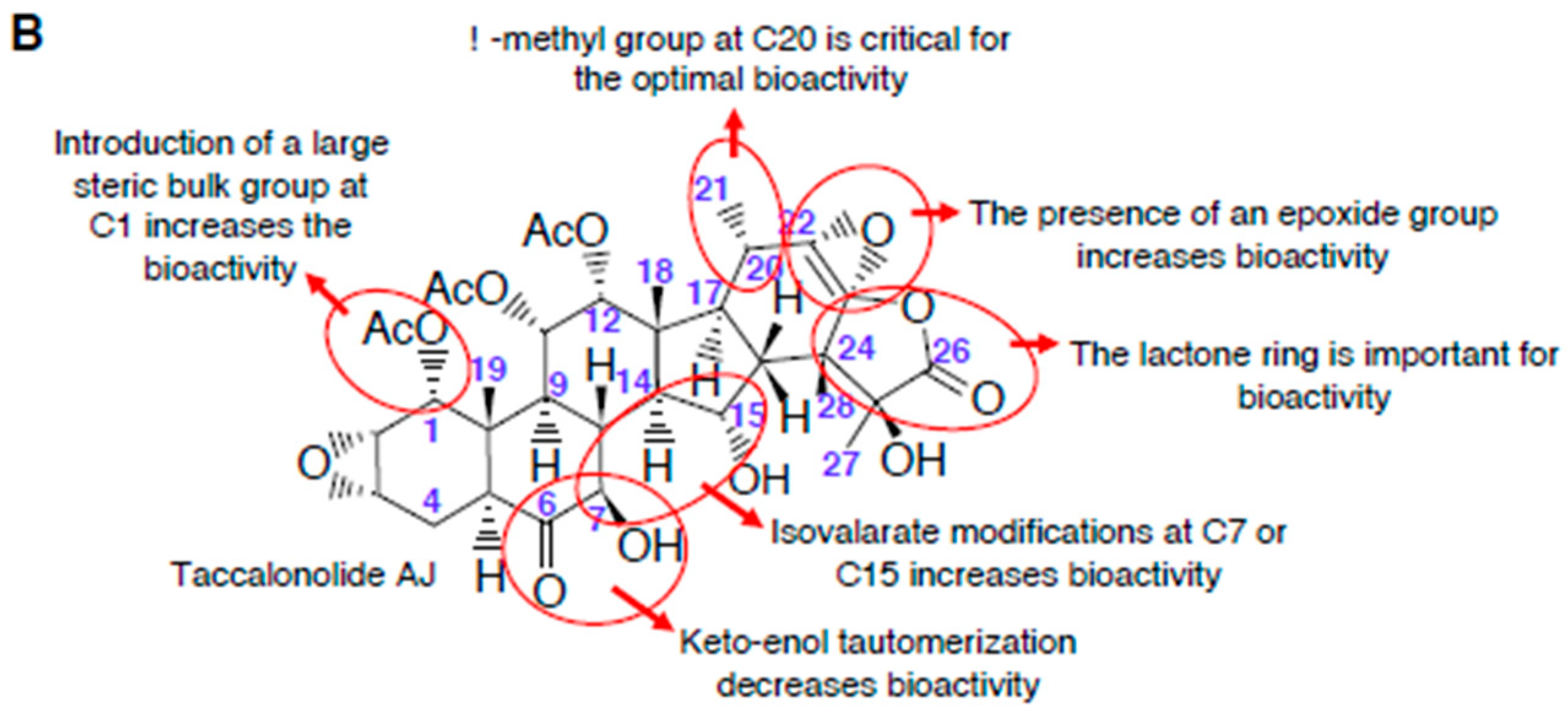
2.1.3. Structure-Activity Relationships

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A) | |||||||||||||||||||||||||||||||||||||||||||||
| Compound | IC50 (μM) | Compound | IC50 (μM) | Compound | IC50 (μM) | Compound | IC50 (μM) | Compound | IC50 | Compound | IC50 (μM) | References: [24,26,30,31,32] | |||||||||||||||||||||||||||||||||
| Taccalonolide A | 5.32 ± 0.23 | Taccalonolide N | 8.5 ± 0.40 | Taccalonolide Z | 0.12 ± 0.008 | Taccalonolide AD | 3.4 ± 0.2 | Taccalonolide AO | >50 | Taccalonolide AN | 1.5 ± 0.1 | ||||||||||||||||||||||||||||||||||
| Taccalonolide B | 3.12 ± 0.18 | Taccalonolide I | 49.2 ± 2.8 | Taccalonolide AA | 0.032 ± 0.002 | Taccalonolide AE | 5.0 ± 0.2 | Taccalonolide AK | >50 | Paclitaxel | 0.0012 ± 0.1 | ||||||||||||||||||||||||||||||||||
| Taccalonolide E | 39.5 ± 4.70 | Taccalonolide R | 13.0 ± 1.0 | Taccalonolide AB | 2.7 ± 0.1 | Taccalonolide AF | 0.023 ± 0.003 | Taccalonolide AL | 34.4 ± 7.5 | ||||||||||||||||||||||||||||||||||||
| Taccalonolide H2 | 0.73 ± 0.02 | Taccalonolide T | 0.34 ± 0.02 | Taccalonolide AC | >50 | Taccalonolide AJ | 0.0042 ± 0.0003 | Taccalonolide AM | 2.0 ± 0.1 | ||||||||||||||||||||||||||||||||||||
| (B) | |||||||||||||||||||||||||||||||||||||||||||||
| Compound | Xenograft Models | Method of Tumor Cell Administration | Treatment Strategy/Dose | References | |||||||||||||||||||||||||||||||||||||||||
| Taccalonolide AF and AJ | MDA-MB-231 breast cancer | intraperitoneal | 1. Taccalonolide AF: 2 mg/kg on Days 1, 4, 8 2. Taccalonolide AF: 2.5 mg/kg on Days 1 and 5 3. Taccalonolide AJ: 0.5 mg/kg on Days 1, 3, 5, and 8 | [27] | |||||||||||||||||||||||||||||||||||||||||
| Taccalonolide AF and AJ | SCC-4 oral cancer cells | subcutaneous | 1. Taccalonolide AF: 80 μg on Days 0 and 3 2. Taccalonolide AJ: 40 μg on Days 0 and 3 3. Taccalonolide AJ: 80 μg on Days 0 and 3 | [34] | |||||||||||||||||||||||||||||||||||||||||
| (C) | |||||||||||||||||||||||||||||||||||||||||||||
| Breast Cancer Cell Lines | Ovarian Cancer Cell Lines | Prostate Cancer Cell Lines | Leukemia Cell Lines | References: [35,36,37] | |||||||||||||||||||||||||||||||||||||||||
| Compound (μM) | MCF-7 | T-47D | MDA-MB-468 | MDA-MB-157 | SK-BR3 | Hs578T | MDA-MB-231 | MCF-10A | OVCAR-3 | IGROV-1 | 1A9 | A2780 | PC-3 | LNCaP | HL-60 | ||||||||||||||||||||||||||||||
| Persin | 15.1 ± 1.3 | 30.3 ± 2.3 | 25.0 ± 2.8 | 12.8 ± 1.2 | 19.7 ± 1.3 | 32.1 ± 2.3 | >39 | >39 | 27.9 ± 4.5 | 15.6 ± 3.6 | 13.7 ± 0.6 | 8.1 ± 1.1 | 30.0 ± 3.0 | 22.0 ± 1.8 | 1.9 ± 0.1 | ||||||||||||||||||||||||||||||
| 1 | 17.1 ± 1.7 | 20.7 ± 3.2 | >39 | >39 | >39 | >39 | >39 | >39 | >39 | >39 | 4.1 ± 0.4 | 8.1 ± 1.4 | >39 | >39 | 0.6 ± 0.03 | ||||||||||||||||||||||||||||||
| 2 | >32 | 18.9 ± 1.3 | 13.7 ± 0.9 | 4.0 ± 0.1 | |||||||||||||||||||||||||||||||||||||||||
| 3 | 27.7 ± 5.5 | 19.4 ± 2.2 | 2.6 ± 0.4 | ||||||||||||||||||||||||||||||||||||||||||
| 4 | >27 | 21.2 ± 1.8 | 7.5 ± 0.2 | ||||||||||||||||||||||||||||||||||||||||||
| 5 | 23.8 ± 2.2 | 34.1 ± 5.3 | 5.8 ± 0.1 | ||||||||||||||||||||||||||||||||||||||||||
| 6 | 29.0 ± 4.2 | 47.6 ± 3.5 | 28.4 ± 0.5 | ||||||||||||||||||||||||||||||||||||||||||
| 7 | >21 | ||||||||||||||||||||||||||||||||||||||||||||
| 8 | >24 | ||||||||||||||||||||||||||||||||||||||||||||
| 9 | 20.1 ± 3.6 | ||||||||||||||||||||||||||||||||||||||||||||
| 10 | >65 | 124 ± 20 | 22.8 ± 1.0 | ||||||||||||||||||||||||||||||||||||||||||
| (D) | |||||||||||||||||||||||||||||||||||||||||||||
| Breast Cancer Cell Lines | Lung Cancer Cell Lines | Squamous Carcinoma Cell Lines | Lymphoma Cell Lines | Ovarian Cancer Cell Line | Cervical Cancer Cell Line | Leukemia Cell Line | Prostate Cancer Cell Line | References | |||||||||||||||||||||||||||||||||||||
| Compound | MCF7 | MDA-MB-231 | BT-474 | SK-BR3 | MDA-MB-435 | A594 | H1299 | H292 | NCI-H358M | Tu212 | Tu686 | BJAB | OVCAR-8 | HeLa | HL60 | LNCap | PC3M | ||||||||||||||||||||||||||||
| Curcumin (μM) | 11.2 | 6.03 | 11.6 | 5.5 | 6.4 | 25.0 | [38,39,40,41,42] | ||||||||||||||||||||||||||||||||||||||
| Maytansine (pM) | 30 | 420 | 44 | 270 | [43,44,45] | ||||||||||||||||||||||||||||||||||||||||
| Combretastatin A4 (nM) | 2.8 | 5.3 | 3.8 | 8 | 0.37 | 0.9 | 2.1 | 4.7 | [46,47] | ||||||||||||||||||||||||||||||||||||
| Noscapine (μM) | 29 | 69 | [48] | ||||||||||||||||||||||||||||||||||||||||||
| Quercetin (μM) | 14 | 1 | 22 | [49] | |||||||||||||||||||||||||||||||||||||||||
2.1.4. Advantages over Paclitaxel
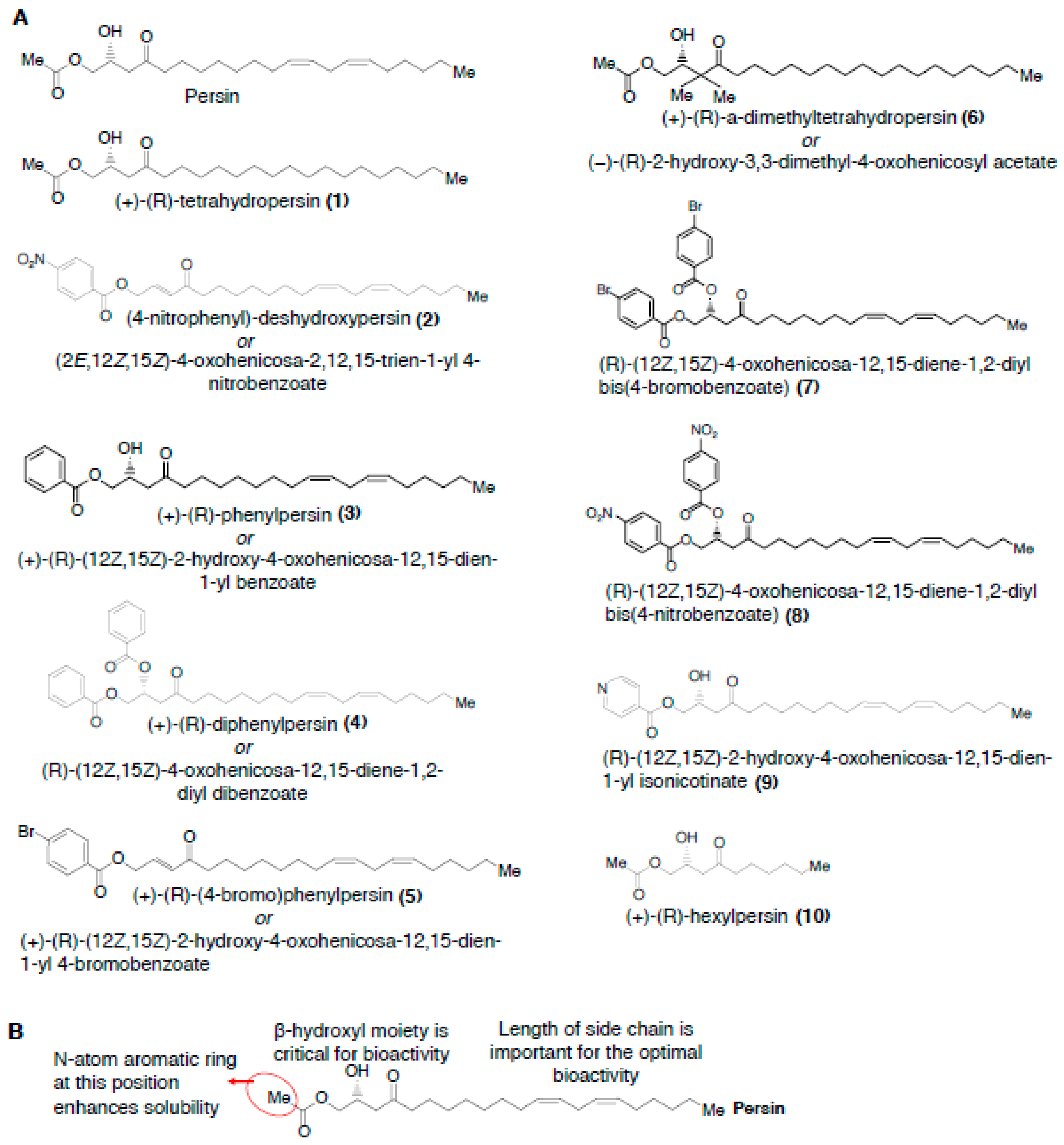
2.2. Persin
2.2.1. Mechanism of Action
2.2.2. Tubulin Binding Sites
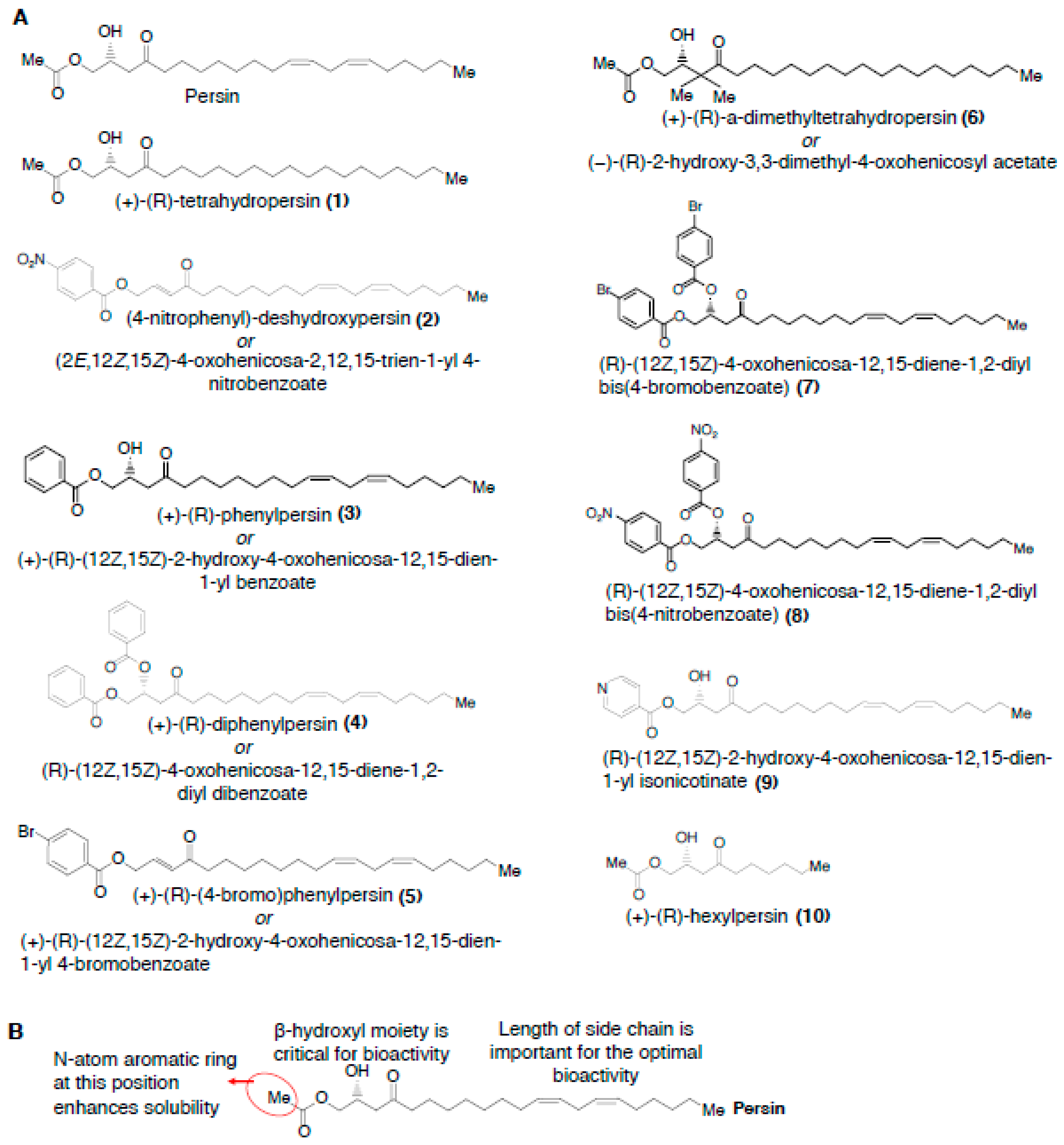
2.2.3. Structure-Activity Relationships
2.2.4. Persin Activity in MTA-Resistant Cells
3. Microtubule Destabilizing Agents
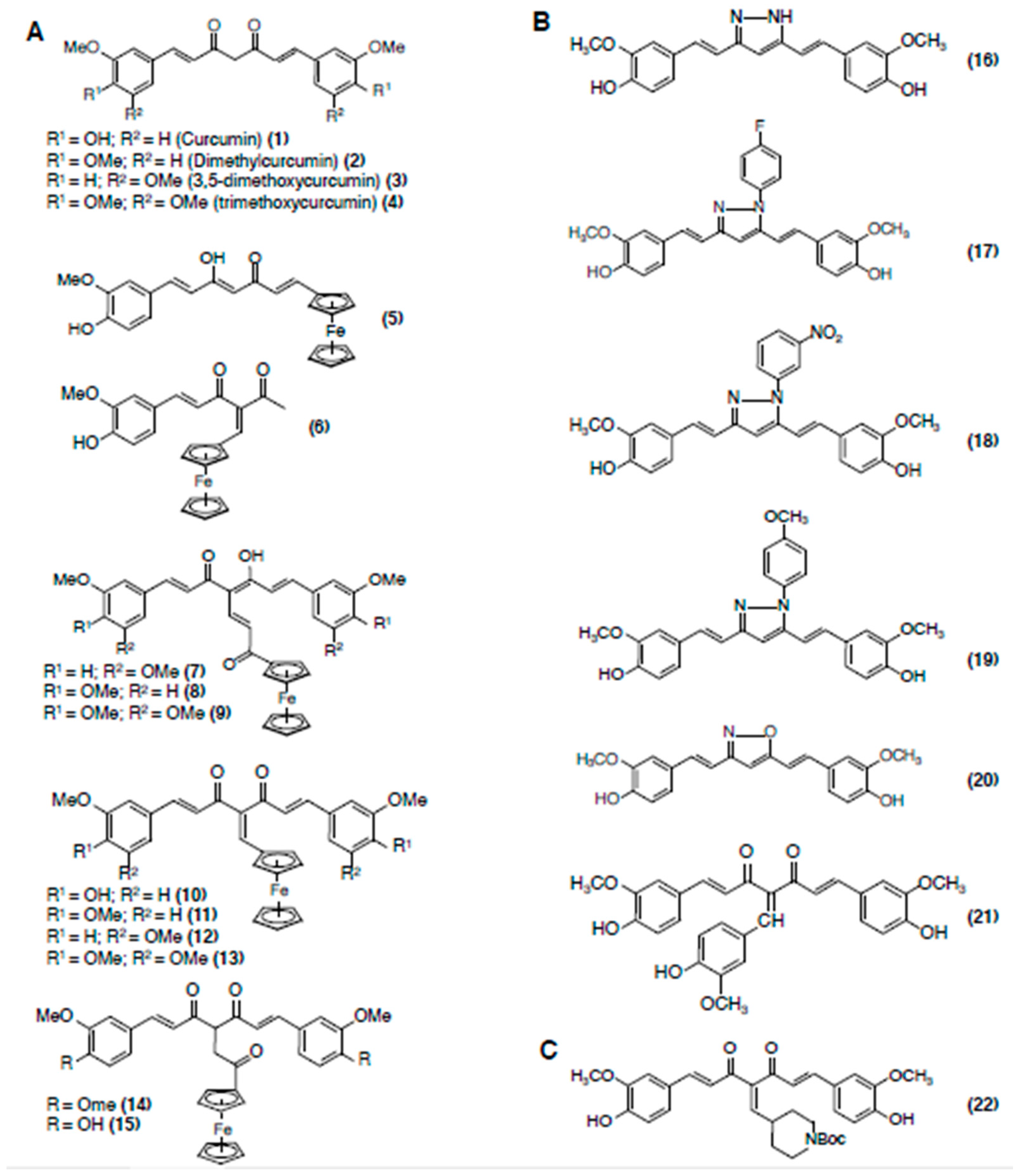
3.1. Curcumin
3.1.1. Mechanism of Action
3.1.2. Tubulin Binding Sites
3.1.3. Structure-Activity Relationships
3.2. Combretastatins
3.2.1. Mechanism of Action and the Tubulin Binding Sites
3.2.2. Structure–Activity Relationships
3.3. Noscapine
3.3.1. Mechanism of Action
3.3.2. Tubulin Binding Sites
3.3.3. Structure–Activity Relationships
3.4. Maytansinoids
3.4.1. Mechanism of Action and the Tubulin Binding Sites
3.4.2. Structure–Activity Relationships
3.5. Chalcones and Quercetin
Mechanism of Action and Tubulin Binding Sites
4. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nogales, E. A structural view of microtubule dynamics. Cell Mol. Life Sci. 1999, 56, 133–142. [Google Scholar] [CrossRef]
- Goodson, H.V.; Jonasson, E.M. Microtubules and Microtubule-Associated Proteins. Cold Spring Harb. Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, M.; Alonso, J.M.; Szymanski, D.B. Microtubule-Dependent Confinement of a Cell Signaling and Actin Polymerization Control Module Regulates Polarized Cell Growth. Curr. Biol. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Field, J.J.; Kanakkanthara, A.; Miller, J.H. Microtubule-targeting agents are clinically successful due to both mitotic and interphase impairment of microtubule function. Bioorg. Med. Chem. 2014, 22, 5050–5059. [Google Scholar] [CrossRef] [PubMed]
- Barbier, P.; Tsvetkov, P.O.; Breuzard, G.; Devred, F. Deciphering the molecular mechanisms of anti-tubulin plant derived drugs. Phytochem. Rev. 2014, 13, 157–169. [Google Scholar] [CrossRef]
- Kanakkanthara, A.; Teesdale-Spittle, H.P.; Miller, H.J. Cytoskeletal alterations that confer resistance to anti-tubulin chemotherapeutics. Anticancer Agents Med. Chem. 2013, 13, 147–158. [Google Scholar] [CrossRef]
- Yang, C.P.H.; Horwitz, S.B. Taxol (R): The First Microtubule Stabilizing Agent. Int. J. Mol. Sci. 2017, 18, 1733. [Google Scholar] [CrossRef] [Green Version]
- Prota, A.E.; Bargsten, K.; Zurwerra, D.; Field, J.J.; Diaz, J.F.; Altmann, K.H.; Steinmetz, M.O. Molecular mechanism of action of microtubule-stabilizing anticancer agents. Science 2013, 339, 587–590. [Google Scholar] [CrossRef] [Green Version]
- Kanakkanthara, A.; Northcote, P.T.; Miller, J.H. Peloruside A: A lead non-taxoid-site microtubule-stabilizing agent with potential activity against cancer, neurodegeneration, and autoimmune disease. Nat. Prod. Rep. 2016, 33, 549–561. [Google Scholar] [CrossRef]
- Cormier, A.; Knossow, M.; Wang, C.; Gigant, B. The binding of vinca domain agents to tubulin: Structural and biochemical studies. Methods Cell Biol. 2010, 95, 373–390. [Google Scholar]
- Li, W.; Sun, H.; Xu, S.; Zhu, Z.; Xu, J. Tubulin inhibitors targeting the colchicine binding site: A perspective of privileged structures. Future Med. Chem. 2017, 9, 1765–1794. [Google Scholar] [CrossRef] [Green Version]
- Prota, A.E.; Bargsten, K.; Diaz, J.F.; Marsh, M.; Cuevas, C.; Liniger, M.; Neuhaus, C.; Andreu, J.M.; Altmann, K.H.; Steinmetz, M.O. A new tubulin-binding site and pharmacophore for microtubule-destabilizing anticancer drugs. Proc. Natl. Acad. Sci. USA 2014, 111, 13817–13821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prota, A.E.; Setter, J.; Waight, A.B.; Bargsten, K.; Murga, J.; Diaz, J.F.; Steinmetz, M.O. Pironetin Binds Covalently to alphaCys316 and Perturbs a Major Loop and Helix of alpha-Tubulin to Inhibit Microtubule Formation. J. Mol. Biol. 2016, 428, 2981–2988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinmetz, M.O.; Prota, A.E. Microtubule-targeting agents: Strategies to hijack the cytoskeleton. Trends Cell Biol. 2018, 28, 776–792. [Google Scholar] [CrossRef]
- Martino, E.; Casamassima, G.; Castiglione, S.; Cellupica, E.; Pantalone, S.; Papagni, F.; Rui, M.; Siciliano, A.M.; Collina, S. Vinca alkaloids and analogues as anti-cancer agents: Looking back, peering ahead. Bioorg. Med. Chem. Lett. 2018, 28, 2816–2826. [Google Scholar] [CrossRef] [PubMed]
- Dasgeb, B.; Kornreich, D.; McGuinn, K.; Okon, L.; Brownell, I.; Sackett, D.L. Colchicine: An ancient drug with novel applications. Br. J. Dermatol. 2018, 178, 350–356. [Google Scholar] [CrossRef]
- Salehi, B.; Zucca, P.; Sharifi-Rad, M.; Pezzani, R.; Rajabi, S.; Setzer, W.N.; Varoni, E.M.; Iriti, M.; Kobarfard, F.; Sharifi-Rad, J. Phytotherapeutics in cancer invasion and metastasis. Phytother. Res. 2018, 32, 1425–1449. [Google Scholar] [CrossRef]
- Li, L.; Jiang, S.; Li, X.; Liu, Y.; Su, J.; Chen, J. Recent advances in trimethoxyphenyl (TMP) based tubulin inhibitors targeting the colchicine binding site. Eur. J. Med. Chem. 2018, 151, 482–494. [Google Scholar] [CrossRef]
- Perez, E.A. Microtubule inhibitors: Differentiating tubulin-inhibiting agents based on mechanisms of action, clinical activity, and resistance. Mol. Cancer Ther. 2009, 8, 2086–2095. [Google Scholar] [CrossRef] [Green Version]
- Bates, D.; Eastman, A. Microtubule destabilising agents: Far more than just antimitotic anticancer drugs. Br. J. Clin. Pharmacol. 2017, 83, 255–268. [Google Scholar] [CrossRef]
- Dall’Acqua, S. Natural Products As Antimitotic Agents. Curr. Top. Med. Chem. 2014, 14, 2272–2285. [Google Scholar] [CrossRef] [PubMed]
- Mukhtar, E.; Adhami, V.M.; Mukhtar, H. Targeting Microtubules by Natural Agents for Cancer Therapy. Mol. Cancer Ther. 2014, 13, 275–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Risinger, A.L.; Mooberry, S.L. Taccalonolide microtubule stabilizers. Bioorg. Med. Chem. 2014, 22, 5091–5096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tinley, T.L.; Randall-Hlubek, D.A.; Leal, R.M.; Jackson, E.M.; Cessac, J.W.; Quada, J.C.; Hemscheidt, T.K.; Mooberry, S.L. Taccalonolides E and A: Plant-derived steroids with microtubule-stabilizing activity. Cancer Res. 2003, 63, 3211–3220. [Google Scholar] [PubMed]
- Wang, Y.; Yu, Y.; Li, G.B.; Li, S.A.; Wu, C.; Gigant, B.; Qin, W.; Chen, H.; Wu, Y.; Chen, Q.; et al. Mechanism of microtubule stabilization by taccalonolide AJ. Nat. Commun. 2017, 8, 15787. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Risinger, A.L.; Peng, J.; Chen, Z.; Hu, L.; Mooberry, S.L. Potent taccalonolides, AF and AJ, inform significant structure-activity relationships and tubulin as the binding site of these microtubule stabilizers. J. Am. Chem. Soc. 2011, 133, 19064–19067. [Google Scholar] [CrossRef] [Green Version]
- Risinger, A.L.; Li, J.; Bennett, M.J.; Rohena, C.C.; Peng, J.; Schriemer, D.C.; Mooberry, S.L. Taccalonolide binding to tubulin imparts microtubule stability and potent in vivo activity. Cancer Res. 2013, 73, 6780–6792. [Google Scholar] [CrossRef] [Green Version]
- Buey, R.M.; Li, J.; Bennett, M.J.; Rohena, C.C.; Peng, J.; Schriemer, D.C.; Mooberry, S.L. Cyclostreptin binds covalently to microtubule pores and lumenal taxoid binding sites. Nat. Chem. Biol. 2007, 3, 117–125. [Google Scholar] [CrossRef] [Green Version]
- Field, J.J.; Pera, B.; Gallego, J.E.; Calvo, E.; Rodríguez-Salarichs, J.; Sáez-Calvo, G.; Zuwerra, D.; Jordi, M.; Andreu, J.M.; Prota, A.E.; et al. Zampanolide Binding to Tubulin Indicates Cross-Talk of Taxane Site with Colchicine and Nucleotide Sites. J. Nat. Prod. 2018, 81, 494–505. [Google Scholar] [CrossRef]
- Li, J.; Peng, J.; Risinger, A.L.; Mooberry, S.L. Hydrolysis reactions of the taccalonolides reveal structure-activity relationships. J. Nat. Prod. 2013, 76, 1369–1375. [Google Scholar] [CrossRef] [Green Version]
- Peng, J.; Risinger, A.L.; Li, J.; Mooberry, S.L. Synthetic reactions with rare taccalonolides reveal the value of C-22,23 epoxidation for microtubule stabilizing potency. J. Med. Chem. 2014, 57, 6141–6149. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Risinger, A.L.; Fest, G.A.; Jackson, E.M.; Helms, G.; Polin, L.A.; Mooberry, S.L. Identification and biological activities of new taccalonolide microtubule stabilizers. J. Med. Chem. 2011, 54, 6117–6124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ola, A.R.B.; Risinger, A.L.; Du, L.; Zammiello, C.L.; Peng, J.; Cichewicz, R.H.; Mooberry, S.L. Taccalonolide Microtubule Stabilizers Generated Using Semisynthesis Define the Effects of Mono Acyloxy Moieties at C-7 or C-15 and Disubstitutions at C-7 and C-25. J. Nat. Prod. 2018, 81, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Risinger, A.L.; Li, J.; Du, L.; Benavides, R.; Robles, A.J.; Cichewicz, R.H.; Kuhn, J.G.; Mooberry, S.L. Pharmacokinetic Analysis and in Vivo Antitumor Efficacy of Taccalonolides AF and AJ. J. Nat. Prod. 2017, 80, 409–414. [Google Scholar] [CrossRef] [Green Version]
- Butt, A.J.; Roberts, C.G.; Seawright, A.A.; Oelrichs, P.B.; MacLeod, J.K.; Liaw, T.Y.; Kavallaris, M.; Somers-Edgar, T.J.; Lehrbach, G.M.; Watts, C.K.; et al. A novel plant toxin, persin, with in vivo activity in the mammary gland, induces Bim-dependent apoptosis in human breast cancer cells. Mol. Cancer Ther. 2006, 5, 2300–2309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Field, J.J.; Kanakkanthara, A.; Brooke, D.G.; Sinha, S.; Pillai, S.D.; Denny, W.A.; Butt, A.J.; Miller, J.H. Microtubule-stabilizing properties of the avocado-derived toxins (+)-(R)-persin and (+)-(R)-tetrahydropersin in cancer cells and activity of related synthetic analogs. Investig. New Drugs 2016, 34, 277–289. [Google Scholar] [CrossRef]
- Brooke, D.G.; Shelley, E.J.; Roberts, C.G.; Denny, W.A.; Sutherland, R.L.; Butt, A.J. Synthesis and in vitro evaluation of analogues of avocado-produced toxin (+)-(R)-persin in human breast cancer cells. Bioorg. Med. Chem. 2011, 19, 7033–7043. [Google Scholar] [CrossRef]
- Imran, M.; Ullah, A.; Saeed, F.; Nadeem, M.; Arshad, M.U.; Suleria, H.A. Cucurmin, anticancer, & antitumor perspectives: A comprehensive review. Crit. Rev. Food Sci. Nutr. 2018, 58, 1271–1293. [Google Scholar]
- Allegra, A.; Innao, V.; Russo, S.; Gerace, D.; Alonci, A.; Musolino, C. Anticancer Activity of Curcumin and Its Analogues: Preclinical and Clinical Studies. Cancer Investig. 2017, 35, 1–22. [Google Scholar] [CrossRef]
- Bava, S.V.; Puliyappadamba, V.T.; Deepti, A.; Nair, A.; Karunagaran, D.; Anto, R.J. Sensitization of taxol-induced apoptosis by curcumin involves down-regulation of nuclear factor- B and the serine/threonine kinase Akt and is independent of tubulin polymerization (vol 280, pg 6301, 2005). J. Biol. Chem. 2018, 293, 12283. [Google Scholar] [CrossRef] [Green Version]
- Chakraborti, S.; Das, L.; Kapoor, N.; Das, A.; Dwivedi, V.; Poddar, A.; Chakraborti, G.; Janik, M.; Basu, G.; Panda, D.; et al. Curcumin Recognizes a Unique Binding Site of Tubulin. J. Med. Chem. 2011, 54, 6183–6196. [Google Scholar] [CrossRef]
- Jackson, S.J.T.; Murphy, L.L.; Venema, R.C.; Singletary, K.W.; Young, A.J. Curcumin binds tubulin, induces mitotic catastrophe, and impedes normal endothelial cell proliferation. Food Chem. Toxicol. 2013, 60, 431–438. [Google Scholar] [CrossRef] [Green Version]
- Rida, P.C.; LiVecche, D.; Ogden, A.; Zhou, J.; Aneja, R. The Noscapine Chronicle: A Pharmaco-Historic Biography of the Opiate Alkaloid Family and its Clinical Applications. Med. Res. Rev. 2015, 35, 1072–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kupchan, S.M.; Komoda, Y.; Court, W.A.; Thomas, G.J.; Smith, R.M.; Karim, A.; Gilmore, C.J.; Haltiwanger, R.C.; Bryan, R.F. Maytansine, a novel antileukemic ansa macrolide from Maytenus ovatus. J. Am. Chem. Soc. 1972, 94, 1354–1356. [Google Scholar] [CrossRef]
- Lopus, M.; Oroudjev, E.; Wilson, L.; Wilhelm, S.; Widdison, W.; Chari, R.; Jordan, M.A. Maytansine and cellular metabolites of antibody-maytansinoid conjugates strongly suppress microtubule dynamics by binding to microtubules. Mol. Cancer Ther. 2010, 9, 2689–2699. [Google Scholar] [CrossRef] [Green Version]
- Jaroch, K.; Karolak, M.; Gorski, P.; Jaroch, A.; Krajewski, A.; Ilnicka, A.; Sloderbach, A.; Stefanski, T.; Sobiak, S. Combretastatins: In vitro structure-activity relationship, mode of action and current clinical status. Pharm. Rep. 2016, 68, 1266–1275. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.R.; Singh, S.B.; Hamel, E.; Lin, C.M.; Alberts, D.S.; Garcia-Kendall, D. Isolation and structure of the strong cell growth and tubulin inhibitor combretastatin A-4. Experientia 1989, 45, 209–211. [Google Scholar] [CrossRef] [PubMed]
- Kocak, C.; Kocak, F.E.; Ozturk, B.; Tekin, G.; Vatansev, H. Cytotoxic, anti-proliferative and apoptotic effects of noscapine on human estrogen receptor positive (MCF-7) and negative (MDA-MB-231) breast cancer cell lines. Bratisl Lek Listy 2020, 121, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.; Panda, D. Perturbation of microtubule polymerization by quercetin through tubulin binding: A novel mechanism of its antiproliferative activity. Biochemistry 2002, 41, 13029–13038. [Google Scholar] [CrossRef] [PubMed]
- Risinger, A.L.; Jackson, E.M.; Polin, L.A.; Helms, G.L.; LeBoeuf, D.A.; Joe, P.A.; Hopper-Borge, E.; Ludueña, R.F.; Kruh, G.D.; Mooberry, S.L. The taccalonolides: Microtubule stabilizers that circumvent clinically relevant taxane resistance mechanisms. Cancer Res. 2008, 68, 8881–8888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Risinger, A.L.; Mooberry, S.L. Cellular studies reveal mechanistic differences between taccalonolide A and paclitaxel. Cell Cycle 2011, 10, 2162–2171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Saona, C.; Trumble, J.T. Biologically active aliphatic acetogenins from specialized idioblast oil cells. Curr. Org. Chem. 2000, 4, 1249–1260. [Google Scholar] [CrossRef]
- Chang, C.F.; Isogai, A.; Kamikado, T.; Murakoshi, S.; Sakurai, A.; Tamura, S. Isolation and Structure Elucidation of Growth-Inhibitors for Silkworm Larvae from Avocado Leaves. Agric. Biol. Chem. 1975, 39, 1167–1168. [Google Scholar] [CrossRef]
- Roberts, C.G.; Gurisik, E.; Biden, T.J.; Sutherland, R.L.; Butt, A.J. Synergistic cytotoxicity between tamoxifen and the plant toxin persin in human breast cancer cells is dependent on Bim expression and mediated by modulation of ceramide metabolism. Mol. Cancer Ther. 2007, 6, 2777–2785. [Google Scholar] [CrossRef] [Green Version]
- Oelrichs, P.B.; Ng, J.C.; Seawright, A.A.; Ward, A.; Schäffeler, L.; Macleod, J.K. IIsolation and identification of a compound from avocado (Persea americana) leaves which causes necrosis of the acinar epithelium of the lactating mammary gland and the myocardium. Nat. Toxins 1995, 3, 344–349. [Google Scholar] [CrossRef]
- Craigmill, A.L.; Seawright, A.A.; Mattila, T.; Frost, A.J. Pathological changes in the mammary gland and biochemical changes in milk of the goat following oral dosing with leaf of the avocado (Persea americana). Aust. Vet. J. 1989, 66, 206–211. [Google Scholar] [CrossRef]
- Field, J.J.; Singh, A.J.; Kanakkanthara, A.; Halafihi, T.I.; Northcote, P.T.; Miller, J.H. Microtubule-stabilizing activity of zampanolide, a potent macrolide isolated from the Tongan marine sponge Cacospongia mycofijiensis. J. Med. Chem. 2009, 52, 7328–7332. [Google Scholar] [CrossRef]
- Kowalski, R.J.; Giannakakou, P.; Gunasekera, S.P.; Longley, R.E.; Day, B.W.; Hamel, E. The microtubule-stabilizing agent discodermolide competitively inhibits the binding of paclitaxel (Taxol) to tubulin polymers, enhances tubulin nucleation reactions more potently than paclitaxel, and inhibits the growth of paclitaxel-resistant cells. Mol. Pharmacol. 1997, 52, 613–622. [Google Scholar] [CrossRef]
- Leon, L.G.; Giovannetti, E.; Alecci, C.; Giancola, F.; Funel, N.; Zucali, P.; Peters, G.J.; Padron, J.M. Antiproliferative Effects of Novel Aliphatic Acetogenin Analogs Against Aggressive Solid Tumor Cell Lines. In Vivo 2011, 25, 203–207. [Google Scholar]
- Padron, J.M.; Miranda, P.O.; Padrón, J.I.; Martín, V.S. Beta’-hydroxy-alpha,beta-unsaturated ketones: A new pharmacophore for the design of anticancer drugs. Bioorg. Med. Chem. Lett. 2006, 16, 2266–2269. [Google Scholar] [CrossRef]
- Leon, L.G.; Carballo, R.M.; Vega-Hernández, M.C.; Miranda, P.O.; Martín, V.S.; Padrón, J.I.; Padrón, J.M. Beta’-Hydroxy-alpha,beta-unsaturated ketones: A new pharmacophore for the design of anticancer drugs. Part 2. ChemMedChem 2008, 3, 1740–1747. [Google Scholar] [CrossRef] [PubMed]
- Kocaadam, B.; Sanlier, N. Curcumin, an active component of turmeric (Curcuma longa), and its effects on health. Crit. Rev. Food Sci. Nutr. 2017, 57, 2889–2895. [Google Scholar] [CrossRef] [PubMed]
- Doello, K.; Ortiz, R.; Alvarez, P.J.; Melguizo, C.; Cabeza, L.; Prados, J. Latest in Vitro and in Vivo Assay, Clinical Trials and Patents in Cancer Treatment using Curcumin: A Literature Review. Nutr. Cancer-An Int. J. 2018, 70, 569–578. [Google Scholar] [CrossRef]
- Gupta, K.; Naik, N.R.; Panda, D. Mechanism of antiproliferative action of curcumin: Perturbation of microtubule assembly and functions through tubulin binding. Mol. Biol. Cell 2004, 15, 395a. [Google Scholar]
- Arezki, A.; Brule, E.; Jaouen, G. Synthesis of the First Ferrocenyl Derivatives of Curcuminoids. Organometallics 2009, 28, 1606–1609. [Google Scholar] [CrossRef]
- Arezki, A.; Chabot, G.G.; Quentin, L.; Scherman, D.; Jaouen, G.; Brule, E. Synthesis and biological evaluation of novel ferrocenyl curcuminoid derivatives. Medchemcomm 2011, 2, 190–195. [Google Scholar] [CrossRef]
- Chakraborti, S.; Dhar, G.; Dwivedi, V.; Das, A.; Poddar, A.; Chakraborti, G.; Basu, G.; Chakrabarti, P.; Surolia, A.; Bhattacharyya, B. Stable and Potent Analogues Derived from the Modification of the Dicarbonyl Moiety of Curcumin. Biochemistry 2013, 52, 7449–7460. [Google Scholar] [CrossRef]
- Srivastava, S.; Mishra, S.; Surolia, A.; Panda, D. C1, a highly potent novel curcumin derivative, binds to tubulin, disrupts microtubule network and induces apoptosis. Biosci. Rep. 2016, 1, 36. [Google Scholar] [CrossRef]
- Khwaja, S.; Fatima, K.; Hasanain, M.; Behera, C.; Kour, A.; Singh, A.; Luqman, S.; Sarkar, J.; Chanda, D.; Shanker, K.; et al. Antiproliferative efficacy of curcumin mimics through microtubule destabilization. Eur. J. Med. Chem. 2018, 151, 51–61. [Google Scholar] [CrossRef]
- Ramya, P.V.S.; Guntuku, L.; Angapelly, S.; Digwal, C.S.; Lakshmi, U.J.; Sigalapalli, D.K.; Babu, B.N.; Naidu, V.G.M.; Kamal, A. Synthesis and biological evaluation of curcumin inspired imidazo [1,2-a]pyridine analogues as tubulin polymerization inhibitors. Eur. J. Med. Chem. 2018, 143, 216–231. [Google Scholar] [CrossRef]
- Ramya, P.V.S.; Angapelly, S.; Guntuku, L.; Digwal, C.S.; Babu, B.N.; Naidu, V.G.M.; Kamal, A. Synthesis and biological evaluation of curcumin inspired indole analogues as tubulin polymerization inhibitors. Eur. J. Med. Chem. 2017, 127, 100–114. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.R.; Singh, S.B.; Niven, M.L.; Hamel, E.; Schmidt, J.M. Isolation, structure, and synthesis of combretastatins A-1 and B-1, potent new inhibitors of microtubule assembly, derived from Combretum caffrum. J. Nat. Prod. 1987, 50, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Mehndiratta, S.; Nepali, K.; Gupta, M.K.; Koul, S.; Sharma, P.R.; Saxena, A.K.; Dhar, K.L. Novel indole-bearing combretastatin analogues as tubulin polymerization inhibitors. Org. Med. Chem. Lett. 2013, 3, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Boyle, N.M.; Greene, L.M.; Keely, N.O.; Wang, S.; Cotter, T.S.; Zisterer, D.M.; Meegan, M.J. Synthesis and biochemical activities of antiproliferative amino acid and phosphate derivatives of microtubule-disrupting beta-lactam combretastatins. Eur. J. Med. Chem. 2013, 62, 705–721. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.R.; Temple, C., Jr.; Narayanan, V.L.; Varma, R.; Simpson, M.J.; Boyd, M.R.; Rener, G.A.; Bansal, N. Antineoplastic agents 322. synthesis of combretastatin A-4 prodrugs. Anticancer Drug Des. 1995, 10, 299–309. [Google Scholar] [PubMed]
- Grisham, R.; Ky, B.; Tewari, K.S.; Chaplin, D.J.; Walker, J. Clinical trial experience with CA4P anticancer therapy: Focus on efficacy, cardiovascular adverse events, and hypertension management. Gynecol. Oncol. Res. Pract. 2018, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Qin, Y.; Wu, L.; Yang, S.; Li, N.; Wang, H.; Xu, H.; Sun, K.; Zhang, S.; Han, X.; et al. A phase I clinical trial assessing the safety and tolerability of combretastatin A4 phosphate injections. Anticancer Drugs 2014, 25, 462–471. [Google Scholar] [CrossRef]
- Gaspari, R.; Prota, A.E.; Bargsten, K.; Cavalli, A.; Steinmetz, M.O. Structural Basis of cis- and trans-Combretastatin Binding to Tubulin. Chem 2017, 2, 102–113. [Google Scholar] [CrossRef] [Green Version]
- Galbraith, S.M.; Chaplin, D.J.; Lee, F.; Stratford, M.R.L.; Locke, R.J.; Vojnovic, B.; Tozer, G.M. Effects of combretastatin A4 phosphate on endothelial cell morphology in vitro and relationship to tumour vascular targeting activity in vivo. Anticancer Res. 2001, 21, 93–102. [Google Scholar]
- Chen, H.; Li, Y.; Sheng, C.; Lv, Z.; Dong, G.; Wang, T.; Liu, J.; Zhang, M.; Li, L.; Zhang, T.; et al. Design and Synthesis of Cyclopropylamide Analogues of Combretastatin-A4 as Novel Microtubule-Stabilizing Agents. J. Med. Chem. 2013, 56, 685–699. [Google Scholar] [CrossRef] [PubMed]
- Ye, K.Q.; Ke, Y.; Keshava, N.; Shanks, J.; Kapp, J.A.; Tekmal, R.R.; Petros, J.; Joshi, H.C. Opium alkaloid noscapine is an antitumor agent that arrests metaphase and induces apoptosis in dividing cells. Proc. Natl. Acad. Sci. USA 1998, 95, 1601–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, Y.; Ye, K.; Grossniklaus, H.E.; Archer, D.R.; Joshi, H.C.; Kapp, J.A. Noscapine inhibits tumor growth with little toxicity to normal tissues or inhibition of immune responses. Cancer Immunol. Immunother. 2000, 49, 217–225. [Google Scholar] [CrossRef]
- Karna, P.; Rida, P.C.; Pannu, V.; Gupta, K.K.; Dalton, W.B.; Joshi, H.; Yang, V.W.; Zhou, J.; Aneja, R. A novel microtubule-modulating noscapinoid triggers apoptosis by inducing spindle multipolarity via centrosome amplification and declustering. Cell Death Differ. 2011, 18, 632–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naik, P.K.; Santoshi, S.; Rai, A.; Joshi, H.C. Molecular modelling and competition binding study of Br-noscapine and colchicine provide insight into noscapinoid-tubulin binding site. J. Mol. Graph. Model. 2011, 29, 947–955. [Google Scholar] [CrossRef] [Green Version]
- Issell, B.F.; Crooke, S.T. Maytansine. Cancer Treat. Rev. 1978, 5, 199–207. [Google Scholar] [CrossRef]
- Oroudjev, E.; Lopus, M.; Wilson, L.; Audette, C.; Provenzano, C.; Erickson, H.; Kovtun, Y.; Chari, R.; Jordan, M.A. Maytansinoid-antibody conjugates induce mitotic arrest by suppressing microtubule dynamic instability. Mol. Cancer Ther. 2010, 9, 2700–2713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Pegram, M.; Oh, D.Y.; Dieras, V.; Guardino, E.; et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N. Engl. J. Med. 2012, 367, 1783–1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kupchan, S.M.; Sneden, A.T.; Branfman, A.R.; Howie, G.A.; Rebhun, L.I.; Mcivor, W.E.; Wang, R.W.; Schnaitman, T.C. Tumor inhibitors. 124. Structural Requirements for Antileukemic Activity among Naturally Occurring and Semisynthetic Maytansinoids. J. Med. Chem. 1978, 21, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Widdison, W.C.; Wilhelm, S.D.; Cavanagh, E.E.; Whiteman, K.R.; Leece, B.A.; Kovtun, Y.; Goldmacher, V.S.; Xie, H.; Steeves, R.M.; Lutz, R.J.; et al. Semisynthetic maytansine analogues for the targeted treatment of cancer. J. Med. Chem. 2006, 49, 4392–4408. [Google Scholar] [CrossRef]
- Cassady, J.M.; Chan, K.K.; Floss, H.G.; Leistner, E. Recent developments in the maytansinoid antitumor agents. Chem. Pharm. Bull. (Tokyo) 2004, 52, 1–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.C.; Huang, R.Z.; Li, L.X.; Gou, S.H.; Wang, H.S. Synthesis and biological evaluation of novel chalcone derivatives as a new class of microtubule destabilizing agents. Eur. J. Med. Chem. 2017, 132, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Klimaszewska-Wisniewska, A.; Halas-Wisniewska, M.; Izdebska, M.; Gagat, M.; Grzanka, A.; Grzanka, D. Antiproliferative and antimetastatic action of quercetin on A549 non-small cell lung cancer cells through its effect on the cytoskeleton. Acta Histochem. 2017, 119, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Kooshyar, M.M.; Mozafari, P.M.; Amirchaghmaghi, M.; Pakfetrat, A.; Karoos, P.; Mohasel, M.R.; Orafai, H.; Azarian, A.A. A Randomized Placebo- Controlled Double Blind Clinical Trial of Quercetin in the Prevention and Treatment of Chemotherapy-Induced Oral Mucositis. J. Clin. Diagn. Res. 2017, 11, ZC46–ZC50. [Google Scholar] [CrossRef]
- De Caralt, S.; Bry, D.; Bontemps, N.; Turon, X.; Uriz, M.J.; Banaigs, B. Sources of secondary metabolite variation in Dysidea avara (Porifera: Demospongiae): The importance of having good neighbors. Mar. Drugs 2013, 11, 489–503. [Google Scholar] [CrossRef] [Green Version]
- Wink, M. Plant secondary metabolism: Diversity, function and its evolution. Nat. Prod. Commun. 2008, 3, 1205–1216. [Google Scholar] [CrossRef] [Green Version]



| Compound | Stage of Development | Clinical Trial | ||
|---|---|---|---|---|
| Active (NCI clinical trial identifier, Study phase, Year of study start) | Completed (NCI clinical trial identifier, Study phase, Year of study start–study completed) | Withdrawn/Terminated/Suspended (NCI clinical trial identifier, Study phase, Year of study start–study withdrawn/terminated/suspended) | ||
| Taccalonolide | Pre-clinical (in vitro cell-based studies and in vivo human tumor xenograft studies in mice) | - | - | - |
| Persin | Pre-clinical (in vitro cell-based studies) | - | - | - |
| Curcumin | Clinical (Total 60 clinical trials: 22 active, 26 completed, and 12 withdrawn/terminated/suspended trials) | NCT04403568, Early Phase 1, 2020 NCT02724202, Early Phase 1, 2016 NCT03980509, Phase 1, 2020 NCT01859858, Phase 1, 2013 NCT01294072, Phase 1, 2011 NCT02598726, Phase 1, 2016 NCT02336087, Phase 1, 2016 NCT04294836, Phase 2, 2020 NCT00745134, Phase 2, 2020 NCT02724618, Phase 2, 2016 NCT04266275, Phase 2, 2020 NCT00745134, Phase 2, 2008 NCT03192059, Phase 2, 2017 NCT03598309, Phase 2, 2019 NCT02782949, Phase 2, 2017 NCT03493997, Phase 2, 2017 NCT03769766, Phase 3, 2019 NCT02064673, Phase 3, 2014 NCT03847623, Phase N/A, 2017 NCT03865992, Phase N/A, 2019 NCT01948661, Phase N/A, 2014 NCT03431896, Phase N/A, 2018 | NCT01160302, Early Phase 1, 2010–2016 NCT01035580, Phase 1, 2010–2012 NCT01333917, Phase 1, 2010–2013 NCT00027495, Phase 1, 2001–2007 NCT01201694, Phase 1, 2011–2014 NCT01042938, Phase 2, 2008–2011 NCT02439385, Phase 2, 2015–2019 NCT03072992, Phase 2, 2017–2019 NCT01490996, Phase 1/2. 2012–2017 NCT00192842, Phase 2, 2004–2010 NCT01740323, Phase 2, 2015–2018 NCT00094445, Phase 2, 2004–2014 NCT02556632, Phase 2, 2015–2016 NCT02017353, Phase 2, 2013–2016 NCT00641147, Phase 2, 2010–2016 NCT00365209, Phase 2, 2006–2011 NCT02100423, Phase 2, 2014–2018 NCT01246973, Phase 2/3, 2011–2015 NCT01712542, Phase N/A, 2012–2013 NCT03290417, Phase N/A, 2017–2019 NCT01975363, Phase N/A, 2013–2016 NCT01917890, Phase N/A, 2011–2013 NCT03211104, Phase N/A, 2007–2015 NCT00113841, Phase N/A, 2004–2009 NCT03482401, Phase N/A, 2017–2019 NCT00927485, Phase N/A, 2007–2016 | NCT01608139, Phase 1, 2012 NCT00247026, Phase 1/2, 2007 NCT02300727, Phase 1/2, 2015–2018 NCT02095717, Phase 2, 2014–2018 NCT00852332, Phase 2, 2009–2017 NCT02944578, Phase 2, 2017 NCT01269203, Phase 2, 2012 NCT00248053, Phase 2, 2005 NCT00969085, Phase 2, 2012 NCT00003365, Phase N/A, 1996–2006 NCT00118989, Phase N/A, 2005–2012 NCT00176618, Phase N/A, 2004–2007 |
| Combretastatin | Clinical (Total 17 clinical trials: 1 active, 11 completed, and 5 withdrawn/terminated/suspended trials) | NCT02576301, Phase 1/2, 2015 | NCT00395434, Phase 1, 2006–2007 NCT00960557, Phase 1, 2009–2010 NCT00003698, Phase 1, 1998–2003 NCT00003768, Phase 1, 1998–2001 NCT01240590, Phase 1/2, 2011–2016 NCT00653939, Phase 2, 2008–2011 NCT00060242, Phase 2, 2003–2008 NCT00113438, Phase 2, 2005–2007 NCT02132468, Phase 2, 2014–2016 NCT02279602, Phase 2, 2014–2016 NCT00699517, Phase 3, 2008–2013 | NCT01085656, Phase 1, 2011–2016 NCT00077103, Phase 1/2, 2003–2007 NCT00507429, Phase 2/3, 2007–2011 NCT02641639, Phase 2/3, 2016–2017 NCT01701349, Phase 3, 2015–2017 |
| Noscapine | Clinical (Total 2 clinical trials: 2 terminated trials) | NCT00912899, Phase 1, 2007–2010 NCT00183950, Phase 1/2, 2000–2006 | ||
| Maytansinoids as ADC | Clinical (Total 92 clinical trials: 42 active, 37 completed, 13 withdrawn/terminated/suspended trials) | NCT04189211, Phase 1, 2017 NCT03364348, Phase 1, 2017 NCT03102320, Phase 1, 2017 NCT04042051, Phase 1, 2019 NCT03552471, Phase 1, 2018 NCT02996825, Phase 1, 2017 NCT04296942, Phase 1, 2020 NCT02390427, Phase 1, 2015 NCT03126630, Phase 1/2, 2018 NCT04298918, Phase 1/2, 2020 NCT03816358, Phase 1/2, 2019 NCT01565200, Phase 2, 2012 NCT03832361, Phase 2, 2020 NCT03418558, Phase 2, 2015 NCT02675829, Phase 2, 2016 NCT01494662, Phase 2, 2012 NCT01904903, Phase 2, 2013 NCT01853748, Phase 2, 2013 NCT04351230, Phase 2, 2020 NCT02452554, Phase 2, 2015 NCT03225937, Phase 2, 2012 NCT04419181, Phase 2, 2020 NCT03894007, Phase 2, 2019 NCT00781612, Phase 2, 2008 NCT04341181, Phase 2, 2020 NCT02314481, Phase 2, 2017 NCT04197687, Phase 2, 2020 NCT04266249, Phase 2, 2020 NCT04274426, Phase 2, 2020 NCT03587311, Phase 2, 2018 NCT02465060, Phase 2, 2015 NCT03784599, Phase 2, 2018 NCT03726879, Phase 3, 2019 NCT01966471, Phase 3, 2014 NCT04296890, Phase 3, 2020 NCT01702571, Phase 3, 2012 NCT01772472, Phase 3, 2013 NCT03084939, Phase 3, 2017 NCT03529110, Phase 3, 2018 NCT04209855, Phase 3, 2019 NCT04185649, Phase 3, 2018 NCT02226276, Phase N/A, 2015 | NCT03153163, Phase 1, 2017–2018 NCT02696642, Phase 1. 2016–2019 NCT01439152, Phase 1, 2011–2019 NCT01513083, Phase 1, 2012–2014 NCT02824042, Phase 1, 2016–2019 NCT02751918, Phase 1, 2016–2019 NCT02254018, Phase 1, 2002–2014 NCT02038010, Phase 1, 2014–2017 NCT01816035, Phase 1, 2014–2017 NCT02605915, Phase 1, 2015–2019 NCT00934856, Phase 1/2, 2009–2013 NCT00875979, Phase 1/2, 2009–2011 NCT00951665, Phase 1/2, 2009–2013 NCT01638936, Phase 1/2, 2012–2018 NCT01001442, Phase 1/2, 2010–2016 NCT01470456, Phase 2, 2011–2014 NCT01472887, Phase 2, 2012–2016 NCT0261014, Phase 2, 2015–2019 NCT03023722, Phase 2, 2017–2019 NCT02924883, Phase 2, 2016–2020 NCT02289833, Phase 2, 2014–2018 NCT03106077, Phase 2, 2017–2019 NCT01975142, Phase 2, 2013–2019 NCT02254005, Phase 2, 2002–2014 NCT00679211, Phase 2, 2008–2011 NCT00509769, Phase 2, 2007–2009 NCT01196052, Phase 2, 2010–2013 NCT02999672, Phase 2, 2016–2018 NCT00679341, Phase 2, 2008–2012 NCT00943670, Phase 2, 2009–2011 NCT02420873, Phase 2, 2015–2017 NCT00829166, Phase 3, 2009–2015 NCT02631876, Phase 3, 2016–2020 NCT0112018, Phase 3, 2010–2016 NCT01419197, Phase 3, 2011–2015 NCT02131064, Phase 3, 2014–2018 NCT02658734, Phase 4, 2016–2019 | NCT02221505, Phase 1, 2014–2015 NCT03045393, Phase 1, 2017–2018 NCT03455556, Phase 1, 2018–2020 NCT02947152, Phase 1, 2016–2017 NCT02318901, Phase 1/2, 2014–2018 NCT02658084, Phase 1/2, 2017–2018 NCT03836157, Phase 2, 2019 NCT02725541, Phase 2, 2016–2019 NCT02839681, Phase 2, 2016–2018 NCT01702558, Phase 2, 2012–2017 NCT01440179, Phase 2, 2011–2014 NCT01641939, Phase 2/3, 2012–2016 NCT02144012, Phase 3, 2014–2016 |
| Chalcones | Pre-clinical (in vitro cell-based studies) | - | - | - |
| Quercetin | Clinical (Total 10 clinical trials: 7 active, 1 completed, and 2 withdrawn/terminated trials | NCT01912820, Phase 1, 2014 NCT03493997, Phase 2, 2017 NCT01961869, Phase 2, 2013 NCT03476330, Phase 2, 2018 NCT04252625, Phase 2, 2020 NCT02195232, Phase 2/3, 2015 NCT04267874, Phase N/A, 2019 | NCT01732393, Phase 1/2, 2010–2012 | NCT02989129, Early Phase 1, 2018–2020 NCT00003365, Phase N/A, 1996–2006 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, D.; Kanakkanthara, A. Beyond the Paclitaxel and Vinca Alkaloids: Next Generation of Plant-Derived Microtubule-Targeting Agents with Potential Anticancer Activity. Cancers 2020, 12, 1721. https://doi.org/10.3390/cancers12071721
Zhang D, Kanakkanthara A. Beyond the Paclitaxel and Vinca Alkaloids: Next Generation of Plant-Derived Microtubule-Targeting Agents with Potential Anticancer Activity. Cancers. 2020; 12(7):1721. https://doi.org/10.3390/cancers12071721
Chicago/Turabian StyleZhang, Dangquan, and Arun Kanakkanthara. 2020. "Beyond the Paclitaxel and Vinca Alkaloids: Next Generation of Plant-Derived Microtubule-Targeting Agents with Potential Anticancer Activity" Cancers 12, no. 7: 1721. https://doi.org/10.3390/cancers12071721
APA StyleZhang, D., & Kanakkanthara, A. (2020). Beyond the Paclitaxel and Vinca Alkaloids: Next Generation of Plant-Derived Microtubule-Targeting Agents with Potential Anticancer Activity. Cancers, 12(7), 1721. https://doi.org/10.3390/cancers12071721