Abstract
This study investigated the efficacy and safety of pimasertib (MEK1/MEK2 inhibitor) versus dacarbazine (DTIC) in patients with untreated NRAS-mutated melanoma. Phase II, multicenter, open-label trial. Patients with unresectable, stage IIIc/IVM1 NRAS-mutated cutaneous melanoma were randomized 2:1 to pimasertib (60 mg; oral twice-daily) or DTIC (1000 mg/m2; intravenously) on Day 1 of each 21-day cycle. Patients progressing on DTIC could crossover to pimasertib. Primary endpoint: investigator-assessed progression-free survival (PFS); secondary endpoints: overall survival (OS), objective response rate (ORR), quality of life (QoL), and safety. Overall, 194 patients were randomized (pimasertib n = 130, DTIC n = 64), and 191 received treatment (pimasertib n = 130, DTIC n = 61). PFS was significantly improved with pimasertib versus DTIC (median 13 versus 7 weeks, respectively; hazard ratio (HR) 0.59, 95% confidence interval (CI) 0.42–0.83; p = 0.0022). ORR was improved with pimasertib (odds ratio 2.24, 95% CI 1.00–4.98; p = 0.0453). OS was similar between treatments (median 9 versus 11 months, respectively; HR 0.89, 95% CI 0.61–1.30); 64% of patients receiving DTIC crossed over to pimasertib. Serious adverse events (AEs) were more frequent for pimasertib (57%) than DTIC (20%). The most common treatment-emergent AEs were diarrhea (82%) and blood creatine phosphokinase (CPK) increase (68%) for pimasertib, and nausea (41%) and fatigue (38%) for DTIC. Most frequent grade ≥3 AEs were CPK increase (34%) for pimasertib and neutropenia (15%) for DTIC. Mean QoL scores (baseline and last assessment) were similar between treatments. Pimasertib has activity in NRAS-mutated cutaneous melanoma and a safety profile consistent with known toxicities of MEK inhibitors. Trial registration: ClinicalTrials.gov, NCT01693068.
1. Introduction
Dacarbazine (DTIC) was the standard of care (SOC) for metastatic melanoma [1] until agents, such as ipilimumab, nivolumab, and pembrolizumab [2,3,4,5], as well as the oncolytic virus therapy talimogene laherparepvec [6], were approved. BRAF and NRAS mutations occur in approximately 40% and 15–20% of cutaneous melanomas, respectively [7,8]. For BRAF-mutated metastatic melanoma, targeted therapies—such as the BRAF inhibitors dabrafenib, vemurafenib, and encorafenib—are available, which in combination with MEK inhibitors—such as trametinib, cobimetinib, and binimetinib—can help overcome acquired resistance and improve survival and response outcomes [9,10,11,12]. For the more aggressive NRAS-mutated melanomas [13,14], however, there are no specifically targeted therapies available; thus, a significant unmet need remains for new treatment options.
Pimasertib is an orally bioavailable, selective small-molecule MEK1/2 inhibitor. At the recommended phase II dose (RP2D) of 60 mg twice-daily (bid) it has an acceptable safety profile in patients with solid tumors and potential efficacy in patients with BRAF- and/or NRAS-mutated melanoma tumors [15].
This phase II study (NCT01693068) aimed to confirm the efficacy and safety of pimasertib at the RP2D in patients with NRAS-mutated cutaneous melanoma, and to compare this with DTIC, the SOC at the time of study initiation.
2. Results
2.1. Patient Population
One hundred and ninety-four patients were randomized (ITT set; DTIC n = 64, pimasertib n = 130) and 191 received treatment (safety population; DTIC n = 61, pimasertib n = 130) (Figure S2). The most common reasons for discontinuation were PD (118 (62%) patients) and TEAEs (69 (36%) patients). At data cut-off, six (3%) patients were still on treatment (DTIC n = 1, crossover n = 1, pimasertib n = 4).
Baseline characteristics (ITT population) were balanced between the DTIC and pimasertib arms (Table 1; Table S1); most patients had an Eastern Cooperative Oncology Group performance (ECOG PS) of 0 (69% versus 69%, respectively), with a similar percentage of patients with M1c melanoma (67% versus 64%, respectively) and lactate dehydrogenase >upper limit of normal (36% versus 42%, respectively).

Table 1.
Patient demographics and baseline characteristics (ITT analysis set).
2.2. Efficacy
The primary objective of the trial was achieved; progression-free survival (PFS) was significantly improved in the pimasertib arm compared with the DTIC arm (median 13 versus 7 weeks, respectively; hazard ratio (HR) 0.59, 95% confidence interval (CI) 0.42–0.83; P = 0.0022) (Figure 1a). Six-month PFS rates were 17% versus 9%, respectively. Independently assessed PFS supported investigator-assessed PFS outcomes (Figure 1b). Sensitivity analyses including all deaths and all scans supported the investigator-assessed PFS findings (Table S2), although this was not statistically significant when based on independent assessment (P = 0.1454). Subgroup analyses indicated a consistent trend in favor of pimasertib treatment (Figure 1c; Figure S3).
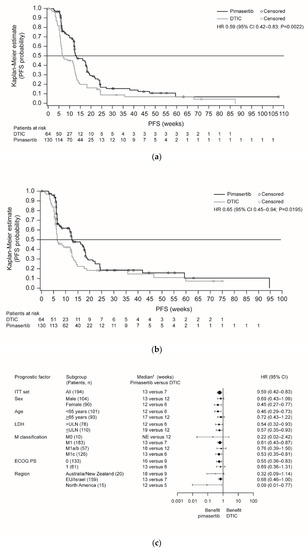
Figure 1.
Kaplan-Meier plots of PFS for pimasertib versus DTIC based on endpoints: (a) read by investigator assessment (primary endpoint); (b) independent assessment of the data (sensitivity analysis); (c) forest plot of PFS for pimasertib versus DTIC based on investigator assessment (ITT analysis set). Abbreviations: CI, confidence interval; DTIC, dacarbazine; ECOG PS, European Cooperative Oncology Group performance status; EU, European Union; HR, hazard ratio; ITT, intent-to-treat; LDH, lactate dehydrogenase; NE, not evaluable; PD, progressive disease; PFS, progression-free survival; ULN, upper limit of normal.
No difference in OS was observed between patients receiving pimasertib and DTIC (median 9 months versus 11 months, respectively; HR 0.89, 95% CI 0.61–1.30) (Figure S4) and no prognostic factors were identified (Figure S5); notably, 64% of patients in the DTIC arm crossed over to pimasertib.
Best overall response data suggested a consistent benefit with pimasertib versus DTIC (Table 2). Investigator-evaluated ORR was 27% for pimasertib and 14% for DTIC (odds ratio 2.24, 95% CI 1.00–4.98, p = 0.0453). Similarly, disease control rate (DCR) was greater for patients receiving pimasertib versus DTIC (odds ratio 2.65, 95% CI 1.23–5.69, p = 0.0106). Independently assessed ORR and DCR data supported these findings, with a trend toward a greater benefit with pimasertib (Table 2).
Table 2.
Best overall response, ORR and DCR based on investigator and independent evaluation of data (ITT analysis set) 1.
2.3. Safety
Patients remained on treatment for a median of 10 versus 7 weeks in the pimasertib and DTIC arms, respectively, and 9 weeks in the crossover group.
The frequency of TEAEs was similar between the arms (100% for pimasertib and crossover versus 98% for DTIC). All patients in the pimasertib and crossover arms and 89% of patients in the DTIC arm had ≥1 treatment-related TEAE.
Serious adverse events (SAEs) (57%, 63%, and 20%) and treatment-related SAEs (45%, 49%, and 7%) were more frequent in the pimasertib and crossover groups than in the DTIC arm, respectively; this was also observed for TEAEs leading to treatment modification (81%, 78%, and 26%) and permanent treatment discontinuations (47%, 39%, and 5%) (Table S3). TEAEs were the primary reason for death in five patients (8%) receiving DTIC and one patient (1%) receiving pimasertib.
The most common TEAEs in the pimasertib and crossover arms were diarrhea (82% and 76%, respectively) and blood CPK increase (68% and 71%, respectively), and, in the DTIC arm, were nausea (41%) and fatigue (38%) (Table S4). More patients receiving pimasertib versus DTIC had grade ≥3 TEAEs (85% versus 41%, respectively; Table 3) and grade ≥3 treatment-related TEAEs (77% versus 28%, respectively). The most frequent treatment-related grade ≥3 TEAEs were increased CPK for pimasertib (34%) and neutropenia (15%) and thrombocytopenia (13%) for DTIC.
Table 3.
Most common grade ≥3 TEAEs (>5% of patients in any treatment group) (safety analysis set) 1.
Ocular events (SRDs) occurred in 58%, 51%, and 31% of patients in the pimasertib, crossover and DTIC groups, respectively; 4%, 5% and 0% of patients, respectively, had retinal vein occlusion (RVO) events (Table S5). Ocular AEs of special interest (AESIs) were largely reversible (Table S5), mild-to-moderate in intensity and had no negative impact on visual acuity.
CPK increases occurred in 57%, 59%, and 28% of patients in the pimasertib, crossover and DTIC groups, respectively, which led to treatment delay/interruption in 31%, 32%, and 0%, and to discontinuation in 13%, 2%, and 0% of patients, respectively. CPK was reversible in 95%, 92%, and 88% of patients, respectively; eight patients had mild-to-moderate muscle pain, otherwise cases were asymptomatic.
At baseline, four patients in the pimasertib arm had a left ventricular ejection fraction (LVEF) <50%; the greatest on-treatment LVEF reduction in these patients was <15%. Of patients with baseline LVEF ≥50% (pimasertib n = 115, crossover n =37), 13 (pimasertib n = 10, crossover n = 3) had a decrease to <45% while on treatment. However, of these 13 patients, most had strong confounding risk factors for LVEF alteration, such as elderly age (65–75 years; n = 8), hypertension (n = 9), decreased hemoglobin (n = 8), and aortic stenosis (n = 1). LVEF alteration led to treatment modification in three patients receiving pimasertib (2%). LVEF was not assessed in patients receiving DTIC for comparison.
No new safety signals for pimasertib were identified.
2.4. Quality of Life
Mean QoL scores were similar between treatment arms. Mean (standard deviation) Functional Assessment of Cancer Therapy (FACT)–General total scores were: 80 (16) and 75 (15) at baseline, and 74 (17) and 70 (15) at end of treatment, for pimasertib and DTIC, respectively (Table S6).
3. Discussion
This study demonstrated a significant improvement in PFS with pimasertib at the RP2D, compared with DTIC, in patients with NRAS-mutated cutaneous melanoma, whether assessed by investigators or independently. The phase III NEMO study observed a similar PFS improvement with the MEK inhibitor binimetinib versus DTIC (HR 0.62; p < 0.001) in treatment-naïve or immunotherapy-pretreated patients with NRAS-mutant metastatic melanoma [16].
Clinically relevant benefits were observed for tumor response with pimasertib over DTIC. This improvement was less pronounced with independent analysis, likely due to differences in PR and SD estimates between these assessments. Notably, a considerable number of patients randomized to DTIC crossed over to pimasertib and may have crossed over too early, which may have influenced not only response outcomes but also OS estimates. Nonetheless, the higher ORR favors pimasertib over DTIC.
The safety profile of pimasertib was consistent with previous studies [15,16,17,18,19] and known MEK inhibitor class effects [16,20,21], with no new safety signals and no impact on QoL. As expected, ocular AESI or CPK increases were seen more frequently in patients who received pimasertib than DTIC; however, the higher than expected incidence of ocular TEAEs with DTIC may reflect the thorough evaluation of ocular toxicity conducted in this study. Although some patients experienced an LVEF decrease while receiving pimasertib, most of these patients had strong confounding factors, such as hypertension, elderly age (≥65 years), and valvular heart disease (aortic stenosis), which are considered major clinical factors for LVEF alteration [22]. Notably, patients were treated for longer with pimasertib than with DTIC and the stringent rules for dose modifications may have resulted in more frequent de-escalation of pimasertib treatment, which is likely reflected in the higher incidence of discontinuation/modification of treatment due to TEAEs with pimasertib. For example, for CPK assessments, treatment discontinuation was based upon laboratory findings/non-resolution, with no integration of further symptomatic findings. However, although dose modifications and interruptions may have affected the overall pimasertib exposure, pimasertib still provided greater efficacy than DTIC.
One limitation of this study is that the comparator, DTIC, is no longer the SOC for metastatic melanoma and has been superseded by targeted and immune therapies [23]. In addition, permanent treatment discontinuations may have affected pimasertib exposure, which, along with the extensive crossover from DTIC to pimasertib, is likely to have affected safety and survival outcomes. Indeed, PD-1 blockade either alone or combined with anti-CLTA-4 as first-line therapy is now considered to be the standard of care of advanced NRAS-mutated melanoma. Anti PD1 alone leads to a best ORR around 40% and a median PFS up to 8 months, while the combination regimen allows for best ORR and a median PFS of 57% and 11.5 months, respectively [24,25].
In conclusion Pimasertib has shown clinical activity in patients with NRAS-mutated cutaneous melanoma and a safety profile that is consistent with the known toxicities of MEK inhibitors. These findings support the potential combination of pimasertib with other agents, such as those targeting the PI3K/mTOR pathway or other MEK pathway components, or immune checkpoint inhibitors.
4. Materials and Methods
4.1. Study Design and Treatment
This phase II, multicenter, randomized, open-label trial was conducted in compliance with the principles of the Declaration of Helsinki, the International Council for Harmonization Note for Guidance on Good Clinical Practice (ICH topic E6, 1996) and all applicable regulatory requirements. this research has been approved by the Quorum Review IRB on 07/03/12 (ethic code: NCT01693068).
Patients were randomized 2:1 (permuted block randomization using an interactive voice response system performed centrally by PPD under the supervision of the Sponsor) to receive pimasertib (60 mg; oral bid) or DTIC (1000 mg/m2; intravenously) on Day 1 of each 21-day cycle (Figure S1). Randomization was stratified by Eastern Cooperative Oncology Group performance status (ECOG PS; 0 versus 1). Patients progressing on DTIC could switch to pimasertib (crossover) if they met the eligibility criteria. After progressive disease (PD), patients were followed up every 6 months to assess survival.
4.2. Patients
Eligible patients were aged ≥18 years and had histologically or cytologically confirmed, unresectable locally advanced or metastatic cutaneous melanoma (stage IIIc or IV (M1a-c)) with a confirmed NRAS mutation, but no prior systemic treatment for locally advanced/metastatic melanoma (see Supplementary information S1 and S2 for full inclusion and exclusion criteria, including protocol amendments).
The study report states the following: For confirmation of entry criteria, centralized testing of N-RAS mutation status was to be done on a tumor sample using the Sanger technique. If the N-RAS status was already known at time of inclusion in the trial, the method used for testing and the date were also to be recorded. N-RAS status had to be known prior to performing trial specific screening assessments and prior to randomization in the trial. Analysis of the presence of different types of genetic variants (e.g., gene mutations, copy number variations, methylations), expression levels pathway associated proteins, and marker of proliferation were to be performed on fresh tumor tissue biopsies or archived tumor tissue.
Patients were recruited from 88 centers in Australia, Europe, Israel, New Zealand, and the USA, from December 2012 to July 2014.
4.3. Assessments
Tumor response was assessed according to Response Evaluation Criteria In Solid Tumors (RECIST, version 1.0), at baseline, Day 1 of Cycles 3, 5, 7, 9, 11, and 13, and every 4 cycles thereafter. Progression-free survival (PFS) was assessed by investigators, and supported by blinded, retrospective, independent central review of all available imaging data. PFS (weeks) was defined as: ([{date of first PD or death or date of censoring} − date of randomization] + 1) / 7. An event was PD or death if it did not occur after 2 or more missed tumor assessments. In patients with only a date of disease progression date or death after 2 or more missed tumor assessments, the PFS time was censored on the minimum of the date of the last tumor assessment and the analysis cut-off date. Median number of weeks rounded to nearest whole number.
Patient reported quality of life (QoL) was assessed by Functional Assessment of Cancer Therapy (FACT)–Melanoma from baseline to last assessment (prior to objective PD).
Safety was monitored throughout the trial and follow-up (30 ± 3 days after last administration). Treatment-emergent adverse events (TEAEs) that started after DTIC subjects had crossed over to pimasertib were counted in the pimasertib (crossover) group. TEAEs were coded according to the most up-to-date version of MedDRA (version 18; protocol amendment September 2015) and graded by investigators according to National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE, version 4.0). The criteria for de-escalation or discontinuation of pimasertib are provided in Supplementary Information S3.
4.4. Statistical Analysis
Trial sample size was calculated based on median PFS with DTIC of 1.6 months for BRAF mutated metastatic melanoma and 2.8 months for unselected patients [17,18]. Assuming median PFS of 2 months under DTIC and a hazard ratio (HR) of 0.57 versus pimasertib, 151 observed events would provide 90% power to detect a difference at a two-sided, 5% significance level. Random (2:1) treatment assignment of 184 patients within 14 months should give a study duration of 34 months (assuming 10% loss to follow-up).
The primary endpoint was investigator-assessed PFS, defined as the duration from randomization to the first documentation of objective PD or death. PD or death were censored if they occurred after ≥2 missed tumor assessments. PFS was compared using a stratified log-rank test, with HR calculated by a Cox proportional hazards model stratified by baseline ECOG PS (0 versus 1). A sensitivity analysis of the primary endpoint was performed using independent central review data, with additional sensitivity analyses including all deaths and scans.
Secondary endpoints included: objective response rate (ORR); disease control rate (DCR; complete response (CR), partial response (PR) and stable disease (SD) for >3 months); PFS at 6 months from randomization; overall survival (OS); change in QoL; TEAEs (occurring within safety follow-up); serious AEs (SAEs); AEs of special interest (AESIs; including serous retinal detachment (SRD) and retinal vein occlusion (RVO), and creatinine phosphokinase (CPK)-related AEs); and deaths. Sensitivity analyses of ORR and DCR were conducted using independent central review data. P-values for secondary endpoints and sensitivity analyses were regarded as exploratory. The primary endpoint, PFS by independent review and OS were also analyzed according to prespecified baseline subgroups.
Efficacy and sensitivity analyses were performed using the intent-to-treat (ITT) population (patients randomized) and safety analyses were carried out in the safety population (patients who received ≥1 dose of study treatment). The cut-off date of the main analysis for the evaluation of primary and secondary objectives was 04 July 2015 (approximately 12 months after randomization of the last patient).
5. Conclusions
Pimasertib has shown clinical activity in patients with NRAS-mutated cutaneous melanoma and a safety profile that is consistent with the known toxicities of MEK inhibitors. These findings support the potential combination of pimasertib with other agents, such as those targeting the PI3K/mTOR pathway or other MEK pathway components, or immune checkpoint inhibitors.
Supplementary Materials
The following are available online at https://www.mdpi.com/2072-6694/12/7/1727/s1, Information S1: Inclusion criteria, Information S2: Exclusion criteria, Information S3: De-escalation or discontinuation criteria, Figure S1: Trial design, Figure S2: Disposition of patients through the trial (CONSORT diagram), Figure S3: Forest plots for PFS: pimasertib versus DTIC (centrally assessed endpoints; ITT analysis set), Figure S4: Kaplan-Meier plot of overall survival: pimasertib versus DTIC, based on investigator reading of data (ITT analysis set), Figure S5: Forest plot of effect of prognostic factors on overall survival (ITT analysis set), Table S1: NRAS mutations identified in enrolled patients (ITT analysis set), Table S2: PFS—primary and sensitivity analyses (ITT analysis set), Table S3: Summary of TEAEs (safety analysis set), Table S4: TEAEs occurring in >25% of patients in any treatment group (safety analysis set), Table S5: TEAEs of special interest (safety analysis set), Table S6: Quality-of-life measures in patients at baseline and end of treatment (ITT analysis set).
Author Contributions
Study concept, C.L. (Celeste Lebbé), C.R., A.S., and G.M.; Study design: C.L. (Celeste Lebbé), C.R., A.S., and G.M.; Data acquisition: W.K., C.L. (Carmen Loquai), C.R., R.C., and E.E.; Quality control of data and algorithms: Data analysis and interpretation: C.L. (Celeste Lebbé), T.L., C.R., A.S., and G.M.; Statistical analysis: A.S.; Manuscript preparation: C.L. (Celeste Lebbé), C.D., T.L., W.K., J.K., L.T., B.G., F.d.B., C.G., J.-J.G., C.L. (Carmen Loquai), V.F., C.R., P.V., R.C., R.I., E.E., A.S., G.M., and B.D.; Manuscript editing: C.L. (Celeste Lebbé), C.D., T.L., W.K., J.K., L.T., B.G., F.d.B., C.G., J.-J.G., C.L. (Carmen Loquai), V.F., C.R., P.V., R.C., R.I., E.E., A.S., G.M., and B.D.; Manuscript review: C.L. (Celeste Lebbé), C.D., T.L., W.K., J.K., L.T., B.G., F.d.B., C.G., J.-J.G., C.L. (Carmen Loquai), V.F., C.R., P.V., R.C., R.I., E.E., A.S., G.M., and B.D. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Merck Serono S.A. (Geneva, Switzerland) and EMD Serono Inc. (Rockland, MA, USA), both businesses of Merck KGaA, Darmstadt, Germany. Representatives of Merck KGaA participated in the study design, the collection, analysis and interpretation of the data, and the development and submission of this manuscript.
Acknowledgments
The authors would like to thank the patients and their families, investigators, co investigators and the study teams at each of the participating centers, and at Merck Healthcare KGaA, Darmstadt, Germany. Trial monitoring, primary quality assurance, data management, statistical analyses and overall operational trial conduct were performed by Pharmaceutical Product Development (PPD), Cambridge, UK. Medical writing assistance was provided by Helen Swainston, Bioscript Science, Macclesfield, UK and funded by Merck Healthcare KGaA, Darmstadt, Germany.
Conflicts of Interest
C.L.(Celeste Lebbé) research grant and personal fees from BMS (advisory board), personal fees from MSD and Merck Serono (advisory boards and symposia), Novartis and Amgen (advisory boards), and participation in advisory boards and symposia for Pierre Fabre. C.D.: personal fees from Amgen, BMS, MSD, Novartis and Roche (advisory boards). T.L.: personal fees from Novartis, Pierre Fabre and MSD, and research funding from Roche. W.K.: No conflicts of interest. J.K.: No conflicts of interest. L.T.: Investigator funds provided to institution. B.G.: No conflicts of interest. F.d.B. personal fees from Pharm Research Associated, Daiichi Sankyo, Ignyta, Novartis, Amgen, Pfizer, Octimet Oncology, Incyte Biosciences, Teofarma, Pierre Fabre, Roche, Nerviano Medical Sciences and Sanofi. C.G. personal fees from Amgen, BMS, LEO, MSD, Novartis and Roche (advisory boards), and research funding from BMS, Novartis and Roche. J.-J.G. personal fees from BMS, MSD, Roche, Novartis, Amgen, Pierre Fabre, Merck Serono, Pfizer and Merck C.L. (Carmen Loquai): personal fees from BMS, Merck, Sanofi, Amgen, Novartis, Roche, Pierre Fabre, Sun Pharma, Biontech, Almiral Hermal and Kiowa Kirin. V.F.: No conflicts of interest. C.R.: Personal fees from Amgen, BMS, Merck, Novartis, Roche, Pierre Fabre, Biothera and MSD (advisory boards). PV No conflicts of interest. C.R.: No conflicts of interest. RI: No conflicts of interest. E.E.: Personal fees from Merck, Novartis, Pierre Fabre and BMS. A.S.: personal fees from Merck KGaA (employee). G.M.: personal fees from Merck KGaA (employee) B.D.: personal fees from BMS, GSK, Novartis and Roche (advisory boards).
References
- Bhatia, S.; Tykodi, S.S.; Thompson, J.A. Treatment of metastatic melanoma: An overview. Oncology 2009, 23, 488–496. [Google Scholar] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus ipilimumab in advanced melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef]
- Rotte, A.; Jin, J.Y.; Lemaire, V. Mechanistic overview of immune checkpoints to support the rational design of their combinations in cancer immunotherapy. Ann. Oncol 2018, 29, 71–83. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Corrigan, P.A.; Beaulieu, C.; Patel, R.B.; Lowe, D.K. Talimogene laherparepvec: An oncolytic virus therapy for melanoma. Ann. Pharmacother. 2017, 51, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Colombino, M.; Capone, M.; Lissia, A.; Cossu, A.; Rubino, C.; De Giorgi, V.; Massi, D.; Fonsatti, E.; Staibano, S.; Nappi, O.; et al. BRAF/NRAS mutation frequencies among primary tumors and metastases in patients with melanoma. J. Clin. Oncol. 2012, 30, 2522–2529. [Google Scholar] [CrossRef] [PubMed]
- Omholt, K.; Karsberg, S.; Platz, A.; Kanter, L.; Ringborg, U.; Hansson, J. Screening of N-RAS codon 61 mutations in paired primary and metastatic cutaneous melanomas: Mutations occur early and persist throughout tumor progression. Clin. Cancer Res. 2002, 8, 3468–3474. [Google Scholar] [PubMed]
- Kakadia, S.; Yarlagadda, N.; Awad, R.; Kundranda, M.; Niu, J.; Naraev, B.; Mina, L.; Dragovich, T.; Gimbel, M.; Mahmoud, F. Mechanisms of resistance to BRAF and MEK inhibitors and clinical update of US Food and Drug Administration-approved targeted therapy in advanced melanoma. Oncol. Targets Ther. 2018, 11, 7095–7107. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Ascierto, P.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Devitt, B.; Liu, W.; Salemi, R.; Wolfe, R.; Kelly, J.; Tzen, C.Y.; Dobrovic, A.; McArthur, G. Clinical outcome and pathological features associated with NRAS mutation in cutaneous melanoma. Pigment Cell Melanoma Res. 2011, 24, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Jakob, J.A.; Bassett, R.L., Jr.; Ng, C.S.; Curry, J.L.; Joseph, R.W.; Alvarado, G.C.; Rohlfs, M.L.; Richard, J.; Gershenwald, J.E.; Kim, K.B.; et al. NRAS mutation status is an independent prognostic factor in metastatic melanoma. Cancer 2012, 118, 4014–4023. [Google Scholar] [CrossRef] [PubMed]
- Awada, A.; Delord, J.P.; Houede, N.; Lebbe, C.; Lesimple, T.; Schellens, J.H.; Rottey, S.; Kefford, R.; Rejeb, N.; Raymond, E. 604 Safety and recommended phase II dose (RP2D) of the selective oral MEK1/2 inhibitor pimasertib (MSC1936369B/AS703026): Results of a phase I trial. Eur. J. Cancer 2012, 48, 185–186. [Google Scholar] [CrossRef]
- Dummer, R.; Schadendorf, D.; Ascierto, P.A.; Arance, A.; Dutriaux, C.; Di Giacomo, A.M.; Rutkowski, P.; Del Vecchio, M.; Gutzmer, R.; Mandala, M.; et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017, 18, 435–445. [Google Scholar] [CrossRef]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef]
- Robert, C.; Thomas, L.; Bondarenko, I.; O’Day, S.; Weber, J.; Garbe, C.; Lebbe, C.; Baurain, J.F.; Testori, A.; Grob, J.J.; et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med. 2011, 364, 2517–2526. [Google Scholar] [CrossRef]
- Ravandi, F.; Pigneux, A.; DeAngelo, D.J.; Raffoux, E.; Delaunay, J.; Thomas, X.; Kadia, T.; Kantarjian, H.; Scheuenpflug, J.; Zhao, C.; et al. Clinical, pharmacokinetic and pharmacodynamic data for the MEK1/2 inhibitor pimasertib in patients with advanced hematologic malignancies. Blood Cancer J. 2015, 5, e375. [Google Scholar] [CrossRef]
- Welsh, S.J.; Corrie, P.G. Management of BRAF and MEK inhibitor toxicities in patients with metastatic melanoma. Ther. Adv. Med. Oncol. 2015, 7, 122–136. [Google Scholar] [CrossRef]
- Duncan, K.E.; Chang, L.Y.; Patronas, M. MEK inhibitors: A new class of chemotherapeutic agents with ocular toxicity. Eye 2015, 29, 1003–1012. [Google Scholar] [CrossRef] [PubMed]
- Bui, A.L.; Horwich, T.B.; Fonarow, G.C. Epidemiology and risk profile of heart failure. Nat. Rev. Cardiol. 2011, 8, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Smalley, K.S.; Eroglu, Z.; Sondak, V.K. Combination therapies for melanoma: A new standard of care? Am. J. Clin. Dermatol. 2016, 17, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Ribas, A.; Schachter, J.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.M.; Lotem, M.; et al. Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): Post-hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study. Lancet Oncol. 2019, 20, 1239–1251. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).