Abstract
Cell-to-cell communication is a fundamental process in every multicellular organism. In addition to membrane-bound and released factors, the sharing of cytosolic components represents a new, poorly explored signaling route. An extraordinary example of this communication channel is the direct transport of mitochondria between cells. In this review, we discuss how intercellular mitochondrial transfer can be used by cancer cells to sustain their high metabolic requirements and promote drug resistance and describe relevant molecular players in the context of current and future cancer therapy.
1. Introduction
The majority of human cells use mitochondria as the main source of energy and metabolites. A typical cancer cell, however, tends to upregulate glycolysis, as postulated by Otto Warburg more than 100 years ago [1,2]. At first glance, this might seem counterproductive, as glycolysis produces fewer ATP molecules and causes constant acidification of the extracellular space by increased production of lactate [3]. On the other hand, an enhanced glycolytic rate contributes to the development of several cancer hallmarks, such as the ability to evade apoptosis by inhibition of oxidative phosphorylation (OXPHOS) [4] and the promotion of metastatic dissemination by the degradation of the extracellular matrix and tissue outgrowth [5]. Moreover, tumor cells often reside in a hypoxic environment that favors the use of anoxygenic production of energy. Therefore, the idea of forcing tumor cells to use OXPHOS instead of glycolysis has emerged as a promising therapeutic strategy [6,7].
Even though most cancers have impaired mitochondrial respiration, recent discoveries indicate that certain solid tumors, such as pancreatic ductal adenocarcinoma and endometrial carcinoma, and many hematological neoplasms rely heavily on OXPHOS and upregulated mitochondrial metabolism [8,9]. In line with these observations, a number of studies highlighted the importance of mitochondria-dependent metabolic reprogramming in boosting proliferation and in the development of drug resistance in several types of cancers [6,10]. Consequently, the clinical relevance of biological processes involving active and healthy mitochondria, initially meant to have a rather tumor suppressive role, is now being revised.
Historically, cancer research has been mostly done by using 2D in vitro models of established cell lines [11,12]. Although this is a powerful and valuable approach, it completely neglects the presence of neighboring non-tumor cells supporting or suppressing the cancer tissue. The influence of the microenvironment on tumor cells is very complex and often includes the direct involvement of tumor mitochondria. Cancer cells can release (e.g., upon necrosis) entire mitochondria or their components, such as mitochondrial DNA (mtDNA), ATP, cytochrome C, or formylated peptides, to the extracellular space [13]. These then serve as Damage-Associated Molecular Patterns (DAMPs) that activate the immune cells [14,15]. Resulting pro-inflammatory and immunosuppressive responses then either inhibit or stimulate the growth and/or metastatic capacity of the tumor [16,17].
The modulation of tumor mitochondria is an important mechanism that aids cancer cells to escape from the immune system control and develop drug resistance [6,10]. In addition to neoplastic and immune cells, the tumor microenvironment contains many different cell types that can control the state of the mitochondria in a tumor both directly, by cell–cell contacts [18], and indirectly, by secretion of soluble factors and a variety of extracellular vesicles [19]. Recently, a novel mechanism of intercellular communication based on a horizontal transfer of mitochondria between non-tumor and malignant cells was described [20,21,22,23,24]. This paradigm-breaking discovery has led to the question of whether the phenomenon of direct mitochondria sharing could also contribute to the aversion of malignant cells to existing drug combinations and possibly further promote tumor growth. We still know very little about this new, exciting way of sharing intracellular molecules and organelles. A deeper understanding of the underlying molecular mechanisms and consequences on cell physiology will likely explain many therapeutic failures and ultimately lead to novel, more efficient drug combinations.
In this review, we provide an overview of the current knowledge of intercellular mitochondrial transfer, with a particular focus on its relevance in cancer initiation, progression, and drug resistance. We present a summary of the known molecular players involved in sharing mitochondria and show examples of mitochondrial exchange in both solid and hematological tumors. Finally, we place all findings in the context of the current therapeutic strategies.
2. Means of Mitochondrial Transfer
The first observation of mitochondrial transfer in 2006 demonstrated that mitochondria from bone marrow stromal cells (BMSCs), but not free mitochondria or mtDNA from the medium, were able to relocate to mitochondria-deficient A549 lung cancer cells and rescue their aerobic respiration [18]. A follow-up study further supported the regulated directionality of the exchange as donor, non-irradiated PC12 cells with faulty mitochondria could not nourish recipient PC12 cells, leading them back to life [25]. Importantly, the transfer of mitochondria was also observed in vivo in mouse melanoma cells injected into mice expressing a fluorescently labelled mitochondrial protein [24].
Fundamental research on the physiological relevance of mitochondrial transfer suggested its importance in the regeneration of damaged or infected tissue [26,27]. The “mito-healing” theory was subsequently supported by additional studies in a variety of tissues, including vascular [28], brain [19], lung [29], cornea [30], and several other tissues. Transfer of mitochondria was also described in the immune system to combat bacterial infections [31]. The presence of pathogens inside immune cells is usually accompanied by a switch from glycolytic metabolism to OXPHOS as a means of triggering a fast anti-microbial response [32]. A striking example of infection-induced metabolic switch is seen in macrophages during acute respiratory distress syndrome, when macrophages are fed additional mitochondria by surrounding mesenchymal stem cells (MSCs), boosting their anti-inflammatory and phagocytic capacity [31]. Thus, the importance of mitochondria exchange in maintaining tissue homeostasis is clearly not disputable. However, the shuttling of mitochondria between cells could also have severe pathological consequences, particularly in cancer, where malignant cells tend to take advantage of the surrounding environment (Figure 1) [23,33,34].
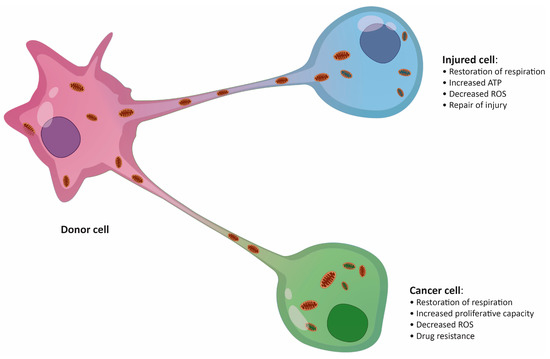
Figure 1.
Putative effects of mitochondrial transfer on an injured cell and a cancer cell. ROS, reactive oxygen species.
The precise mechanism of mitochondrial transfer remains unknown, and so far, only a few crucial molecules involved in the process have been described. High OXPHOS demand and/or severe mitochondria damage are typical features of the recipient cells [23,35,36]. To initiate the transfer, the providing cells should not only possess non-damaged, healthy mitochondria [37] but also be specifically activated [22]. Metalloproteinase-1 (MMP-1), nestin, and proinflammatory cytokines have been identified as essential factors stimulating donor cells to dispatch mitochondria [22]. When cultured with leukemic cells, donor BMSCs exhibited increased levels of PGC1α, a master regulator of mitochondrial biogenesis necessary for efficient mitochondrial transfer [38]. The activation of donor functions often correlates with a rise in intracellular reactive oxygen species (ROS) [22,37] generated by the recipient cells [33]. The main trigger regulating the directional release of mitochondria is still unknown, but the signal is likely multifactorial and, at least partially, mediated by ROS.
3. Tunneling Nanotubes Are the Main Delivery Route for Mitochondria
Neighboring cells can share mitochondria through several mechanisms, including (i) the formation of extracellular vehicles (EVs), (ii) tunneling nanotubes (TNTs) formed at the sites of physical contact, (iii) mitochondrial ejection, or (iv) cytoplasmic fusion [39]. Multiple studies have shown that TNTs, ultrafine cytoplasmic bridges between cells, are the main delivery system for mitochondria in both healthy and tumor tissues (Figure 2) [23,33]. TNT-independent mitochondria sharing in certain tumors was also described [19,29,37]. However, there seems to be a rather scarce number of examples, and more investigation of the transfer mechanisms is needed.
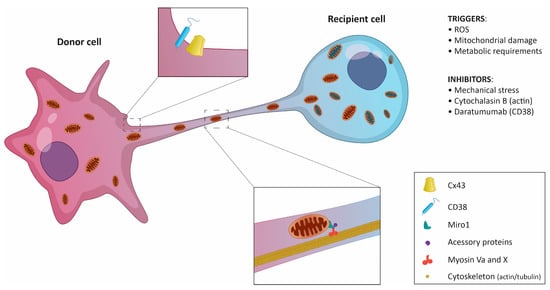
Figure 2.
Schematic representation of mitochondrial transfer via tunneling nanotubes. The donor cell, usually a non-cancerous cell, sends mitochondria to the recipient cell. In certain cases, the transfer is possible in both directions. Key regulatory molecules and triggers are listed.
TNTs, discovered by Rustom et al. in 2004, serve as direct communication channels between neighboring cells to exchange a wide variety of molecules and organelles [40]. Their diameter ranges from 50 to 200 nm, and their length may reach up to 150 μm. TNTs are formed de novo in a matter of minutes and can connect multiple cells at once. Specific structural features or biomarkers unique to TNTs have not been identified. TNTs lack any attachment to the substrate, but their structure is enforced by actin [40] and microtubule filaments [25,41]. In experimental settings, the actin-disrupting drug cytochalasin B is often used as an efficient inhibitor of TNTs formation [23,33].
For the efficient transport of mitochondria, the presence of functional microtubules and associated molecular motors seems to be required [25]. Specifically, Myosin X and Myosin Va have been co-localized with mitochondria inside TNTs [40,42,43]. Some donor cells exhibit high expression of the small GTPase Miro1 localized on the outer mitochondrial membrane [44]. When Miro1 was depleted in MSCs cultured with LA-4 epithelial cells, mitochondrial transfer was ineffective, whereas Miro1 overexpression increased the ability of MSCs to donate mitochondria [45]. Mechanistically, Miro1 seems to co-ordinate mitochondrial movement along microtubules by promoting the assembly of a complex molecular motor machinery [46].
The role of TNT-localized actin in mitochondria exchange is less clear. It is known that filamentous actin impedes the passive transfer of soluble cytoplasmic molecules through TNTs [40] and serves as a scaffold that stabilizes TNT structure [47]. The actin-binding protein M-Sec has been shown to be important for the formation of TNTs and the intercellular propagation of calcium (Ca2+) in macrophages [48,49]. Increased levels of Ca2+ activate mitochondria-localized Miro1 that further binds to microtubule-associated motor proteins [46].
Recently, CD38, an ectoenzyme involved in transmembrane signaling and cell adhesion, was identified as one of the key players in mitochondrial transfer [19,23]. In addition to its receptor function, CD38 modulates intracellular Ca2+ levels by generating cyclic ADP-ribose [50,51]. The Ca2+ regulatory role of CD38 seems important for mitochondrial delivery from BMSCs to myeloma cells [23] and from astrocytes to neurons in brain tissue damaged by stroke [19], even though the transfer occurs via a different mechanism (TNTs vs. EVs, respectively). Thus, an increase in intracellular Ca2+ might be a general mechanism priming mitochondria for transfer. However, it is unclear how CD38 enzymatic activity is initiated to raise cytosolic Ca2+ levels and further promote mitochondrial transfer. It is possible that an increase in extracellular NAD+, the substrate of CD38, induced by changes in the cellular redox state could play a role [52].
Interestingly, the presence of gap junction (GJ) proteins, especially connexin 43 [53], in TNTs has been confirmed [54]. Connexins are important for Ca2+ propagation between neighboring cells via TNTs [48,55,56]. Similarities in the composition of GJ and TNTs have also been pointed out [40]. In spite of this, GJ have been functionally distinguished from TNTs [55]. While GJ perform short-range cell-to-cell interactions and allow the transfer of molecules up to 1.2 kDa only [57,58], TNTs mediate long-range cell-to-cell interactions and allow the transfer of significantly bigger cargos [59,60]. More research will be needed to fully understand the contribution of GJ proteins to TNT formation and mitochondrial transfer.
4. Mitochondrial Transfer in Solid Cancers
The current knowledge on mitochondrial transfer in solid cancers is limited, and the results of various studies are often difficult to compare due to the use of different experimental systems (Table 1). Cells of mesenchymal origin or fibroblasts are the most commonly used mitochondria donors [18,21,36]. However, the tumor tissue is a complex environment composed of many different cell types, and competition between cells could have a significant effect on mitochondrial transfer [61]. Indeed, in the co-culture of BMSCs, endothelial cells, and MCF7 cells, the formation of TNTs between the three cell types was observed, but mitochondria were sent only from endothelial cells to MCF7 cells [21]. Recently, TNT-mediated mitochondrial transfer was also observed between natural killer T cells and breast cancer cells [62]. These studies indicate that the ability to donate mitochondria might be affected by the presence of a particular cell type in the tumor microenvironment. Therefore, results of mitochondrial transfer experiments using cells usually not present in the solid tumor microenvironment (e.g., BMSCs or umbilical cord Wharton’s jelly cells) should be carefully interpreted. Nevertheless, the current data still provide interesting views of the general mechanisms of mitochondria shuttling.

Table 1.
Mitochondrial transfer studies.
Investigations of the mechanism of intercellular mitochondrial transfer in solid tumors point to TNTs as the main delivery route [21,45,63]. The very first mitochondrial transfer was seen between mesenchymal and lung cancer cells with severely damaged or completely missing mitochondria [18]. To promote mitochondrial transfer, many studies used inhibitors such as rotenone to block the electron transport in the respiratory chain [23,33,37,45,65]. Thus, the OXPHOS status in the recipient cells seems to be a crucial factor mediating mitochondrial transfer. However, no mitochondrial transfer occurred between BMSCs and osteosarcoma cells with pathogenic mutations in mtDNA, which encodes critical components of the OXPHOS system [35]. Moreover, BMSCs were shown to donate mitochondria to ovarian and breast cancer cells [21], lung adenocarcinoma cells [45], and prostate cancer cells [63] with no apparent mitochondrial damage. This suggests that not only the state of mitochondria but also specific metabolic requirements could play a role in promoting mitochondrial transfer.
5. Mitochondrial Transfer in Hematological Malignancies
The tumor microenvironment is critical for progression and drug resistance also in hematological cancers [11,59,60]. Acquiring new mitochondria in the bone marrow niche was recently suggested as a way by which leukemic cells can achieve drug resistance [22,23,37]. Up to date, mitochondrial transfer was observed in several types of hematological malignancies, where it seems to have a pro-tumor function [32,33,61].
6. Mitochondrial Transfer in Acute Lymphoblastic Leukemia
The cross-talk between acute lymphoblastic leukemia (ALL) cells and their niche proved that mitochondria are delivered via TNTs from MSCs to primary B-cell precursor ALL cells [22]. The presence of TNTs facilitated the signaling from ALL cells towards MSCs, affecting the release of cytokines and chemokines in the microenvironment, supporting the survival and chemoresistance of ALL cells [34]. In addition to B-cell ALL, transport of mitochondria from MSCs was later shown in T-cell ALL (T-ALL), where mitochondrial transfer was mediated by T-ALL cell/MSC adhesion and occurred through TNTs. However, in contrast to B-ALL cells, mitochondria were exported from malignant T-ALL cells to the surrounding MSCs, probably due to the preferential use of glycolysis in T-ALL cells [64]. This example provides a unique model that can help to uncover the signals and mechanisms driving the transfer directionality. Moreover, it would be interesting to see whether T-ALL cells could donate mitochondria to other cell types and whether the increase in mitochondria number in MSCs would directly support the tumoral properties of T-ALL cells.
7. Mitochondrial Transfer in Acute Myeloid Leukemia
Acute myeloid leukemia (AML) is a typical hematological malignancy highly dependent on OXPHOS [66]. Not surprisingly, AML cells are more prone to receive new mitochondria as compared to healthy CD34+ hematopoietic stem/progenitors or lymphoid CD3+ cells [37]. When co-cultured with human BMSCs, AML cells can gain additional mitochondria in a TNT-dependent process [33]. However, in other study, endocytic inhibitors blocked the mitochondria exchange between murine MS-5 BMSCs and human AML cells [37], suggesting a TNT-independent delivery.
Commonly used chemotherapeutics such as cytarabine [67], etoposide [68], and doxorubicin [69] are factors that promote mitochondrial uptake by AML cells [70]. Consequently, the treatment could have a pro-tumor effect by stimulating oxidative metabolism in resistant AML clones [12]. The surface molecule CD38 is another clinically relevant target in AML, critical for the transport of mitochondria from BMSCs to AML cells [71]. Daratumumab, a monoclonal anti-CD38 antibody approved for the treatment of multiple myeloma (MM) [72,73], was shown to block the delivery of mitochondria to AML cells under both in vitro and in vivo conditions, decrease the oxygen consumption rate (OCR), and inhibit the growth of leukemic cells [74,75]. These studies suggest a novel, previously unexpected anti-tumor mechanism of anti-CD38 therapy.
8. Mitochondrial Transfer in Multiple Myeloma
Aberrant myeloma cells reside in the hypoxic environment of the bone marrow [76]. Unexpectedly, primary multiple myeloma (MM) cells isolated from patients’ biopsies were shown to have a higher basal OCR compared to long-term in vitro cultured MM cell lines [23]. Similarly, when established MM cell lines were injected into a mouse or co-cultured with BMSCs, their OCR and ATP production significantly increased [23]. Moreover, the presence of BMSCs enhanced mitochondrial metabolism and drug resistance in MM cells [23]. This suggests that the bone marrow microenvironment might stimulate aerobic respiration in MM cells. A deeper investigation of the responsible mechanism indicated the presence of TNT-mediated mitochondrial transfer from BMSCs to MM cells, and ROS-inducing compounds, including commonly used proteasome inhibitors, significantly potentiated this process [23,77].
CD38, a crucial player in mitochondrial transfer, is currently one of the most attractive molecules for targeted therapy in MM patients [74,78,79,80]. Treatment with an anti-CD38 antibody or genetic deletion of CD38 were shown to inhibit mitochondrial transfer from BMSCs to MM cells and induce tumor shrinkage in xenografts models [23]. Lower mitochondrial activity, particularly a drop in OXPHOS, was previously associated with increased sensitivity of MM cells to proteasome inhibitors [77]. In line with these observations are clinical data indicating high efficacy of a combinatory therapy using an anti-CD38 antibody and proteasome inhibitors [81]. On the other hand, proteasome inhibitors were shown to downregulate the expression of vascular cell adhesion molecule 1 (VCAM-1) on BMSCs [82], a major ligand for VLA-4 on MM cells, and thus impede the binding of BMSCs and MMs [83], that is required for efficient mitochondrial transfer.
9. Conclusions
The direct transfer of mitochondria from one cell to another has emerged as a thrilling mechanism, whose potential targeting offers great opportunities for both cancer therapy and tissue regeneration. The fact that mitochondrial transfer seems to be executed in a similar way in both solid and hematological cancers further multiplies the importance of this process. It also underlines the significance of tumor microenvironment and cellular plasticity in cancer progression and drug resistance. Furthermore, the involvement of mitochondrial transfer may offer an explanation for the yet unclear mechanisms of action of certain anti-cancer drugs. Although the entire signaling machinery driving mitochondrial transfer is still unknown, the discovery of key molecular players such as Miro1, connexin 43, and CD38 has already opened the doors for possible therapeutic targeting. Future research of the molecular processes governing mitochondria shuttling in both normal and pathological settings will likely bring many exciting discoveries and provide new therapeutic possibilities to improve tissue regeneration and cancer therapy.
Author Contributions
H.S. and M.S. wrote the original draft and designed the figures. M.T. prepared the figures. T.J.; M.H.; T.S.; A.K.-K., J.R.B. and R.H. reviewed and edited the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by The Czech Science Foundation (GA CR 19-25354Y), Institutional Development Plan of University of Ostrava (project no. IRP03_2018-2020), ERDF-Project ENOCH (No. CZ.02.1.01/0.0/0.0/16_019/0000868), and New Directions of Biomedical Research in the Ostrava Region (No. CZ.02.1.01/0.0/0.0/18_069/0010060), SGS03/LF/2019-2020, SGS03/PrF/2019-2020.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Warburg, O. On the Origin of Cancer Cells. Am. Assoc. Adv. Sci. 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Potter, M.; Newport, E.; Morten, K.J. The Warburg effect: 80 years on. Biochem. Soc. Trans. 2016, 44, 1499–1505. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Sanchez, R.; Rodriguez-Enriquez, S.; Saavedra, E.; Marin-Hernandez, A.; Gallardo-Perez, J.C. The bioenergetics of cancer: Is glycolysis the main ATP supplier in all tumor cells? Biofactors 2009, 35, 209–225. [Google Scholar] [CrossRef] [PubMed]
- Michelakis, E.D.; Webster, L.; Mackey, J.R. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br. J. Cancer 2008, 99, 989–994. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Detappe, A.; Cai, K.; Keys, H.R.; Brune, Z.; Ying, W.; Thiru, P.; Reidy, M.; Kugener, G.; Rossen, J.; et al. Mitochondrial metabolism promotes adaptation to proteotoxic stress. Nat. Chem. Biol. 2019, 15, 681–689. [Google Scholar] [CrossRef]
- Kuntz, E.M.; Baquero, P.; Michie, A.M.; Dunn, K.; Tardito, S.; Holyoake, T.L.; Helgason, G.V.; Gottlieb, E. Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nat. Med. 2017, 23, 1234–1240. [Google Scholar] [CrossRef]
- Moreno-Sanchez, R.; Rodriguez-Enriquez, S.; Marin-Hernandez, A.; Saavedra, E. Energy metabolism in tumor cells. FEBS J. 2007, 274, 1393–1418. [Google Scholar] [CrossRef]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. 2018, 24, 2482–2491. [Google Scholar] [CrossRef]
- Song, I.S.; Kim, H.K.; Lee, S.R.; Jeong, S.H.; Kim, N.; Ko, K.S.; Rhee, B.D.; Han, J. Mitochondrial modulation decreases the bortezomib-resistance in multiple myeloma cells. Int. J. Cancer 2013, 133, 1357–1367. [Google Scholar] [CrossRef]
- Chauncey, T.R. Drug resistance mechanisms in acute leukemia. Curr. Opin. Oncol. 2001, 13, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Zhan, J.; Gu, Y.; Chen, B. Mechanisms of drug resistance in acute myeloid leukemia. OncoTargets Ther. 2019, 12, 1937–1945. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.S.; Sohn, D.H. Damage-associated molecular patterns in inflammatory diseases. Immune Netw. 2018, 18, 1–14. [Google Scholar] [CrossRef]
- Rubartelli, A.; Lotze, M.T. Inside, outside, upside down: Damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol. 2007, 28, 429–436. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, C.; Huebener, P.; Schwabe, R. Damage-associated molecular patterns in cancer: A double edged sword. Oncogene 2016, 35, 5931–5941. [Google Scholar] [CrossRef] [PubMed]
- Krysko, O.; Aaes, T.L.; Bachert, C.; Vandenabeele, P.; Krysko, D.V. Many faces of DAMPs in cancer therapy. Cell Death Dis. 2013, 4, e631. [Google Scholar] [CrossRef]
- Spees, J.L.; Olson, S.D.; Whitney, M.J.; Prockop, D.J. Mitochondrial transfer between cells can rescue aerobic respiration. Proc. Natl. Acad. Sci. USA 2006, 103, 1283–1288. [Google Scholar] [CrossRef]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef]
- Lu, J.; Zheng, X.; Li, F.; Yu, Y.; Chen, Z.; Liu, Z.; Xu, H.; Yang, W. Tunneling nanotubes promote intercellular mitochondria transfer followed by increased invasiveness in bladder cancer cells. Oncotarget 2017, 8, 15539–15552. [Google Scholar] [CrossRef]
- Pasquier, J.; Guerrouahen, B.S.; Al Thawadi, H.; Ghiabi, P.; Maleki, M.; Abu-Kaoud, N.; Jacob, A.; Mirshahi, M.; Galas, L.; Rafii, S.; et al. Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance. J. Transl. Med. 2013, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Burt, R.; Dey, A.; Aref, S.; Aguiar, M.; Akarca, A.; Bailey, K.; Day, W.; Hooper, S.; Kirkwood, A.; Kirschner, K.; et al. Activated stromal cells transfer mitochondria to rescue acute lymphoblastic leukemia cells from oxidative stress. Blood 2019, 134, 1415–1429. [Google Scholar] [CrossRef] [PubMed]
- Marlein, C.R.; Piddock, R.E.; Mistry, J.J.; Zaitseva, L.; Hellmich, C.; Horton, R.H.; Zhou, Z.; Auger, M.J.; Bowles, K.M.; Rushworth, S.A. CD38-driven mitochondrial trafficking promotes bioenergetic plasticity in multiple myeloma. Cancer Res. 2019, 79, 2285–2297. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.F.; Kovarova, J.; Bajzikova, M.; Bezawork-Geleta, A.; Svec, D.; Endaya, B.; Sachaphibulkij, K.; Coelho, A.R.; Sebkova, N.; Ruzickova, A.; et al. Horizontal transfer of whole mitochondria restores tumorigenic potential in mitochondrial DNA-deficient cancer cells. eLife 2017, 6, e22187. [Google Scholar] [CrossRef]
- Wang, X.; Gerdes, H.H. Transfer of mitochondria via tunneling nanotubes rescues apoptotic PC12 cells. Cell Death Differ. 2015, 22, 1181–1191. [Google Scholar] [CrossRef]
- Liu, K.; Ji, K.; Guo, L.; Wu, W.; Lu, H.; Shan, P.; Yan, C. Mesenchymal stem cells rescue injured endothelial cells in an in vitro ischemia-reperfusion model via tunneling nanotube like structure-mediated mitochondrial transfer. Microvasc. Res. 2014, 92, 10–18. [Google Scholar] [CrossRef]
- Li, X.; Zhang, Y.; Yeung, S.C.; Liang, Y.; Liang, X.; Ding, Y.; Ip, M.S.M.; Tse, H.F.; Mak, J.C.W.; Lian, Q. Mitochondrial transfer of induced pluripotent stem cell-derived mesenchymal stem cells to airway epithelial cells attenuates cigarette smoke-induced damage. Am. J. Respir. Cell Mol. Biol. 2014, 51, 455–465. [Google Scholar] [CrossRef]
- Vallabhaneni, K.C.; Haller, H.; Dumler, I. Vascular smooth muscle cells initiate proliferation of mesenchymal stem cells by mitochondrial transfer via tunneling nanotubes. Stem Cells Dev. 2012, 21, 3104–3113. [Google Scholar] [CrossRef]
- Islam, M.N.; Das, S.R.; Emin, M.T.; Wei, M.; Sun, L.; Westphalen, K.; Rowlands, D.J.; Quadri, S.K.; Bhattacharya, S.; Bhattacharya, J. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 2012, 18, 759–765. [Google Scholar] [CrossRef]
- Jiang, D.; Gao, F.; Zhang, Y.; Wong, D.S.H.; Li, Q.; Tse, H.F.; Xu, G.; Yu, Z.; Lian, Q. Mitochondrial transfer of mesenchymal stem cells effectively protects corneal epithelial cells from mitochondrial damage. Cell Death Dis. 2016, 7, e2467. [Google Scholar] [CrossRef]
- Jackson, M.V.; Morrison, T.J.; Doherty, D.F.; McAuley, D.F.; Matthay, M.A.; Kissenpfennig, A.; O’Kane, C.M.; Krasnodembskaya, A.D. Mitochondrial Transfer via Tunneling Nanotubes is an Important Mechanism by Which Mesenchymal Stem Cells Enhance Macrophage Phagocytosis in the in vitro and in vivo Models of ARDS. Stem Cells 2016, 34, 2210–2223. [Google Scholar] [CrossRef] [PubMed]
- Mistry, J.J.; Marlein, C.R.; Moore, J.A.; Hellmich, C.; Wojtowicz, E.E.; Smith, J.G.W.; Macaulay, I.; Sun, Y.; Morfakis, A.; Patterson, A.; et al. ROS-mediated PI3K activation drives mitochondrial transfer from stromal cells to hematopoietic stem cells in response to infection. Proc. Natl. Acad. Sci. USA 2019, 116, 24610–24619. [Google Scholar] [CrossRef] [PubMed]
- Marlein, C.R.; Zaitseva, L.; Piddock, R.E.; Robinson, S.D.; Edwards, D.R.; Shafat, M.S.; Zhou, Z.; Lawes, M.; Bowles, K.M.; Rushworth, S.A. NADPH oxidase-2 derived superoxide drives mitochondrial transfer from bone marrow stromal cells to leukemic blasts. Blood 2017, 130, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Polak, R.; Rooij, B.D.; Pieters, R.; Boer, M.L. Den B-cell precursor acute lymphoblastic leukemia cells use tunneling nanotubes to orchestrate their microenvironment. Blood 2015, 126, 2404–2414. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.M.; Kim, J.H.; Kim, M.; Park, S.J.; Koh, S.H.; Ahn, H.S.; Kang, G.H.; Lee, J.-B.; Park, K.S.; Lee, H.K. Mesenchymal stem cells transfer mitochondria to the cells with virtually no mitochondrial function but not with pathogenic mtDNA mutations. PLoS ONE 2012, 7, e32778. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Liou, C.; Chen, S.; Hsu, T.; Chuang, J.; Wang, P.; Huang, S.; Tiao, M.; Chen, J.; Lin, T.; et al. Mitochondrial transfer from Wharton’s jelly-derived mesenchymal stem cells to mitochondria-defective cells recaptures impaired mitochondrial function. Mitochondrion 2015, 22, 31–34. [Google Scholar] [CrossRef]
- Moschoi, R.; Imbert, V.; Nebout, M.; Chiche, J.; Mary, D.; Prebet, T.; Saland, E.; Castellano, R.; Pouyet, L.; Collette, Y.; et al. Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy. Blood 2016, 128, 253–264. [Google Scholar] [CrossRef]
- Marlein, C.R.; Zaitseva, L.; Piddok, R.; Schafat, M.; Collins, A.; Bowles, K.; Rushworth, S. PGC1α Driven Mitochondrial Biogenesis within the Bone Marrow Stromal Cells of the Acute Myeloid Leukemia Micro-Environment Is a Pre-Requisite for Mitochondrial Transfer to Leukemic Blasts. Blood 2017, 130 (Suppl. 1), 3927. [Google Scholar]
- Torralba, D.; Baixauli, F.; Sánchez-Madrid, F. Mitochondria know no boundaries: Mechanisms and functions of intercellular mitochondrial transfer. Front. Cell Dev. Biol. 2016, 4, 107. [Google Scholar] [CrossRef]
- Rustom, A.; Saffrich, R.; Markovic, I.; Walther, P.; Gerdes, H.H. Nanotubular Highways for Intercellular Organelle Transport. Science 2004, 303, 1007–1010. [Google Scholar] [CrossRef]
- Schapman, D.; Lebon, A.; Monterroso, B.; Bellenger, M.; Foll, F.L.; Pasquier, J.; Vaudry, H.; Vaudry, D.; Galas, L. Structural and functional analysis of tunneling nanotubes (TnTs) using g CW STED and g confocal approaches. Biol. Cell 2015, 107, 419–425. [Google Scholar] [CrossRef]
- Gousset, K.; Marzo, L.; Commere, P.; Zurzolo, C. Myo10 is a key regulator of TNT formation in neuronal cells. J. Cell Sci. 2013, 126, 4424–4435. [Google Scholar] [CrossRef] [PubMed]
- Tardivel, M.; Bégard, S.; Bousset, L.; Dujardin, S.; Coens, A.; Melki, R. Tunneling nanotube (TNT)-mediated neuron-to neuron transfer of pathological Tau protein assemblies. Acta Neuropathol. Commun. 2016, 4, 117. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yu, Z.; Jiang, D.; Liang, X.; Liao, S.; Zhang, Z.; Yue, W.; Li, X.; Chiu, S.M.; Chai, Y.H.; et al. iPSC-MSCs with High Intrinsic MIRO1 and Sensitivity to TNF-α Yield Efficacious Mitochondrial Transfer to Rescue Anthracycline-Induced Cardiomyopathy. Stem Cell Rep. 2016, 7, 749–763. [Google Scholar] [CrossRef]
- Ahmad, T.; Mukherjee, S.; Pattnaik, B.; Kumar, M.; Singh, S.; Rehman, R.; Tiwari, B.K.; Jha, K.A.; Barhanpurkar, A.P.; Wani, M.R.; et al. Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. EMBO J. 2014, 33, 994–1010. [Google Scholar] [CrossRef]
- López-Doménech, G.; Covill-Cooke, C.; Ivankovic, D.; Halff, E.F.; Sheehan, D.F.; Norkett, R.; Birsa, N.; Kittler, J.T. Miro proteins coordinate microtubule- and actin-dependent mitochondrial transport and distribution. EMBO J. 2018, 37, 321–336. [Google Scholar] [CrossRef]
- Zhang, Y. Tunneling-nanotube. Commun. Integr. Biol. 2011, 4, 324–325. [Google Scholar] [CrossRef]
- Hase, K.; Kimura, S.; Takatsu, H.; Ohmae, M.; Kawano, S.; Kitamura, H.; Ito, M.; Watarai, H.; Hazelett, C.; Yeaman, C.; et al. M-Sec promotes membrane nanotube formation by interacting with Ral and the exocyst complex. Nat. Cell Biol. 2009, 12, 1427–1432. [Google Scholar] [CrossRef]
- Ohno, H.; Hase, K.; Kimura, S. M-Sec: Emerging secrets of tunneling nanotube formation. Commun. Integr. Biol. 2010, 3, 231–233. [Google Scholar] [CrossRef]
- Aarhus, R.; Graeff, R.M.; Dickey, D.M.; Walseth, T.F.; Lee, H.C. ADP-ribosyl cyclase and CD38 catalyze the synthesis of a calcium-mobilizing metabolite from NADP. J. Biol. Chem. 1995, 270, 30327–30333. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C. Physiologival functions of cyclic ADP-ribose and NAADP as calcium messangers. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 317–345. [Google Scholar] [CrossRef] [PubMed]
- Bruzzone, S.; Moreschi, I.; Guida, L.; Usai, C.; Zocchi, E.; De Flora, A. Extracellular NAD+ regulates intracellular calcium levels and induces activation of human granulocytes. Biochem. J. 2006, 393, 697–704. [Google Scholar] [CrossRef]
- Ariazi, J.; Benowitz, A.; De Biasi, V.; Den Boer, M.L.; Cherqui, S.; Cui, H.; Douillet, N.; Eugenin, E.A.; Favre, D.; Goodman, S.; et al. Tunneling Nanotubes and Gap Junctions—Their Role in Long-Range Intercellular Communication during Development, Health, and Disease Conditions. Front. Mol. Neurosci. 2017, 10, 333. [Google Scholar] [CrossRef] [PubMed]
- Osswald, M.; Jung, E.; Sahm, F.; Solecki, G.; Venkataramani, V.; Blaes, J.; Weil, S.; Horstmann, H.; Wiestler, B.; Syed, M.; et al. Brain tumour cells interconnect to a functional and resistant network. Nature 2015, 528, 93–98. [Google Scholar] [CrossRef]
- Wang, X.; Veruki, M.L.; Bukoreshtliev, N.V.; Hartveit, E.; Gerdes, H. Animal cells connected by nanotubes can be electrically coupled through interposed gap-junction channels. Proc. Natl. Acad. Sci. USA 2010, 107, 17194–17199. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Shi, X.; Zhang, X.; Dang, S.; Ma, X.; Liu, F.; Xu, M.; Lv, Z.; Han, D.; Fang, X.; et al. Long-distance intercellular connectivity between cardiomyocytes and cardiofibroblasts mediated by membrane nanotubes. Cardiovasc. Res. 2011, 92, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Eugenin, E.A. Role of Connexin/Pannexin containing channels in infectious diseases. FEBS Lett. 2014, 588, 1389–1395. [Google Scholar] [CrossRef] [PubMed]
- Aasen, T. Connexins: Junctional and non-junctional modulators of proliferation. Cell Tissue Res. 2015, 360, 685–699. [Google Scholar] [CrossRef]
- Gerdes, H.H.; Carvalho, R.N. Intercellular transfer mediated by tunneling nanotubes. Curr. Opin. Cell Biol. 2008, 20, 470–475. [Google Scholar] [CrossRef]
- Gerdes, H.H.; Rustom, A.; Wang, X. Tunneling nanotubes, an emerging intercellular communication route in development. Mech. Dev. 2013, 130, 381–387. [Google Scholar] [CrossRef]
- Whiteside, T. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [PubMed]
- Saha, T.; Dash, C.; Khiste, S.; Sengupta, S. A novel mechanism of immunosuppression via nanotube mediated mitochondrial trafficking between cancer cell and immune cell. Cancer Res. 2019. [Google Scholar] [CrossRef]
- Ippolito, L.; Morandi, A.; Taddei, M.L.; Parri, M.; Comito, G.; Iscaro, A.; Raspollini, M.R.; Magherini, F.; Rapizzi, E.; Masquelier, J.; et al. Cancer-associated fibroblasts promote prostate cancer malignancy via metabolic rewiring and mitochondrial transfer. Oncogene 2019, 38, 5339–5355. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, X.; Qiu, Y.; Shi, Y.; Cai, J.; Wang, B.; Wei, X.; Ke, Q.; Sui, X.; Wang, Y.; et al. Cell adhesion-mediated mitochondria transfer contributes to mesenchymal stem cell-induced chemoresistance on T cell acute lymphoblastic leukemia cells. J. Hematol. Oncol. 2018, 11, 11. [Google Scholar] [CrossRef]
- Poburko, D.; Santo-Domingo, J.; Demaurex, N. Dynamic regulation of the mitochondrial proton gradient during cytosolic calcium elevations. J. Biol. Chem. 2011, 286, 11672–11684. [Google Scholar] [CrossRef]
- Basak, N.P.; Banerjee, S. Mitochondrial dependency in progression of acute myeloid leukemia. Mitochondrion 2015, 21, 41–48. [Google Scholar] [CrossRef]
- Putten, W.V.; Sc, M.; Schouten, H.C.; Graux, C.; Ferrant, A.; Sonneveld, P.; Biemond, B.J.; Gratwohl, A.; Greef, G.E.D.; Verdonck, L.F.; et al. Cytarabine Dose for Acute Myeloid Leukemia. N. Engl. J. Med. 2011, 1027–1036. [Google Scholar]
- Greenberg, P.L.; Lee, S.J.; Advani, R.; Tallman, M.S.; Sikic, B.I.; Letendre, L.; Dugan, K.; Lum, B.; Chin, D.L.; Dewald, G.; et al. Mitoxantrone, Etoposide, and Cytarabine with or without Valspodar in Patients with Relapsed or Refractory Acute Myeloid Leukemia and High-Risk Myelodysplastic Syndrome: A Phase III Trial (E2995). J. Clin. Oncol. 2004, 22, 1078. [Google Scholar] [CrossRef]
- Teuffel, O.; Leibundgut, K.; Lehrnbecher, T.; Alonzo, T.A.; Beyene, J.; Sung, L. Anthracyclines during induction therapy in acute myeloid leukaemia: A systematic review and meta-analysis. Br. J. Haematol. 2013, 161, 192–203. [Google Scholar] [CrossRef]
- Hole, P.S.; Zabkiewicz, J.; Munje, C.; Newton, Z.; Pearn, L.; White, P.; Marquez, N.; Hills, R.K.; Burnett, A.K.; Tonks, A.; et al. Overproduction of NOX-derived ROS in AML promotes proliferation and is associated with defective oxidative stress signaling. Blood 2013, 122, 3322–3330. [Google Scholar] [CrossRef]
- Naik, J.; Themeli, M.; de Jong-Korlaar, R.; Ruiter, R.W.J.; Poddighe, P.J.; Yuan, H.; de Bruijin, J.D.; Ossenkoppele, G.J.; Zweegman, S.; Smit, L.; et al. CD38 as a therapeutic target for adult acute myeloid leukemia and T-cell acute lymphoblastic leukemia Acute. Haematologica 2019, 14, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, N.; Kumar, S.K. Daratumumab in untreated newly diagnosed multiple myeloma. Ther. Adv. Hematol. 2019, 10. [Google Scholar] [CrossRef]
- Mateos, M.; Spencer, A.; Nooka, A.K.; Pour, L.; Weisel, K.; Cavo, M.; Laubach, J.P.; Cook, G. Daratumumab-based regimens are highly effective and well tolerated in relapsed or refractory multiple myeloma regardless of patient age: Subgroup analysis of the phase 3 CASTOR and POLLUX studies. Haematologica 2020, 105, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Mistry, J.; Hellmich, C.; Moore, J.A.; Marlein, C.; Pillinger, G.; Collings, A.; Bowles, K.; Rushworth, S. Daratumumab Inhibits AML Metabolic Capacity and Tumor Growth through Inhibition of CD38 Mediated Mitochondrial Transfer from Bone Marrow Stromal Cells to Blasts in the Leukemic Microenvironment. Blood 2019. [Google Scholar] [CrossRef]
- Farber, M.; Arnold, L.; Chen, Y.; Mollmann, M.; Duehrsen, U.; Hanoun, M. Inhibition of CD38 Shows Anti-Leukemic Activity in Acute Myeloid Leukemia. Blood 2018, 132, 1456. [Google Scholar] [CrossRef]
- Saba, F.; Soleimani, M.; Abroun, S. New role of hypoxia in pathophysiology of multiple myeloma through miR-210. EXCLI J. 2018, 17, 647–662. [Google Scholar] [CrossRef]
- Lipchick, B.; Fink, E.; Nikiforoc, M. Oxidative Stress and Proteasome Inhibitors in Multiple Myeloma. Pharmacol. Res. 2016, 105, 210–215. [Google Scholar] [CrossRef]
- Krejcik, J.; Casneuf, T.; Nijhof, I.S.; Verbist, B.; Bald, J.; Plesner, T.; Syed, K.; Liu, K.; van de Donk, N.W.C.J.; Weiss, B.M.; et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood 2016, 128, 384–394. [Google Scholar] [CrossRef]
- Moreno, A.L.; Perez, C.; Zabaleta, A.; Manrique, I.; Garate, S.; Jelinek, T.; Segura, V.; Moreno, C. The Mechanism of Action of The Anti-CD38 Monoclonal Antibody Isatuximab In Multiple Myeloma. Clin. Cancer Res. 2019, 25, 3176–3187. [Google Scholar] [CrossRef]
- Martin, T.; Strickland, S.; Glenn, M.; Charpentier, E.; Guillemin, H.; Hsu, K.; Mikhael, J. Phase I trial of isatuximab monotherapy in the treatment of refractory multiple myeloma. Blood Cancer J. 2019, 9, 1–10. [Google Scholar] [CrossRef]
- Spencer, A.; Lentzsch, S.; Weisel, K.; Avet-Loiseau, H.; Mark, T.M.; Spicka, I.; Masszi, T.; Lauri, B.; Levin, M.D.; Bosi, A.; et al. Daratumumab plus bortezomib and dexamethasone versus bortezomib and dexamethasone in relapsed or refractory multiple myeloma: Updated analysis of CASTOR. Haematologica 2018, 103, 2079–2087. [Google Scholar] [CrossRef] [PubMed]
- Read, M.A.; Neish, A.S.; Luscinskas, F.W.; Palombella, V.J.; Yaniatis, T.; Collins, T. The Proteasome Pathway is Required for Cytokine-Induced Endothelial-Leukocyte Adhesion Molecule Expression. Immunity 1995, 2, 493–506. [Google Scholar] [CrossRef]
- Chauhan, D.; Uchiyama, H.; Akbarali, Y.; Urashima, M.; Yamamoto, K.I.; Libermann, T.A.; Anderson, K.C. Multiple myeloma cell adhesion-induced interleukin-6 expression in bone marrow stromal cells involves activation of NF-κB. Blood 1996, 87, 1104–1112. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).