Tumor Cell-Intrinsic Immunometabolism and Precision Nutrition in Cancer Immunotherapy

 , ,
, ,  and
and 
Abstract
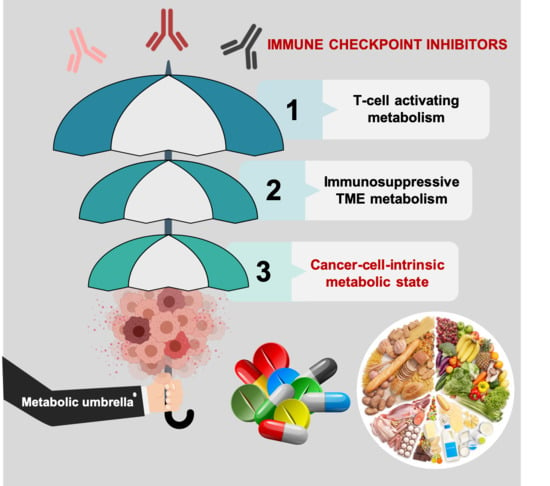
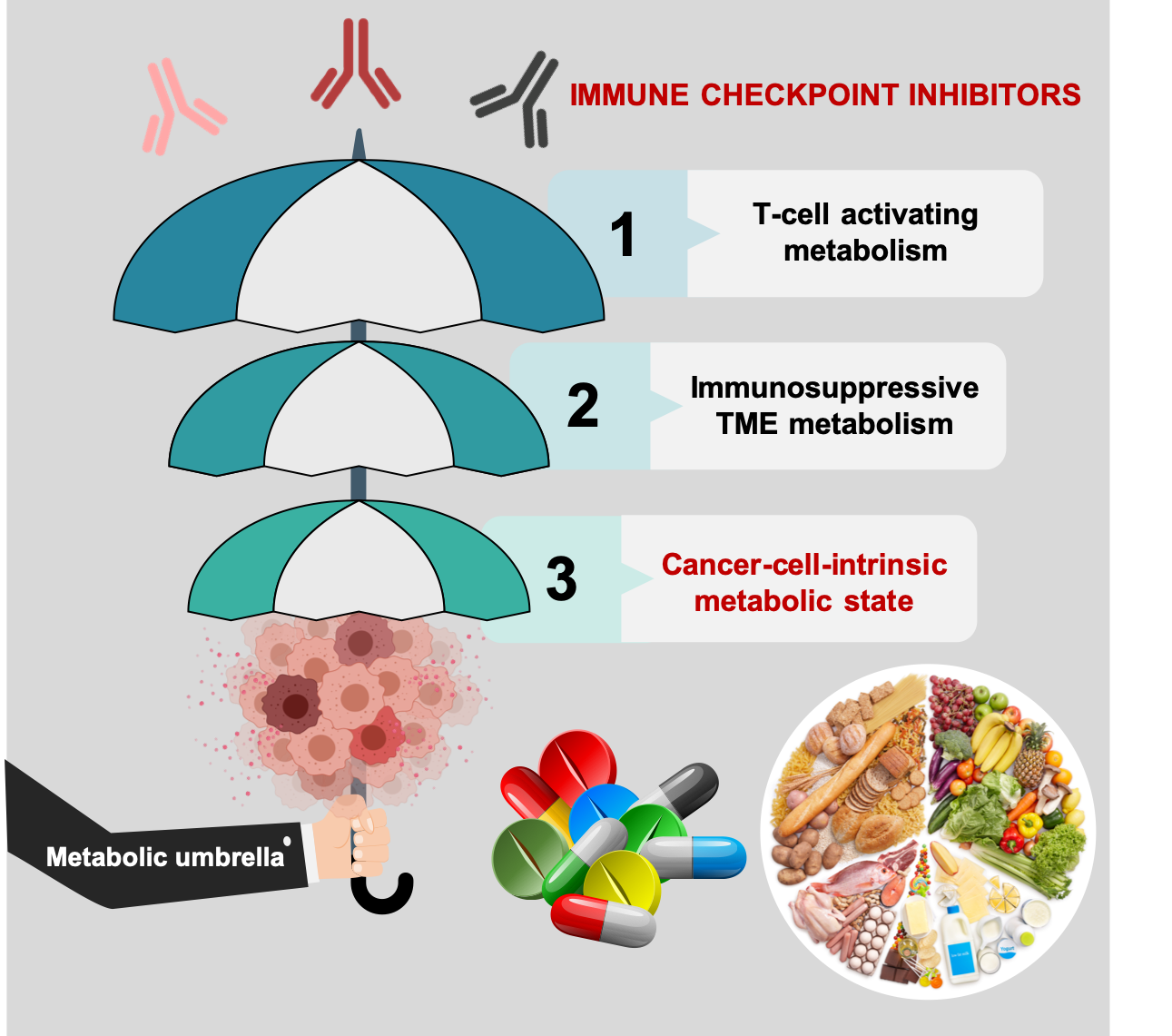
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
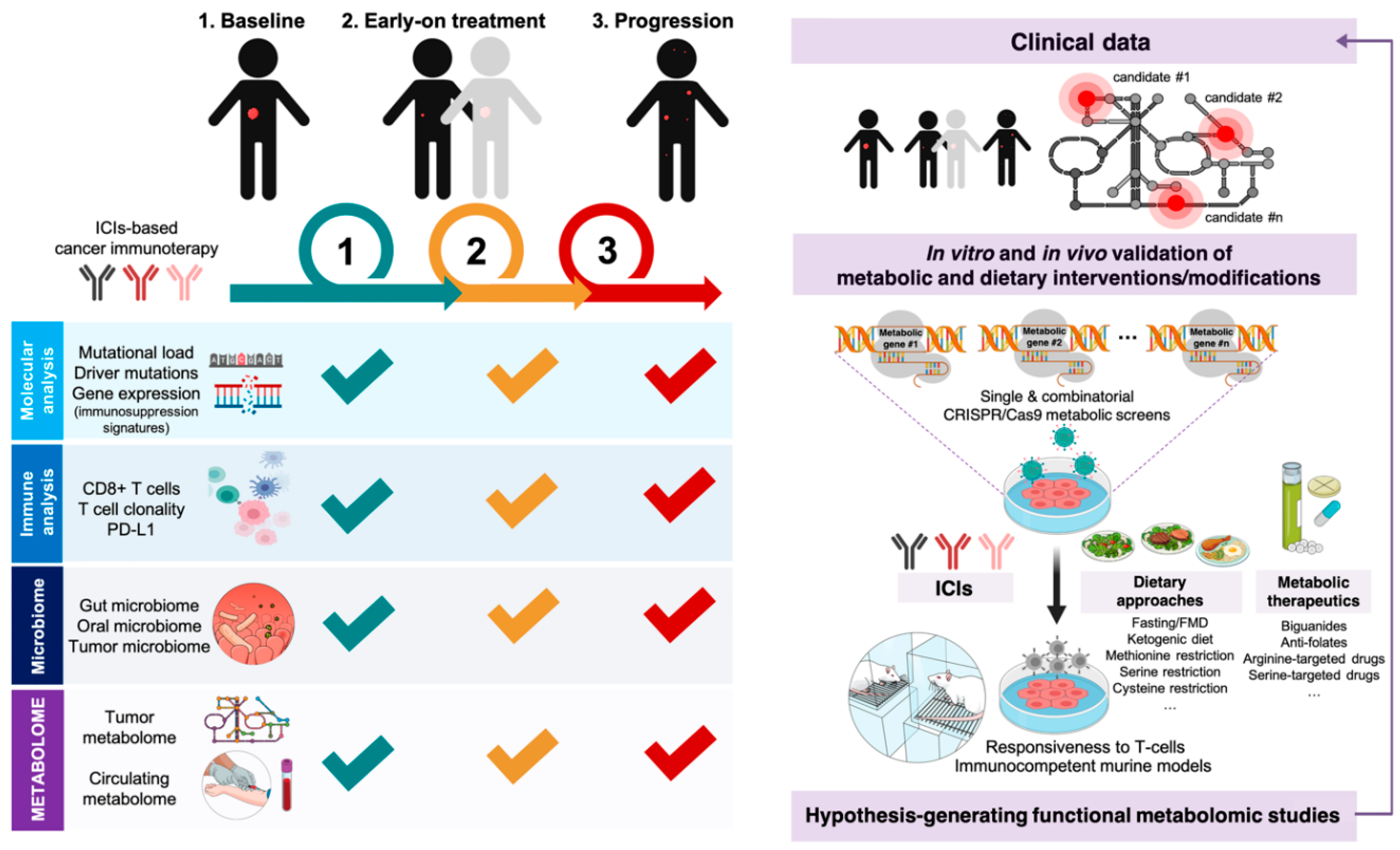{kind=link}
1. Introduction
2. Metabolic Functions of Immune Checkpoints and the Metabolic Nature of Immunosuppressive TME: Battling over Metabolic Resources
2.1. Metabolic Drugs Targeting the Immunosuppressive Nature of TME: A First-in-Class Success of the Cancer Metabolism Pipeline
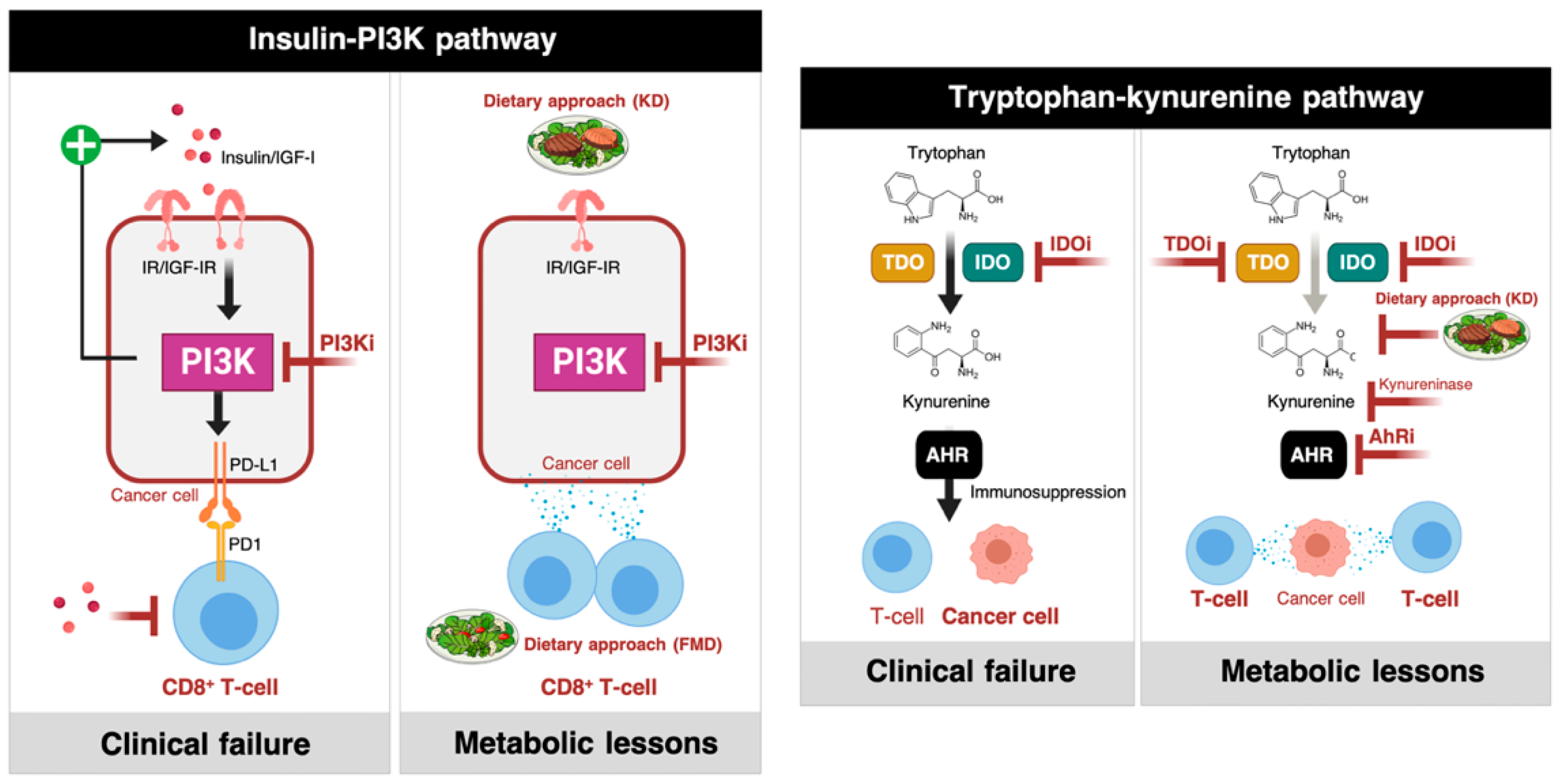
2.1.1. Phosphatidylinositol 3-Kinase (PI3K) Inhibitors and Insulin Signaling
2.1.2. IDO Inhibitors and the Trypophan-Kynurenine Pathway
2.2. Dietary Interventions/Modifications Impacting the Immunosuppressive Traits of TME
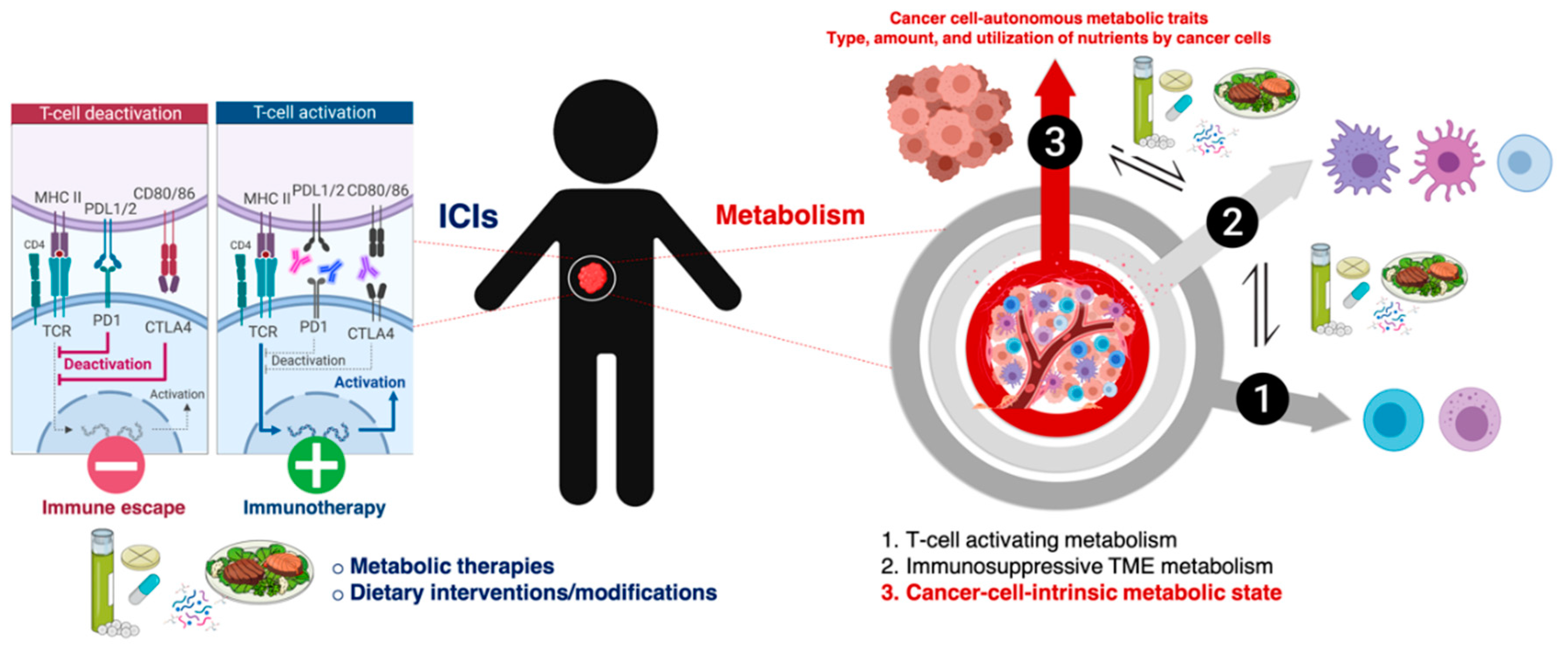
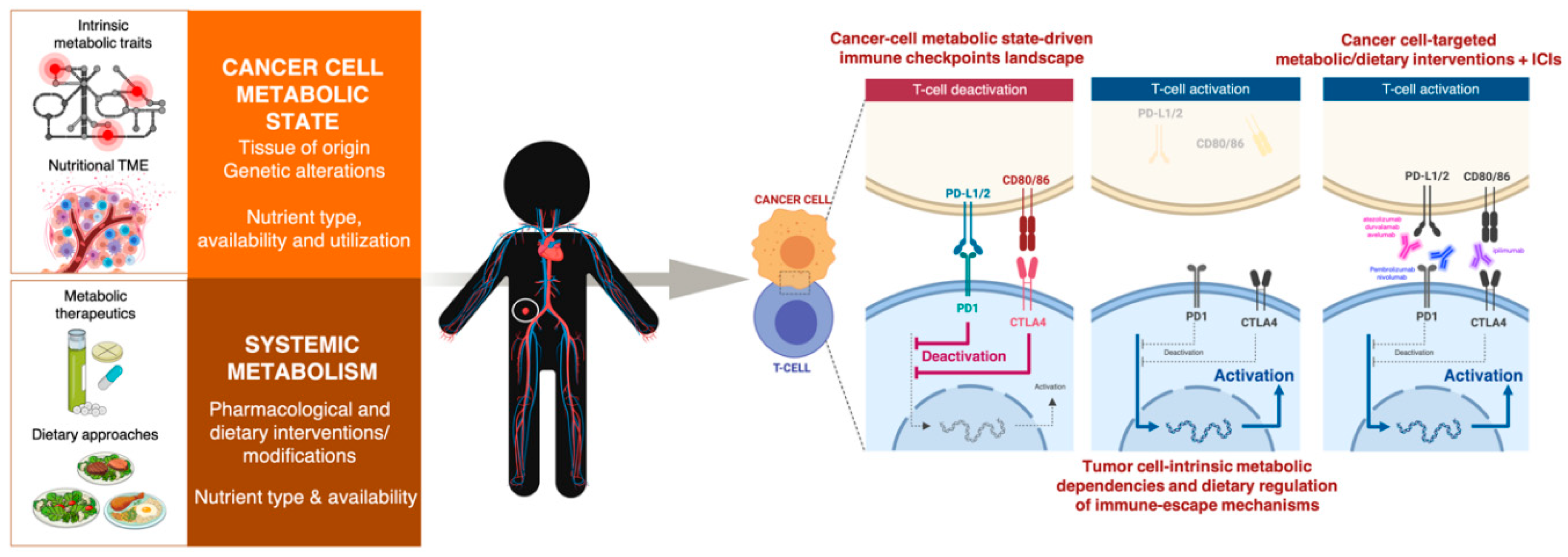
3. Cell-Intrinsic Metabolic Traits and The Immune Checkpoint Composition of Tumor Cells: A Forgotten Dimension of Cancer Immunometabolism
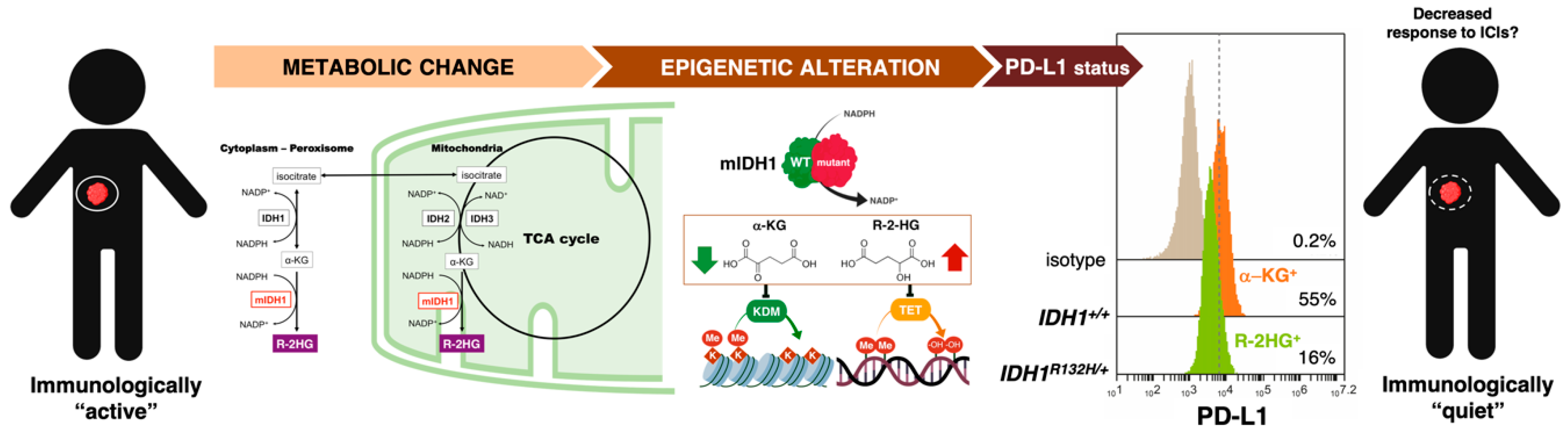
3.1. How Can We Examine the Cancer Cell-Autonomous Metabolic Dependencies of the Immune Checkpoint Landscape?
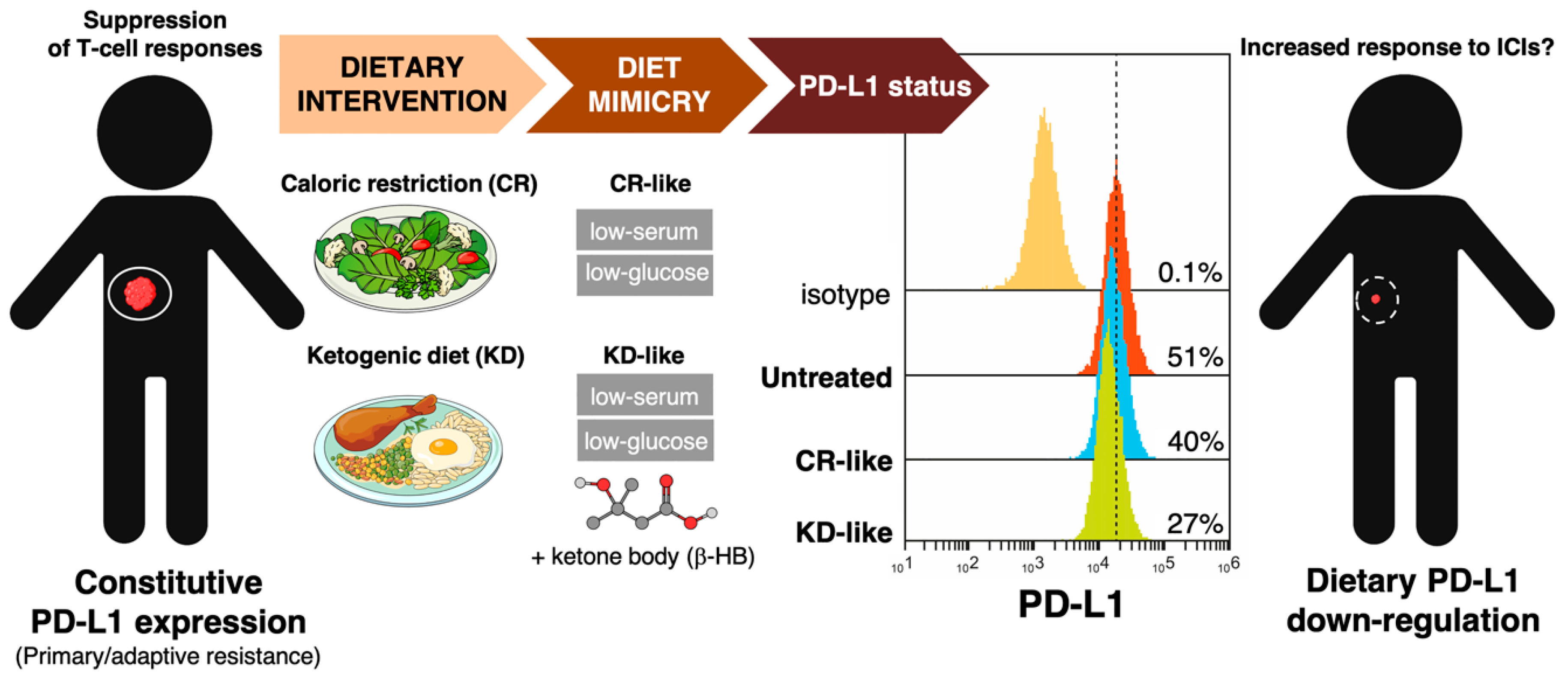
3.2. How Can We Examine the Impact of Dietary Interventions on the Efficacy of Cancer Immunotherapy?
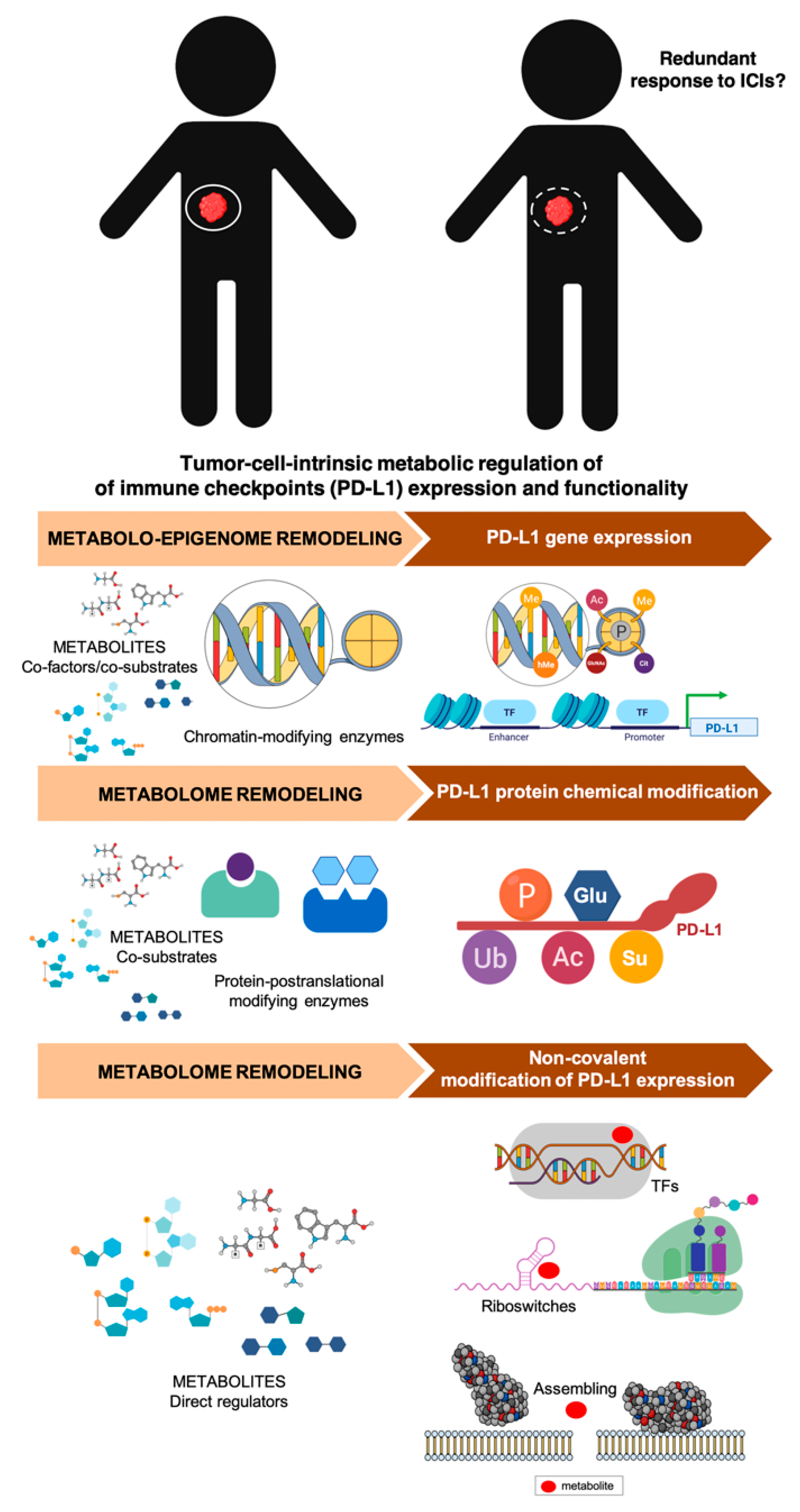
4. Clinical and Molecular Monitoring of Tumor Cell-Intrinsic Metabolic Resistance to Cancer Immunotherapy
5. Gaps and Limitations of Immunotherapy-Boosting Metabolic and Dietary Approaches
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.F.; Sadelain, M. The journey from discoveries in fundamental immunology to cancer immunotherapy. Cancer Cell 2015, 27, 439–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. Immune checkpoint targeting in cancer therapy: Toward combination strategies with curative potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [Green Version]
- Syn, N.L.; Teng, M.W.L.; Mok, T.S.K.; Soo, R.A. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol. 2017, 18, 731–741. [Google Scholar] [CrossRef]
- Li, X.; Shao, C.; Shi, Y.; Han, W. Lessons learned from the blockade of immune checkpoints in cancer immunotherapy. J. Hematol. Oncol. 2018, 11, 31. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Thummalapalli, R.; Carter, J.; Cañadas, I.; Barbie, D.A. Molecular and Genomic Determinants of Response to Immune Checkpoint Inhibition in Cancer. Annu. Rev. Med. 2018, 69, 333–347. [Google Scholar] [CrossRef]
- Conway, J.R.; Kofman, E.; Mo, S.S.; Elmarakeby, H.; Van Allen, E. Genomics of response to immune checkpoint therapies for cancer: Implications for precision medicine. Genome Med. 2018, 10, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegde, P.S.; Chen, D.S. Top 10 Challenges in Cancer Immunotherapy. Immunity 2020, 52, 17–35. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, H.; Ohno, Y.; Toyoshima, Y.; Ohtake, J.; Homma, S.; Kawamura, H.; Takahashi, N.; Taketomi, A. Interleukin-6/STAT3 signaling as a promising target to improve the efficacy of cancer immunotherapy. Cancer Sci. 2017, 108, 1947–1952. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, H.; Fujieda, K.; Miyashita, A.; Fukushima, S.; Ikeda, T.; Kubo, Y.; Senju, S.; Ihn, H.; Nishimura, Y.; Oshiumi, H. Combined Blockade of IL6 and PD-1/PD-L1 Signaling Abrogates Mutual Regulation of Their Immunosuppressive Effects in the Tumor Microenvironment. Cancer Res. 2018, 78, 5011–5022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J.; Ganesh, K. TGF- Inhibition and Immunotherapy: Checkmate. Immunity 2018, 48, 626–628. [Google Scholar]
- Kaplanov, I.; Carmi, Y.; Kornetsky, R.; Shemesh, A.; Shurin, G.V.; Shurin, M.R.; Dinarello, C.A.; Voronov, E.; Apte, R.N. Blocking IL-1β Reverses the Immunosuppression in Mouse Breast Cancer and Synergizes With anti-PD-1 for Tumor Abrogation. Proc. Natl. Acad. Sci. USA 2019, 116, 1361–1369. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Maddocks, O.D.K.; Athineos, D.; Cheung, E.C.; Lee, P.; Zhang, T.; van den Broek, N.J.F.; Mackay, G.M.; Labuschagne, C.F.; Gay, D.; Kruiswijk, F.; et al. Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature 2017, 544, 372–376. [Google Scholar] [CrossRef]
- Hopkins, B.D.; Pauli, C.; Du, X.; Wang, D.G.; Li, X.; Wu, D.; Amadiume, S.C.; Goncalves, M.D.; Hodakoski, C.; Lundquist, M.R.; et al. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 2018, 560, 499–503. [Google Scholar] [CrossRef] [Green Version]
- Nencioni, A.; Caffa, I.; Cortellino, S.; Longo, V.D. Fasting and cancer: Molecular mechanisms and clinical application. Nat. Rev. Cancer 2018, 18, 707–719. [Google Scholar] [CrossRef]
- Allison, K.E.; Coomber, B.L.; Bridle, B.W. Metabolic reprogramming in the tumour microenvironment: A hallmark shared by cancer cells and T lymphocytes. Immunology 2017, 152, 175–184. [Google Scholar] [CrossRef]
- Sugiura, A.; Rathmell, J.C. Metabolic Barriers to T Cell Function in Tumors. J. Immunol. 2018, 200, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Singer, K.; Cheng, W.C.; Kreutz, M.; Ho, P.C.; Siska, P.J. Immunometabolism in cancer at a glance. Dis. Model Mech. 2018, 11. pii: dmm034272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer metabolism pipeline breaks new ground. Nat. Rev. Drug Discov. 2016, 15, 735–737. [CrossRef] [PubMed]
- Li, X.; Wenes, M.; Romero, P.; Huang, S.C.; Fendt, S.M.; Ho, P.C. Navigating metabolic pathways to enhance antitumour immunity and immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 425–441. [Google Scholar] [CrossRef] [PubMed]
- Souza-Fonseca-Guimaraes, F.; Cursons, J.; Huntington, N.D. The Emergence of Natural Killer Cells as a Major Target in Cancer Immunotherapy. Trends Immunol. 2019, 40, 142–158. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Sun, C. The Rise of NK Cell Checkpoints as Promising Therapeutic Targets in Cancer Immunotherapy. Front. Immunol. 2019, 10, 2354. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.; Arooj, S.; Wang, H. NK Cell-Based Immune Checkpoint Inhibition. Front. Immunol. 2020, 11, 167. [Google Scholar] [CrossRef]
- Van den Berg, T.; Valerius, T. Myeloid Immune-Checkpoint Inhibition Enters the Clinical Stage. Nat. Rev. Clin. Oncol. 2019, 16, 275–276. [Google Scholar] [CrossRef]
- Nakamura, K.; Smyth, M.J. Myeloid Immunosuppression and Immune Checkpoints in the Tumor Microenvironment. Cell Mol. Immunol. 2020, 17, 1–12. [Google Scholar] [CrossRef]
- Strauss, L.; Mahmoud, M.A.A.; Weaver, J.D.; Tijaro-Ovalle, N.M.; Christofides, A.; Wang, Q.; Pal, R.; Yuan, M.; Asara, J.; Patsoukis, N.; et al. Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci. Immunol. 2020, 5, eaay1863. [Google Scholar] [CrossRef]
- Kishton, R.J.; Sukumar, M.; Restifo, N.P. Metabolic Regulation of T Cell Longevity and Function in Tumor Immunotherapy. Cell Metab. 2017, 26, 94–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dugnani, E.; Pasquale, V.; Bordignon, C.; Canu, A.; Piemonti, L.; Monti, P. Integrating T cell metabolism in cancer immunotherapy. Cancer Lett. 2017, 411, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Lim, A.R.; Rathmell, W.K.; Rathmell, J.C. The tumor microenvironment as a metabolic barrier to effector T cells and immunotherapy. Elife 2020, 9, e55185. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Phillips, J.B.; Madeira da Silva, L.; Zhou, M.; Fodstad, O.; Owen, L.B.; Tan, M. Interplay between Immune Checkpoint Proteins and Cellular Metabolism. Cancer Res. 2017, 77, 1245–1249. [Google Scholar] [CrossRef] [Green Version]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 6692. [Google Scholar] [CrossRef] [Green Version]
- Qorraj, M.; Böttcher, M.; Mougiakakos, D. PD-L1/PD-1: New kid on the “immune metabolic” block. Oncotarget 2017, 8, 73364–73365. [Google Scholar] [CrossRef]
- Wang, Z.; Aguilar, E.G.; Luna, J.I.; Dunai, C.; Khuat, L.T.; Le, C.T.; Mirsoian, A.; Minnar, C.M.; Stoffel, K.M.; Sturgill, I.R.; et al. Paradoxical effects of obesity on T cell function during tumor progression and PD-1 checkpoint blockade. Nat. Med. 2019, 25, 141–151. [Google Scholar] [CrossRef]
- Cortellini, A.; Bersanelli, M.; Buti, S.; Cannita, K.; Santini, D.; Perrone, F.; Giusti, R.; Tiseo, M.; Michiara, M.; Marino, P.D.; et al. A multicenter study of body mass index in cancer patients treated with anti-PD-1/PD-L1 immune checkpoint inhibitors: When overweight becomes favorable. J. Immunother. Cancer 2019, 7, 57. [Google Scholar] [CrossRef]
- Chaker, M.; Minden, A.; Chen, S.; Weiss, R.H.; Chini, E.N.; Mahipal, A.; Azmi, A.S. Rho GTPase effectors and NAD metabolism in cancer immune suppression. Expert Opin. Ther. Targets 2018, 22, 9–17. [Google Scholar] [CrossRef]
- Mu, L.; Long, Y.; Yang, C.; Jin, L.; Tao, H.; Ge, H.; Chang, Y.E.; Karachi, A.; Kubilis, P.S.; De Leon, G.; et al. The IDH1 Mutation-Induced Oncometabolite, 2-Hydroxyglutarate, May Affect DNA Methylation and Expression of PD-L1 in Gliomas. Front. Mol. Neurosci. 2018, 11, 82. [Google Scholar] [CrossRef] [Green Version]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Yang, T.H.O.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kepp, O.; Loos, F.; Liu, P.; Kroemer, G. Extracellular nucleosides and nucleotides as immunomodulators. Immunol. Rev. 2017, 280, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Soldati, L.; Di Renzo, L.; Jirillo, E.; Ascierto, P.A.; Marincola, F.M.; De Lorenzo, A. The influence of diet on anti-cancer immune responsiveness. J. Transl. Med. 2018, 16, 75. [Google Scholar] [CrossRef] [PubMed]
- Pietrocola, F.; Pol, J.; Vacchelli, E.; Rao, S.; Enot, D.P.; Baracco, E.E.; Levesque, S.; Castoldi, F.; Jacquelot, N.; Yamazaki, T.; et al. Caloric Restriction Mimetics Enhance Anticancer Immunosurveillance. Cancer Cell 2016, 30, 147–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Biase, S.; Lee, C.; Brandhorst, S.; Manes, B.; Buono, R.; Cheng, C.W.; Cacciottolo, M.; Martin-Montalvo, A.; Cabo, R.; Wei, M.; et al. Fasting-Mimicking Diet Reduces HO-1 to Promote T Cell-Mediated Tumor Cytotoxicity. Cancer Cell 2016, 30, 136–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renner, K.; Singer, K.; Koehl, G.E.; Geissler, E.K.; Peter, K.; Siska, P.J.; Kreutz, M. Metabolic Hallmarks of Tumor and Immune Cells in the Tumor Microenvironment. Front. Immunol. 2017, 8, 248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijayan, D.; Young, A.; Teng, M.W.L.; Smyth, M.J. Targeting immunosuppressive adenosine in cancer. Nat. Rev. Cancer 2017, 17, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Allard, D.; Allard, B.; Gaudreau, P.O.; Chrobak, P.; Stagg, J. CD73-adenosine: A next-generation target in immuno-oncology. Immunotherapy 2016, 8, 145–163. [Google Scholar] [CrossRef]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Angelin, A.; Gil-de-Gómez, L.; Dahiya, S.; Jiao, J.; Guo, L.; Levine, M.H.; Wang, Z.; Quinn, W.J., III; Kopinski, P.K.; Wang, L.; et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab. 2017, 25, 1282–1293. [Google Scholar] [CrossRef] [Green Version]
- Bohn, T.; Rapp, S.; Luther, N.; Klein, M.; Bruehl, T.J.; Kojima, N.; Lopez, P.A.; Hahlbrock, J.; Muth, S.; Endo, S.; et al. Tumor immunoevasion via acidosis-dependent induction of regulatory tumor-associated macrophages. Nat. Immunol. 2018, 19, 1319–1329. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Kroemer, G. Potent immunosuppressive effects of the oncometabolite R-2-hydroxyglutarate. Oncoimmunology 2018, 7, e1528815. [Google Scholar] [CrossRef] [PubMed]
- Mockler, M.B.; Conroy, M.J.; Lysaght, J. Targeting T cell immunometabolism for cancer immunotherapy; understanding the impact of the tumor microenvironment. Front. Oncol. 2014, 4, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buck, M.D.; Sowell, R.T.; Kaech, S.M.; Pearce, E.L. Metabolic Instruction of Immunity. Cell 2017, 169, 570–586. [Google Scholar] [CrossRef]
- Kouidhi, S.; Ben Ayed, F.; Benammar Elgaaied, A. Targeting Tumor Metabolism: A New Challenge to Improve Immunotherapy. Front. Immunol. 2018, 9, 353. [Google Scholar] [CrossRef] [Green Version]
- Cheong, J.E.; Sun, L. Targeting the IDO1/TDO2-KYN-AhR Pathway for Cancer Immunotherapy—Challenges and Opportunities. Trends Pharmacol. Sci. 2018, 39, 307–325. [Google Scholar] [CrossRef]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.J.; Kim, T.M.; et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): A phase 3, randomised, double-blind study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef]
- Labadie, B.W.; Bao, R.; Luke, J.J. Reimagining IDO Pathway Inhibition in Cancer Immunotherapy via Downstream Focus on the Tryptophan-Kynurenine-Aryl Hydrocarbon Axis. Clin. Cancer Res. 2019, 25, 1462–1471. [Google Scholar] [CrossRef] [Green Version]
- Muller, A.J.; Manfredi, M.G.; Zakharia, Y.; Prendergast, G.C. Inhibiting IDO Pathways to Treat Cancer: Lessons From the ECHO-301 Trial and Beyond. Semin Immunopathol. 2019, 41, 41–48. [Google Scholar] [CrossRef]
- Goodpaster, B.H.; Sparks, L.M. Metabolic Flexibility in Health and Disease. Cell Metab. 2017, 25, 1027–1036. [Google Scholar] [CrossRef] [Green Version]
- Kreuzaler, P.; Panina, Y.; Segal, J.; Yuneva, M. Adapt and conquer: Metabolic flexibility in cancer growth, invasion and evasion. Mol. Metab. 2020, 33, 83–101. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.L.; Soeters, M.R.; Wüst, R.C.I.; Houtkooper, R.H. Metabolic Flexibility as an Adaptation to Energy Resources and Requirements in Health and Disease. Endocr. Rev. 2018, 39, 489–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herst, P.M.; Berridge, M.V. Cell Hierarchy, Metabolic Flexibility and Systems Approaches to Cancer Treatment. Curr. Pharm. Biotechnol. 2013, 14, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Nanda, C.S.; Venkateswaran, S.V.; Patani, N.; Yuneva, M. Defining a Metabolic Landscape of Tumours: Genome Meets Metabolism. Br. J. Cancer 2020, 122, 136–149. [Google Scholar] [CrossRef]
- Méndez-Lucas, A.; Lin, W.; Driscoll, P.C.; Legrave, N.; Novellasdemunt, L.; Xie, C.; Charles, M.; Wilson, Z.; Jones, N.P.; Rayport, S.; et al. Identifying strategies to target the metabolic flexibility of tumours. Nat. Metab. 2020, 2, 335–350. [Google Scholar] [CrossRef]
- Kanarek, N.; Petrova, B.; Sabatini, D.M. Dietary modifications for enhanced cancer therapy. Nature 2020, 579, 507–517. [Google Scholar] [CrossRef]
- Tajan, M.; Vousden, K.H. Dietary Approaches to Cancer Therapy. Cancer Cell. 2020, 27. [Google Scholar] [CrossRef]
- Hopkins, B.D.; Goncalves, M.D.; Cantley, L.C. Insulin-PI3K Signalling: An Evolutionarily Insulated Metabolic Driver of Cancer. Nat. Rev. Endocrinol. 2020, 16, 276–283. [Google Scholar] [CrossRef]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef]
- Bendell, J.C.; Rodon, J.; Burris, H.A.; de Jonge, M.; Verweij, J.; Birle, D.; Demanse, D.; De Buck, S.S.; Ru, Q.C.; Peters, M.; et al. Dose-Escalation Study of BKM120, an Oral pan-Class I PI3K Inhibitor, in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2012, 30, 282–290. [Google Scholar] [CrossRef]
- Lussier, D.M.; Woolf, E.C.; Johnson, J.L.; Brooks, K.S.; Blattman, J.N.; Scheck, A.C. Enhanced immunity in a mouse model of malignant glioma is mediated by a therapeutic ketogenic diet. BMC Cancer 2016, 16, 310. [Google Scholar] [CrossRef]
- Heischmann, S.; Gano, L.B.; Quinn, K.; Liang, L.-P.; Klepacki, J.; Christians, U.; Reisdorph, N.; Patel, M. Regulation of Kynurenine Metabolism by a Ketogenic Diet. J. Lipid. Res. 2018, 59, 958–966. [Google Scholar] [CrossRef] [Green Version]
- Żarnowska, I.; Wróbel-Dudzińska, D.; Tulidowicz-Bielak, M.; Kocki, T.; Mitosek-Szewczyk, K.; Gasior, M.; Turski, W.A. Changes in Tryptophan and Kynurenine Pathway Metabolites in the Blood of Children Treated with Ketogenic Diet for Refractory Epilepsy. Seizure 2019, 69, 265–272. [Google Scholar] [CrossRef]
- Lettieri-Barbato, D.; Aquilano, K. Pushing the Limits of Cancer Therapy: The Nutrient Game. Front. Oncol. 2018, 8, 148. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Patiño, C.; Bossowski, J.P.; De Donatis, G.M.; Mondragón, L.; Villa, E.; Aira, L.E.; Chiche, J.; Mhaidly, R.; Lebeaupin, C.; Marchetti, S.; et al. Low-Protein Diet Induces IRE1α-Dependent Anticancer Immunosurveillance. Cell Metab. 2018, 27, 828–842.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orillion, A.; Damayanti, N.P.; Shen, L.; Adelaiye-Ogala, R.; Affronti, H.; Elbanna, M.; Chintala, S.; Ciesielski, M.; Fontana, L.; Kao, C.; et al. Dietary Protein Restriction Reprograms Tumor-Associated Macrophages and Enhances Immunotherapy. Clin. Cancer Res. 2018, 24, 6383–6395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, M.R.; Danai, L.V.; Lewis, C.A.; Chan, S.H.; Gui, D.Y.; Kunchok, T.; Dennstedt, E.A.; Vander Heiden, M.G.; Muir, A. Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. Elife 2019, 8, e44235. [Google Scholar] [CrossRef]
- Yin, J.; Ren, W.; Huang, X.; Li, T.; Yin, Y. Protein Restriction and Cancer. Biochim. Biophys. Acta Rev. Cancer 2018, 1869, 256–262. [Google Scholar] [CrossRef]
- Ron-Harel, N.; Ghergurovich, J.M.; Notarangelo, G.; LaFleur, M.W.; Tsubosaka, Y.; Sharpe, A.H.; Rabinowitz, J.D.; Haigis, M.C. T Cell Activation Depends on Extracellular Alanine. Cell Rep. 2019, 28, 3011–3021.e4. [Google Scholar] [CrossRef] [Green Version]
- Ma, E.H.; Bantug, G.; Griss, T.; Condotta, S.; Johnson, R.M.; Samborska, B.; Mainolfi, N.; Suri, V.; Guak, H.; Balmer, M.L.; et al. Serine Is an Essential Metabolite for Effector T Cell Expansion. Cell Metab. 2017, 25, 482. [Google Scholar] [CrossRef] [Green Version]
- Keshet, R.; Szlosarek, P.; Carracedo, A.; Erez, A. Rewiring urea cycle metabolism in cancer to support anabolism. Nat. Rev. Cancer 2018, 18, 634–645. [Google Scholar] [CrossRef] [PubMed]
- Szlosarek, P.W.; Steele, J.P.; Nolan, L.; Gilligan, D.; Taylor, P.; Spicer, J.; Lind, M.; Mitra, S.; Shamash, J.; Phillips, M.M.; et al. Arginine Deprivation With Pegylated Arginine Deiminase in Patients With Argininosuccinate Synthetase 1-Deficient Malignant Pleural Mesothelioma: A Randomized Clinical Trial. JAMA Oncol. 2017, 3, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Feng, Y.; Zhang, Y.; Zhu, X.; Jin, F. L-Arginine Supplementation Inhibits the Growth of Breast Cancer by Enhancing Innate and Adaptive Immune Responses Mediated by Suppression of MDSCs in Vivo. BMC Cancer 2016, 16, 343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 2016, 167, 829–842.e13. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, M.; Ramirez, M.E.; Sierra, R.A.; Raber, P.; Thevenot, P.; Al-Khami, A.A.; Sanchez-Pino, D.; Hernandez, C.; Wyczechowska, D.D.; Ochoa, A.C.; et al. L-Arginine depletion blunts antitumor T-cell responses by inducing myeloid-derived suppressor cells. Cancer Res. 2015, 75, 275–283. [Google Scholar] [CrossRef] [Green Version]
- Mestas, J.; Hughes, C.C. Of mice and not men: Differences between mouse and human immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef] [Green Version]
- Zschaler, J.; Schlorke, D.; Arnhold, J. Differences in innate immune response between man and mouse. Crit. Rev. Immunol. 2014, 34, 433–454. [Google Scholar] [CrossRef] [Green Version]
- Ip, B.C.; Hogan, A.E.; Nikolajczyk, B.S. Lymphocyte roles in metabolic dysfunction: Of men and mice. Trends Endocrinol. Metab. 2015, 26, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Klevorn, L.E.; Teague, R.M. Adapting Cancer Immunotherapy Models for the Real World. Trends Immunol. 2016, 37, 354–363. [Google Scholar] [CrossRef] [Green Version]
- Thomas, A.C.; Mattila, J.T. “Of mice and men”: Arginine metabolism in macrophages. Front. Immunol. 2014, 5, 479. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, P.C.; Ochoa, A.C.; Al-Khami, A.A. Arginine Metabolism in Myeloid Cells Shapes Innate and Adaptive Immunity. Front. Immunol. 2017, 8, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Routy, B.; Gopalakrishnan, V.; Daillère, R.; Zitvogel, L.; Wargo, J.A.; Kroemer, G. The gut microbiota influences anticancer immunosurveillance and general health. Nat. Rev. Clin. Oncol. 2018, 15, 382–396. [Google Scholar] [CrossRef]
- Yi, M.; Yu, S.; Qin, S.; Liu, Q.; Xu, H.; Zhao, W.; Chu, Q.; Wu, K. Gut microbiome modulates efficacy of immune checkpoint inhibitors. J. Hematol. Oncol. 2018, 11, 47. [Google Scholar] [CrossRef] [Green Version]
- Olson, C.A.; Vuong, H.E.; Yano, J.M.; Liang, Q.Y.; Nusbaum, D.J.; Hsiao, E.Y. The Gut Microbiota Mediates the Anti-Seizure Effects of the Ketogenic Diet. Cell 2018, 173, 1728–1741.e13. [Google Scholar] [CrossRef] [Green Version]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [Green Version]
- Wallis, K.F.; Melnyk, S.B.; Miousse, I.R. Sex-Specific Effects of Dietary Methionine Restriction on the Intestinal Microbiome. Nutrients 2020, 12, 781. [Google Scholar] [CrossRef] [Green Version]
- Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Cheng, J.; Duncan, A.E.; Kau, A.L.; Griffin, N.W.; Lombard, V.; Henrissat, B.; Bain, J.R.; et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 2013, 341, 1241214. [Google Scholar] [CrossRef] [Green Version]
- Harel, M.; Ortenberg, R.; Varanasi, S.K.; Mangalhara, K.C.; Mardamshina, M.; Markovits, E. Proteomics of Melanoma Response to Immunotherapy Reveals Mitochondrial Dependence. Cell 2019, 179, 236–250. [Google Scholar] [CrossRef]
- Longo, V.D.; Fontana, L. Calorie restriction and cancer prevention: Metabolic and molecular mechanisms. Trends Pharmacol. Sci. 2010, 31, 89–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanarek, N.; Keys, H.R.; Cantor, J.R.; Lewis, C.A.; Chan, S.H.; Kunchok, T. Histidine catabolism is a major determinant of methotrexate sensitivity. Nature 2018, 559, 632–636. [Google Scholar] [CrossRef] [PubMed]
- Lien, E.C.; Vander Heiden, M.G. A framework for examining how diet impacts tumour metabolism. Nat. Rev. Cancer 2019, 19, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Alarcón, T. Metabostemness: A new cancer hallmark. Front. Oncol. 2014, 4, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; Fillmore, C.M.; Koyama, S.; Wu, H.; Zhao, Y.; Chen, Z.; Herter-Sprie, G.S.; Akbay, E.A.; Tchaicha, J.H.; Altabef, A.; et al. Loss of Lkb1 and Pten leads to lung squamous cell carcinoma with elevated PD-L1 expression. Cancer Cell 2014, 25, 590–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casey, S.C.; Tong, L.; Li, Y.; Do, R.; Walz, S.; Fitzgerald, K.N.; Gouw, A.M.; Baylot, V.; Gütgemann, I.; Eilers, M.; et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 2016, 352, 227–231. [Google Scholar] [CrossRef] [Green Version]
- Marzec, M.; Zhang, Q.; Goradia, A.; Raghunath, P.N.; Liu, X.; Paessler, M.; Wang, H.Y.; Wysocka, M.; Cheng, M.; Ruggeri, B.A.; et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc. Natl. Acad. Sci. USA 2008, 105, 20852–20857. [Google Scholar] [CrossRef] [Green Version]
- Lastwika, K.J.; Wilson, W., 3rd; Li, Q.K.; Norris, J.; Xu, H.; Ghazarian, S.R.; Kitagawa, H.; Kawabata, S.; Taube, J.M.; Yao, S.; et al. Control of PD-L1 Expression by Oncogenic Activation of the AKT-mTOR Pathway in Non-Small Cell Lung Cancer. Cancer Res. 2016, 76, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Palsson-McDermott, E.M.; Dyck, L.; Zasłona, Z.; Menon, D.; McGettrick, A.F.; Mills, K.H.G.; O’Neill, L.A. Pyruvate Kinase M2 Is Required for the Expression of the Immune Checkpoint PD-L1 in Immune Cells and Tumors. Front. Immunol. 2017, 8, 1300. [Google Scholar] [CrossRef] [Green Version]
- Coleman, M.F.; Cozzo, A.J.; Pfeil, A.J.; Etigunta, S.K.; Hursting, S.D. Cell Intrinsic and Systemic Metabolism in Tumor Immunity and Immunotherapy. Cancers (Basel) 2020, 12, 852. [Google Scholar] [CrossRef] [Green Version]
- Bunse, L.; Pusch, S.; Bunse, T.; Sahm, F.; Sanghvi, K.; Friedrich, M.; Alansary, D.; Sonner, J.K.; Green, E.; Deumelandt, K.; et al. Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat. Med. 2018, 24, 1192–1203. [Google Scholar] [CrossRef] [PubMed]
- Micevic, G.; Thakral, D.; McGeary, M.; Bosenberg, M.W. PD-L1 methylation regulates PD-L1 expression and is associated with melanoma survival. Pigment Cell Melanoma Res. 2019, 32, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Verdura, S.; Cuyàs, E.; Lozano-Sánchez, J.; Bastidas-Velez, C.; Llorach-Parés, L.; Fernández-Arroyo, S.; Hernández-Aguilera, A.; Joven, J.; Nonell-Canals, A.; Bosch-Barrera, J.; et al. An olive oil phenolic is a new chemotype of mutant isocitrate dehydrogenase 1 (IDH1) inhibitors. Carcinogenesis 2019, 40, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Catalán, E.; Charni, S.; Jaime, P.; Aguiló, J.I.; Enríquez, J.A.; Naval, J.; Pardo, J.; Villalba, M.; Anel, A. MHC-I modulation due to changes in tumor cell metabolism regulates tumor sensitivity to CTL and NK cells. Oncoimmunology 2015, 4, e985924. [Google Scholar] [CrossRef] [Green Version]
- Marijt, K.A.; Sluijter, M.; Blijleven, L.; Tolmeijer, S.H.; Scheeren, F.A.; van der Burg, S.H.; van Hall, T. Metabolic stress in cancer cells induces immune escape through a PI3K-dependent blockade of IFNγ receptor signaling. J. Immunother. Cancer 2019, 7, 152. [Google Scholar] [CrossRef] [Green Version]
- Rodig, S.J.; Gusenleitner, D.; Jackson, D.G.; Gjini, E.; Giobbie-Hurder, A.; Jin, C.; Chang, H.; Lovitch, S.B.; Horak, C.; Weber, J.S.; et al. MHC proteins confer differential sensitivity to CTLA-4 and PD-1 blockade in untreated metastatic melanoma. Sci. Transl. Med. 2018, 10, eaar3342. [Google Scholar] [CrossRef] [Green Version]
- Garrido, F.; Aptsiauri, N.; Doorduijn, E.M.; Garcia Lora, A.M.; van Hall, T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr. Opin. Immunol. 2016, 39, 44–51. [Google Scholar] [CrossRef] [Green Version]
- Oliveras-Ferraros, C.; Cufí, S.; Vazquez-Martin, A.; Menendez, O.J.; Bosch-Barrera, J.; Martin-Castillo, B.; Joven, J.; Menendez, J.A. Metformin rescues cell surface major histocompatibility complex class I (MHC-I) deficiency caused by oncogenic transformation. Cell Cycle 2012, 11, 865–870. [Google Scholar] [CrossRef] [Green Version]
- Verdura, S.; Cuyàs, E.; Martin-Castillo, B.; Menendez, J.A. Metformin as an archetype immuno-metabolic adjuvant for cancer immunotherapy. Oncoimmunology 2019, 8, e1633235. [Google Scholar] [CrossRef]
- Ho, P.C.; Bihuniak, J.D.; Macintyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Yao-Chen, T.; Cui, G.; Micevic, G.; Perales, T.C.; et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef] [Green Version]
- Choi, I.Y.; Lee, C.; Longo, V.D. Nutrition and fasting mimicking diets in the prevention and treatment of autoimmune diseases and immunosenescence. Mol. Cell. Endocrinol. 2017, 455, 4–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rom-Jurek, E.M.; Kirchhammer, N.; Ugocsai, P.; Ortmann, O.; Wege, A.K.; Brockhoff, G. Regulation of Programmed Death Ligand 1 (PD-L1) Expression in Breast Cancer Cel Lines In Vitro and in Immunodeficient and Humanized Tumor Mice. Int. J. Mol. Sci. 2018, 19, 563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y. Regulatory mechanisms of PD-L1 expression in cancer cells. Cancer Immunol. Immunother. 2018, 67, 1481–1489. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.M.; Li, C.W.; Lai, Y.J.; Hung, M.C. Posttranslational Modifications of PD-L1 and Their Applications in Cancer Therapy. Cancer Res. 2018, 78, 6349–6353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Dang, F.; Ren, J.; Wei, W. Biochemical Aspects of PD-L1 Regulation in Cancer Immunotherapy. Trends Biochem. Sci. 2018, 43, 1014–1032. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Zhang, L.; Li, J.; Li, Y.; Wang, Y.; Xu, Z.X. Recent findings in the Regulation of Programmed Death Ligand 1 Expression. Front. Immunol. 2019, 10, 1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuyàs, E.; Corominas-Faja, B.; Menendez, J.A. The nutritional phenome of EMT-induced cancer stem-like cells. Oncotarget 2014, 5, 3970–3982. [Google Scholar] [CrossRef] [Green Version]
- Sciacovelli, M.; Frezza, C. Metabolic reprogramming and epithelial-to-mesenchymal transition in cancer. FEBS J. 2017, 284, 3132–3144. [Google Scholar] [CrossRef]
- Sciacovelli, M.; Gonçalves, E.; Johnson, T.I.; Zecchini, V.R.; da Costa, A.S.; Gaude, E.; Drubbel, V.A.; Theobald, S.J.; Abbo, S.R.; Tran, M.G.B.; et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 2016, 537, 544–547. [Google Scholar] [CrossRef]
- Menendez, J.A.; Corominas-Faja, B.; Cuyàs, E.; García, M.G.; Fernández-Arroyo, S.; Fernández, A.F.; Joven, J.; Fraga, M.F.; Alarcón, T. Oncometabolic Nuclear Reprogramming of Cancer Stemness. Stem. Cell Rep. 2016, 6, 273–283. [Google Scholar] [CrossRef] [Green Version]
- Colvin, H.; Nishida, N.; Konno, M.; Haraguchi, N.; Takahashi, H.; Nishimura, J.; Hata, T.; Kawamoto, K.; Asai, A.; Tsunekuni, K.; et al. Oncometabolite D-2-Hydroxyglutarate Directly Induces Epithelial-Mesenchymal Transition and is Associated with Distant Metastasis in Colorectal Cancer. Sci. Rep. 2016, 6, 36289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terunuma, A.; Putluri, N.; Mishra, P.; Mathé, E.A.; Dorsey, T.H.; Yi, M.; Stevenson, H.S.; Karoly, E.D.; Chan, K.; Samanta, S.; et al. MYC-driven accumulation of 2-hydroxyglutarate is associated with breast cancer prognosis. J. Clin. Investig. 2014, 124, 398–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, P.; Tang, W.; Putluri, V.; Dorsey, T.H.; Jin, F.; Wang, F.; Minas, T.Z.; Yfantis, H.G.; Lee, D.H.; Sreekumar, A.; et al. ADHFE1 is a breast cancer oncogene and induces metabolic reprogramming. J. Clin. Investig. 2018, 128, 323–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wangpaichitr, M.; Kandemir, H.; Li, Y.Y.; Wu, C.; Nguyen, D.; Feun, L.G.; Kuo, M.T.; Savaraj, N. Relationship of Metabolic Alterations and PD-L1 Expression in Cisplatin Resistant Lung Cancer. Cell Dev. Biol. 2017, 6, 183. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Janji, B.; Abdou, A.; Hasmim, M.; Terry, S.; Tan, T.Z.; Mami-Chouaib, F.; Thiery, J.P.; Chouaib, S. The immune checkpoint ligand PD-L1 is upregulated in EMT-activated human breast cancer cells by a mechanism involving ZEB-1 and miR-200. Oncoimmunology 2017, 6, e1263412. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Van Moer, K.; Marani, V.; Gemmill, R.M.; Tranchevent, L.C.; Azuaje, F. CD47 is a direct target of SNAI1 and ZEB1 and its blockade activates the phagocytosis of breast cancer cells undergoing EMT. Oncoimmunology 2018, 7, e1345415. [Google Scholar] [CrossRef] [Green Version]
- Asgarova, A.; Asgarov, K.; Godet, Y.; Peixoto, P.; Nadaradjane, A.; Boyer-Guittaut, M. PD-L1 expression is regulated by both DNA methylation and NF-kB during EMT signaling in non-small cell lung carcinoma. Oncoimmunology 2018, 7, e1423170. [Google Scholar] [CrossRef] [Green Version]
- Terry, S.; Savagner, P.; Ortiz-Cuaran, S.; Mahjoubi, L.; Saintigny, P.; Thiery, J.P.; Chouaib, S. New insights into the role of EMT in tumor immune escape. Mol. Oncol. 2017, 11, 824–826. [Google Scholar] [CrossRef] [Green Version]
- McLaughlin, J.; Han, G.; Schalper, K.A.; Carvajal-Hausdorf, D.; Pelekanou, V.; Rehman, J. Quantitative Assessment of the Heterogeneity of PD-L1 Expression in Non-Small-Cell Lung Cancer. JAMA Oncol. 2016, 2, 46–54. [Google Scholar] [CrossRef]
- Yue, C.; Jiang, Y.; Li, P.; Wang, Y.; Xue, J.; Li, N. Dynamic change of PD-L1 expression on circulating tumor cells in advanced solid tumor patients undergoing PD-1 blockade therapy. Oncoimmunology 2018, 7, e1438111. [Google Scholar] [CrossRef]
- Rinschen, M.M.; Ivanisevic, J.; Giera, M.; Siuzdak, G. Identification of bioactive metabolites using activity metabolomics. Nat. Rev. Mol. Cell Biol. 2019, 20, 353–367. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.H.; Yang, W.H.; Xia, W.; Wei, Y.; Chan, L.C.; Lim, S.O.; Li, C.-W.; Kim, T.; Chang, S.-S.; Lee, H.-H.; et al. Metformin Promotes Antitumor Immunity via Endoplasmic-Reticulum-Associated Degradation of PD-L1. Mol. Cell 2018, 71, 606–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdura, S.; Cuyàs, E.; Cortada, E.; Brunet, J.; Lopez-Bonet, E.; Martin-Castillo, B.; Bosch-Barrera, J.; Encina, J.A.; Menendez, J.A. Resveratrol targets PD-L1 glycosylation and dimerization to enhance antitumor T-cell immunity. Aging (Albany N. Y.) 2020, 12, 8–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, C.P.; Ferré-D’Amaré, A.R. Long-Range Interactions in Riboswitch Control of Gene Expression. Annu. Rev. Biophys. 2017, 46, 455–481. [Google Scholar] [CrossRef]
- Serganov, A.; Patel, D.J. Molecular recognition and function of riboswitches. Curr. Opin. Struct. Biol. 2012, 22, 279–286. [Google Scholar] [CrossRef] [Green Version]
- Arroyo, J.D.; Jourdain, A.A.; Calvo, S.E.; Ballarano, C.A.; Doench, J.G.; Root, D.E.; Mootha, V.K. A Genome-wide CRISPR Death Screen Identifies Genes Essential for Oxidative Phosphorylation. Cell Metab. 2016, 24, 875–885. [Google Scholar] [CrossRef] [Green Version]
- Reczek, C.R.; Birsoy, K.; Kong, H.; Martínez-Reyes, I.; Wang, T.; Gao, P.; Sabatini, D.M.; Chandel, N.S. A CRISPR screen identifies a pathway required for paraquat-induced cell death. Nat. Chem. Biol. 2017, 13, 1274–1279. [Google Scholar] [CrossRef]
- Birsoy, K.; Wang, T.; Chen, W.W.; Freinkman, E.; Abu-Remaileh, M.; Sabatini, D.M. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 2015, 162, 540–551. [Google Scholar] [CrossRef] [Green Version]
- Cantor, J.R.; Abu-Remaileh, M.; Kanarek, N.; Freinkman, E.; Gao, X.; Louissaint, A., Jr.; Lewis, C.A.; Sabatini, D.M. Physiologic Medium Rewires Cellular Metabolism and Reveals Uric Acid as an Endogenous Inhibitor of UMP Synthase. Cell 2017, 169, 258–272. [Google Scholar] [CrossRef] [Green Version]
- Vande Voorde, J.; Ackermann, T.; Pfetzer, N.; Sumpton, D.; Mackay, G.; Kalna, G.; Nixon, C.; Blyth, K.; Gottlieb, E.; Tardito, S. Improving the metabolic fidelity of cancer models with a physiological cell culture medium. Sci. Adv. 2019, 5, eaau7314. [Google Scholar] [CrossRef] [Green Version]
- Muir, A.; Vander Heiden, M.G. The nutrient environment affects therapy. Science 2018, 360, 962–963. [Google Scholar] [CrossRef] [PubMed]
- Muir, A.; Danai, L.V.; Gui, D.Y.; Waingarten, C.Y.; Lewis, C.A.; Vander Heiden, M.G. Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition. Elife 2017, 6, e27713. [Google Scholar] [CrossRef] [PubMed]
- Tardito, S.; Oudin, A.; Ahmed, S.U.; Fack, F.; Keunen, O.; Zheng, L.; Miletic, H.; Sakariassen, P.Ø.; Weinstock, A.; Wagner, A.; et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat. Cell Biol. 2015, 17, 1556–1568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birsoy, K.; Possemato, R.; Lorbeer, F.K.; Bayraktar, E.C.; Thiru, P.; Yucel, B.; Wang, T.; Chen, W.W.; Clish, C.B.; Sabatin, D.M. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 2014, 508, 108–112. [Google Scholar] [CrossRef] [Green Version]
- Minton, D.R.; Nam, M.; McLaughlin, D.J.; Shin, J.; Bayraktar, E.C.; Alvarez, S.W. Serine Catabolism by SHMT2 Is Required for Proper Mitochondrial Translation Initiation and Maintenance of Formylmethionyl-tRNAs. Mol. Cell 2018, 69, 610–621. [Google Scholar] [CrossRef] [Green Version]
- Zhao, D.; Badur, M.G.; Luebeck, J.; Magaña, J.H.; Birmingham, A.; Sasik, R. Combinatorial CRISPR-Cas9 Metabolic Screens Reveal Critical Redox Control Points Dependent on the KEAP1-NRF2 Regulatory Axis. Mol. Cell 2018, 69, 699–708. [Google Scholar] [CrossRef] [Green Version]
- Daemen, A.; Peterson, D.; Sahu, N.; McCord, R.; Du, X.; Liu, B. Metabolite profiling stratifies pancreatic ductal adenocarcinomas into subtypes with distinct sensitivities to metabolic inhibitors. Proc. Natl. Acad. Sci. USA 2015, 112, E4410–E4417. [Google Scholar] [CrossRef] [Green Version]
- Daemen, A.; Liu, B.; Song, K.; Kwong, M.; Gao, M.; Hong, R.; Nannini, M.; Peterson, D.; Liederer, B.M.; la Cruz, C.; et al. Pan-Cancer Metabolic Signature Predicts Co-Dependency on Glutaminase and De Novo Glutathione Synthesis Linked to a High-Mesenchymal Cell State. Cell Metab. 2018, 28, 383–399. [Google Scholar] [CrossRef] [Green Version]
- Viswanathan, V.S.; Ryan, M.J.; Dhruv, H.D.; Gill, S.; Eichhoff, O.M.; Seashore-Ludlow, B.; Kaffenberger, S.D.; Eaton, J.K.; Shimada, K.; Aguirre, A.J.; et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017, 547, 453–457. [Google Scholar] [CrossRef]
- Schaer, D.A.; Geeganage, S.; Amaladas, N.; Lu, Z.H.; Rasmussen, E.R.; Sonyi, A.; Chin, D.; Capen, A.; Li, Y.; Meyer, C.M.; et al. The Folate Pathway Inhibitor Pemetrexed Pleiotropically Enhances Effects of Cancer Immunotherapy. Clin. Cancer Res. 2019, 25, 7175–7188. [Google Scholar] [CrossRef]
- Corominas-Faja, B.; Quirantes-Piné, R.; Oliveras-Ferraros, C.; Vazquez-Martin, A.; Cufí, S.; Martin-Castillo, B.; Micol, V.; Joven, J.; Segura-Carretero, A.; Menendez, J.A. Metabolomic fingerprint reveals that metformin impairs one-carbon metabolism in a manner similar to the antifolate class of chemotherapy drugs. Aging (Albany N. Y.) 2012, 4, 480–498. [Google Scholar] [CrossRef] [Green Version]
- Cuyàs, E.; Fernández-Arroyo, S.; Buxó, M.; Pernas, S.; Dorca, J.; Álvarez, I.; Martínez, S.; Pérez-Garcia, J.M.; Batista-López, N.; Rodríguez-Sánchez, C.A.; et al. Metformin induces a fasting- and antifolate-mimicking modification of systemic host metabolism in breast cancer patients. Aging (Albany N. Y.) 2019, 11, 2874–2888. [Google Scholar] [CrossRef] [PubMed]
- Brunner, K.T.; Mauel, J.; Cerottini, J.C.; Chapuis, B. Quantitative assay of the lytic action of immune lymphoid cells on 51-Cr-labelled allogeneic target cells in vitro; inhibition by isoantibody and by drugs. Immunology 1968, 14, 181–196. [Google Scholar]
- Peper, J.K.; Schuster, H.; Löffler, M.W.; Schmid-Horch, B.; Rammensee, H.G.; Stevanović, S. An impedance-based cytotoxicity assay for real-time and label-free assessment of T-cell-mediated killing of adherent cells. J. Immunol. Methods 2014, 405, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Henle, A.M.; Erskine, C.L.; Benson, L.M.; Clynes, R.; Knutson, K.L. Enzymatic discovery of a HER-2/neu epitope that generates cross-reactive T cells. J. Immunol. 2013, 190, 479–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erskine, C.L.; Henle, A.M.; Knutson, K.L. Determining optimal cytotoxic activity of human Her2neu specific CD8 T cells by comparing the Cr51 release assay to the xCELLigence system. J. Vis. Exp. 2012, e3683. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [Green Version]
- Nowicki, T.S.; Hu-Lieskovan, S.; Ribas, A. Mechanisms of Resistance to PD-1 and PD-L1 Blockade. Cancer J. 2018, 24, 47–53. [Google Scholar] [CrossRef]
- Kalbasi, A.; Ribas, A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat. Rev. Immunol. 2020, 20, 25–39. [Google Scholar] [CrossRef]
- Blank, C.U.; Haanen, J.B.; Ribas, A.; Schumacher, T.N. Cancer Immunology. The “Cancer Immunogram”. Science 2016, 352, 658–660. [Google Scholar] [CrossRef]
- Pitt, J.M.; Vétizou, M.; Daillère, R.; Roberti, M.P.; Yamazaki, T.; Routy, B.; Lepage, P.; Boneca, I.G.; Chamaillard, M.; Kroemer, G.; et al. Resistance Mechanisms to Immune-Checkpoint Blockade in Cancer: Tumor-Intrinsic and –Extrinsic Factors. Immunity 2016, 44, 1255–1269. [Google Scholar] [CrossRef] [Green Version]
- Wellenstein, M.D.; de Visser, K.E. Cancer-Cell-Intrinsic Mechanisms Shaping the Tumor Immune Landscape. Immunity 2018, 48, 399–416. [Google Scholar] [CrossRef] [Green Version]
- Gibney, G.T.; Weiner, L.M.; Atkins, M.B. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016, 17, e542–e551. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.L.; Roh, W.; Reuben, A.; Cooper, Z.A.; Spencer, C.N.; Prieto, P.A.; Miller, J.P.; Bassett, R.L.; Gopalakrishnan, V.; Wani, K.; et al. Analysis of Immune Signatures in Longitudinal Tumor Samples Yields Insight into Biomarkers of Response and Mechanisms of Resistance to Immune Checkpoint Blockade. Cancer Discov. 2016, 6, 827–837. [Google Scholar] [CrossRef] [Green Version]
- Wargo, J.A.; Reddy, S.M.; Reuben, A.; Sharma, P. Monitoring immune responses in the tumor microenvironment. Curr. Opin. Immunol. 2016, 41, 23–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, S.H.; Newgard, C.B. Integrated metabolomics and genomics: Systems approaches to biomarkers and mechanisms of cardiovascular disease. Circ. Cardiovasc. Genet. 2015, 8, 410–419. [Google Scholar] [CrossRef] [Green Version]
- Burr, M.L.; Sparbier, C.E.; Chan, Y.C.; Williamson, J.C.; Woods, K.; Beavis, P.A.; Lam, E.Y.N.; Henderson, M.A.; Bell, C.C.; Stolzenburg, S.; et al. CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature 2017, 549, 101–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muir, A.; Danai, L.V.; Vander Heiden, M.G. Microenvironmental regulation of cancer cell metabolism: Implications for experimental design and translational studies. Dis. Model Mech. 2018, 11, dmm035758. [Google Scholar] [CrossRef] [Green Version]
- Alsabeeh, N.; Chausse, B.; Kakimoto, P.A.; Kowaltowski, A.J.; Shirihai, O. Cell culture models of fatty acid overload: Problems and solutions. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 143–151. [Google Scholar] [CrossRef]
- Imbert, C.; Montfort, A.; Fraisse, M.; Marcheteau, E.; Gilhodes, J.; Martin, E.; Bertrand, F.; Marcellin, M.; Burlet-Schiltz, O.; Peredo, A.G.; et al. Resistance of melanoma to immune checkpoint inhibitors is overcome by targeting the sphingosine kinase-1. Nat. Commun. 2020, 11, 437. [Google Scholar] [CrossRef]
- Rosario, S.R.; Long, M.D.; Affronti, H.C.; Rowsam, A.M.; Eng, K.H.; Smiraglia, D.J. Pan-cancer analysis of transcriptional dysregulation using The Cancer Genome Atlas. Nat. Commun. 2018, 9, 5330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruggeri, R.M.; Campennì, A.; Giuffrida, G.; Trimboli, P.; Giovanella, L.; Trimarchi, F.; Cannavò, S. Endocrine and metabolic adverse effects of immune checkpoint inhibitors: An overview (what endocrinologists should know). J. Endocrinol, Invest. 2019, 42, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Parthymos, I.; Liamis, G.; Dounousi, E.; Pentheroudakis, G.; Mauri, D.; Zarkavelis, G.; Florentin, M. Metabolic consequences of immune checkpoint inhibitors: A new challenge in clinical practice. Crit. Rev. Oncol. Hematol. 2020, 151, 102979. [Google Scholar] [CrossRef] [PubMed]
- Vajaitu, C.; Draghici, C.C.; Solomon, I.; Lisievici, C.V.; Popa, A.V.; Lupu, M.; Caruntu, C.; Constantin, M.M.; Voiculescu, V.M. The Central Role of Inflammation Associated with Checkpoint Inhibitor Treatments. J. Immunol. Res. 2018, 2018, 4625472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naik, A.; Monjazeb, A.M.; Decock, J. The Obesity Paradox in Cancer, Tumor Immunology, and Immunotherapy: Potential Therapeutic Implications in Triple Negative Breast Cancer. Front. Immunol. 2019, 10, 1940. [Google Scholar] [CrossRef]
- Woodall, M.J.; Neumann, S.; Campbell, K.; Pattison, S.T.; Young, S.L. The Effects of Obesity on Anti-Cancer Immunity and Cancer Immunotherapy. Cancers (Basel) 2020, 12, 1230. [Google Scholar] [CrossRef]
- Rogado, J.; Sánchez-Torres, J.M.; Romero-Laorden, N.; Ballesteros, A.I.; Pacheco-Barcia, V.; Ramos-Leví, A.; Arranz, R.; Lorenzo, A.; Gullón, P.; Donnay, O.; et al. Immune-related adverse events predict the therapeutic efficacy of anti-PD-1 antibodies in cancer patients. Eur. J. Cancer 2019, 109, 21–27. [Google Scholar] [CrossRef]
- Rogado, J.; Romero-Laorden, N.; Sanchez-Torres, J.M.; Ramos-Levi, A.M.; Pacheco-Barcia, V.; Ballesteros, A.I.; Arranz, R.; Lorenzo, A.; Gullon, P.; Garrido, A.; et al. Effect of excess weight and immune-related adverse events on the efficacy of cancer immunotherapy with anti-PD-1 antibodies. Oncoimmunology 2020, 9, 1751548. [Google Scholar] [CrossRef] [Green Version]
- Hotamisligil, G.S.; Erbay, E. Nutrient sensing and inflammation in metabolic diseases. Nat. Rev. Immunol. 2008, 8, 923–934. [Google Scholar] [CrossRef] [Green Version]
- Johnson, A.R.; Milner, J.J.; Makowski, L. The inflammation highway: Metabolism accelerates inflammatory traffic in obesity. Immunol. Rev. 2012, 249, 218–238. [Google Scholar] [CrossRef] [Green Version]
- Hotamisligil, G.S. Foundations of Immunometabolism and Implications for Metabolic Health and Disease. Immunity 2017, 47, 406–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotamisligil, G.S. Inflammation, metaflammation and immunometabolic disorders. Nature 2017, 542, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Lagoudas, G.K.; Zhao, C.; Bullman, S.; Bhutkar, A.; Hu, B.; Ameh, S.; Sandel, D.; Liang, X.S.; Mazzilli, S.; et al. Commensal Microbiota Promote Lung Cancer Development via γδ T Cells. Cell 2019, 176, 998–1013.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xavier, J.B.; Young, V.B.; Skufca, J.; Ginty, F.; Testerman, T.; Pearson, A.T.; Macklin, P.; Mitchell, A.; Shmulevich, I.; Xie, L.; et al. The Cancer Microbiome: Distinguishing Direct and Indirect Effects Requires a Systemic View. Trends Cancer 2020, 6, 192–204. [Google Scholar] [CrossRef] [Green Version]
- Helmink, B.A.; Khan, M.A.W.; Hermann, A.; Gopalakrishnan, V.; Wargo, J.A. The microbiome, cancer, and cancer therapy. Nat. Med. 2019, 25, 377–388. [Google Scholar] [CrossRef]
- Shats, I.; Williams, J.G.; Liu, J.; Makarov, M.V.; Wu, X.; Lih, F.B.; Deterding, L.J.; Lim, C.; Xu, X.; Randall, T.A.; et al. Bacteria Boost Mammalian Host NAD Metabolism by Engaging the Deamidated Biosynthesis Pathway. Cell Metab. 2020, 31, 564–579.e7. [Google Scholar] [CrossRef]
- Bullman, S.; Pedamallu, C.S.; Sicinska, E.; Clancy, T.E.; Zhang, X.; Cai, D.; Neuberg, D.; Huang, K.; Guevara, F.; Nelson, T.; et al. Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science 2017, 358, 1443–1448. [Google Scholar] [CrossRef] [Green Version]
- Bose, S.; Allen, A.E.; Locasale, J.W. The Molecular Link from Diet to Cancer Cell Metabolism. Mol. Cell. 2020, 78, 1034–1044. [Google Scholar] [CrossRef]
- Ke, W.; Saba, J.A.; Yao, C.H.; Hilzendeger, M.A.; Drangowska-Way, A.; Joshi, C.; Mony, V.K.; Benjamin, S.B.; Zhang, S.; Locasale, J.; et al. Dietary serine-microbiota interaction enhances chemotherapeutic toxicity without altering drug conversion. Nat. Commun. 2020, 11, 2587. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuyàs, E.; Verdura, S.; Martin-Castillo, B.; Alarcón, T.; Lupu, R.; Bosch-Barrera, J.; Menendez, J.A. Tumor Cell-Intrinsic Immunometabolism and Precision Nutrition in Cancer Immunotherapy. Cancers 2020, 12, 1757. https://doi.org/10.3390/cancers12071757
Cuyàs E, Verdura S, Martin-Castillo B, Alarcón T, Lupu R, Bosch-Barrera J, Menendez JA. Tumor Cell-Intrinsic Immunometabolism and Precision Nutrition in Cancer Immunotherapy. Cancers. 2020; 12(7):1757. https://doi.org/10.3390/cancers12071757
Chicago/Turabian StyleCuyàs, Elisabet, Sara Verdura, Begoña Martin-Castillo, Tomás Alarcón, Ruth Lupu, Joaquim Bosch-Barrera, and Javier A. Menendez. 2020. "Tumor Cell-Intrinsic Immunometabolism and Precision Nutrition in Cancer Immunotherapy" Cancers 12, no. 7: 1757. https://doi.org/10.3390/cancers12071757
APA StyleCuyàs, E., Verdura, S., Martin-Castillo, B., Alarcón, T., Lupu, R., Bosch-Barrera, J., & Menendez, J. A. (2020). Tumor Cell-Intrinsic Immunometabolism and Precision Nutrition in Cancer Immunotherapy. Cancers, 12(7), 1757. https://doi.org/10.3390/cancers12071757