Induction of Lysosome Membrane Permeabilization as a Therapeutic Strategy to Target Pancreatic Cancer Stem Cells

 , , , , and
, , , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
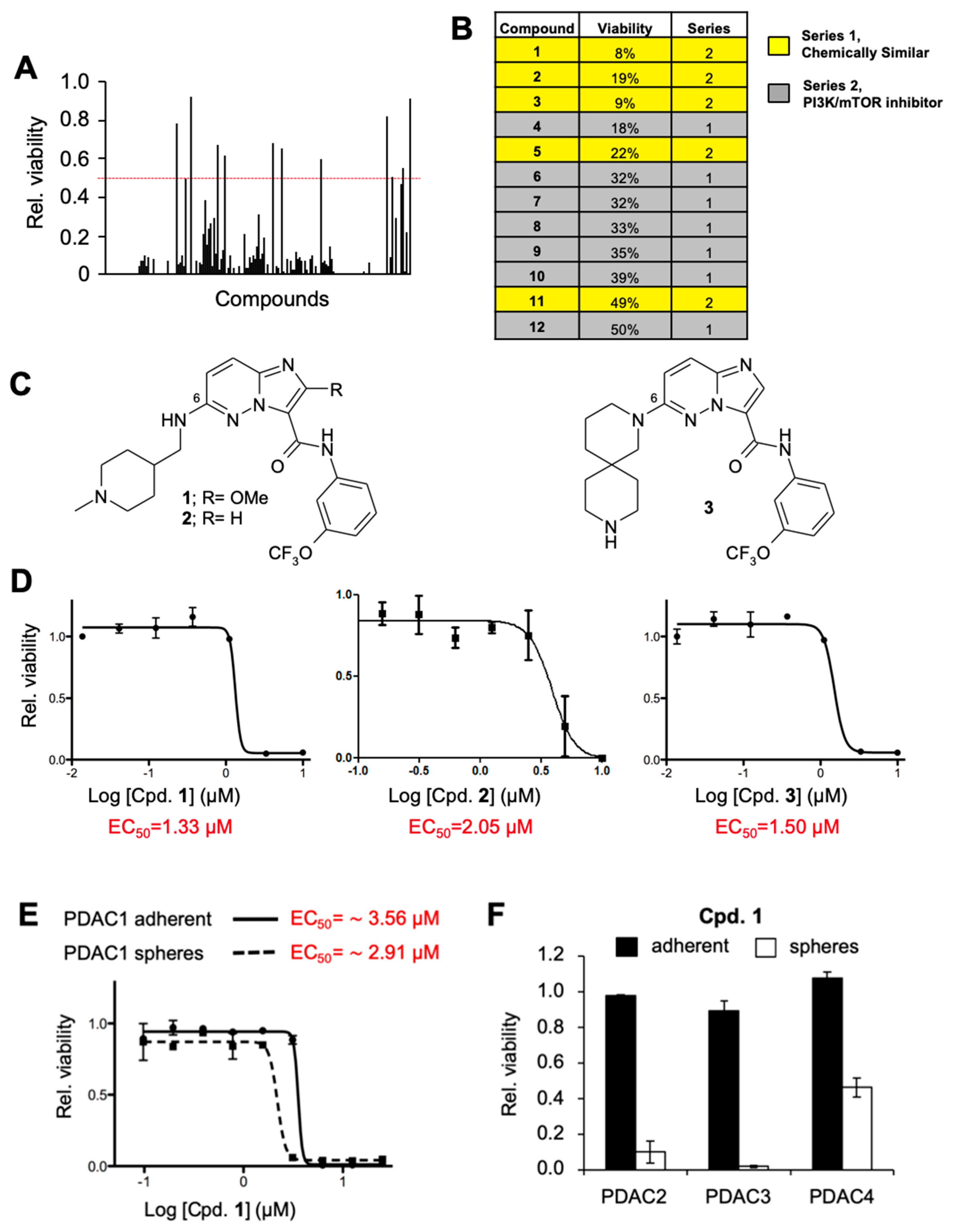
2.1. Identification of Novel Compounds That Compromise CSC Viability
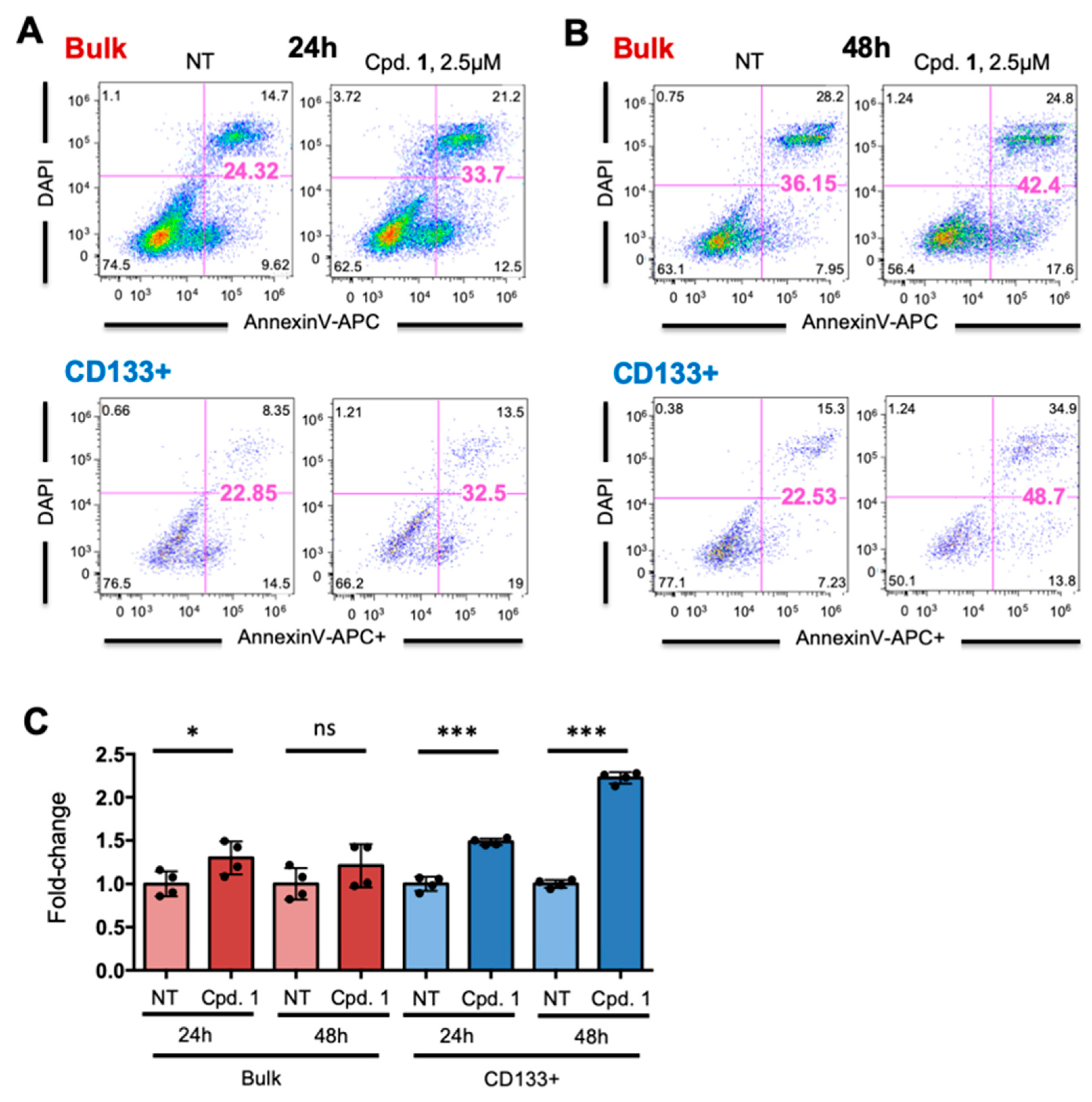
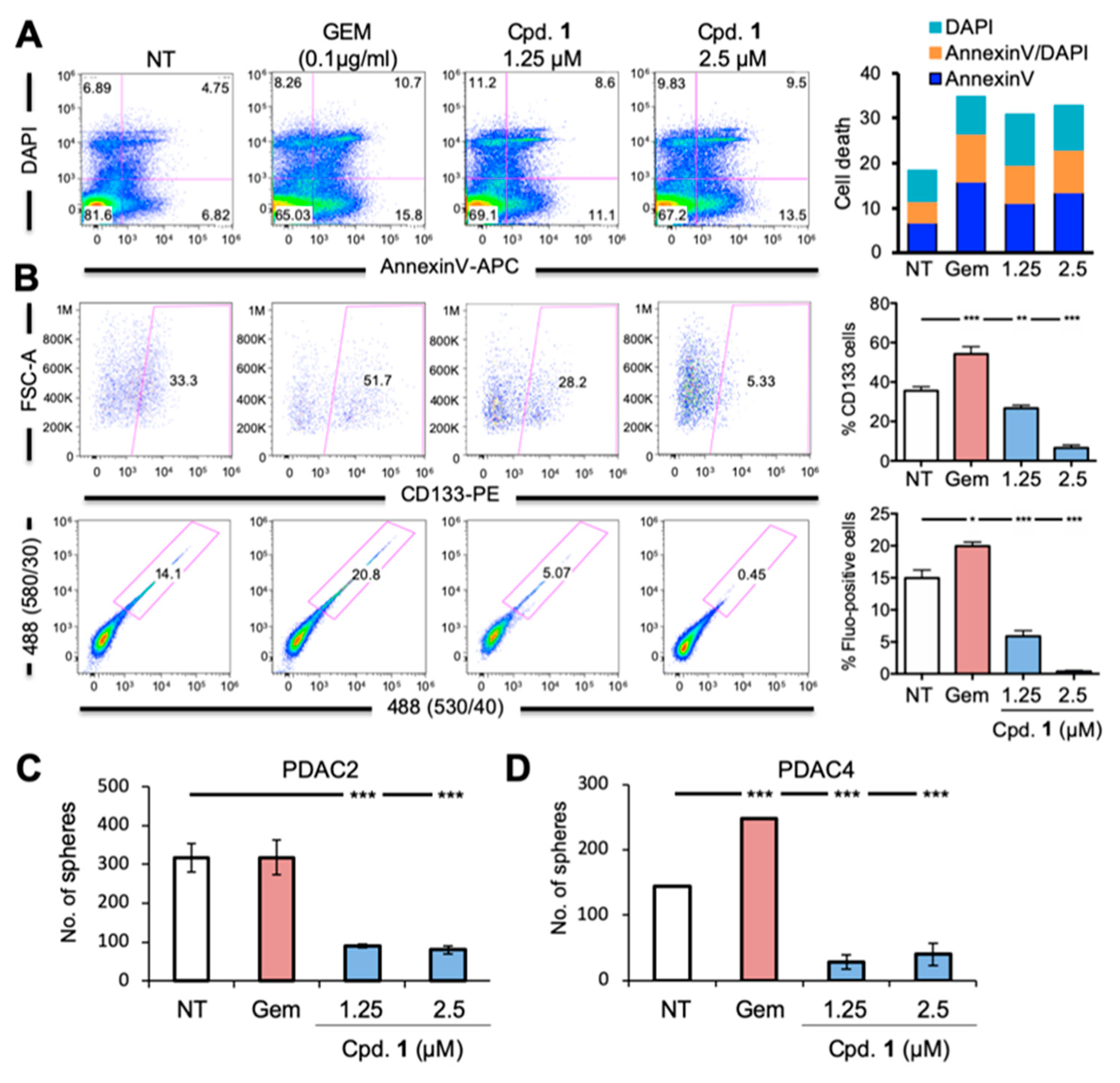
2.2. Compound 1 Impairs the Self-Renewal and Tumor Initiating Capacity of PDAC Cells by Depleting the CSC Compartment
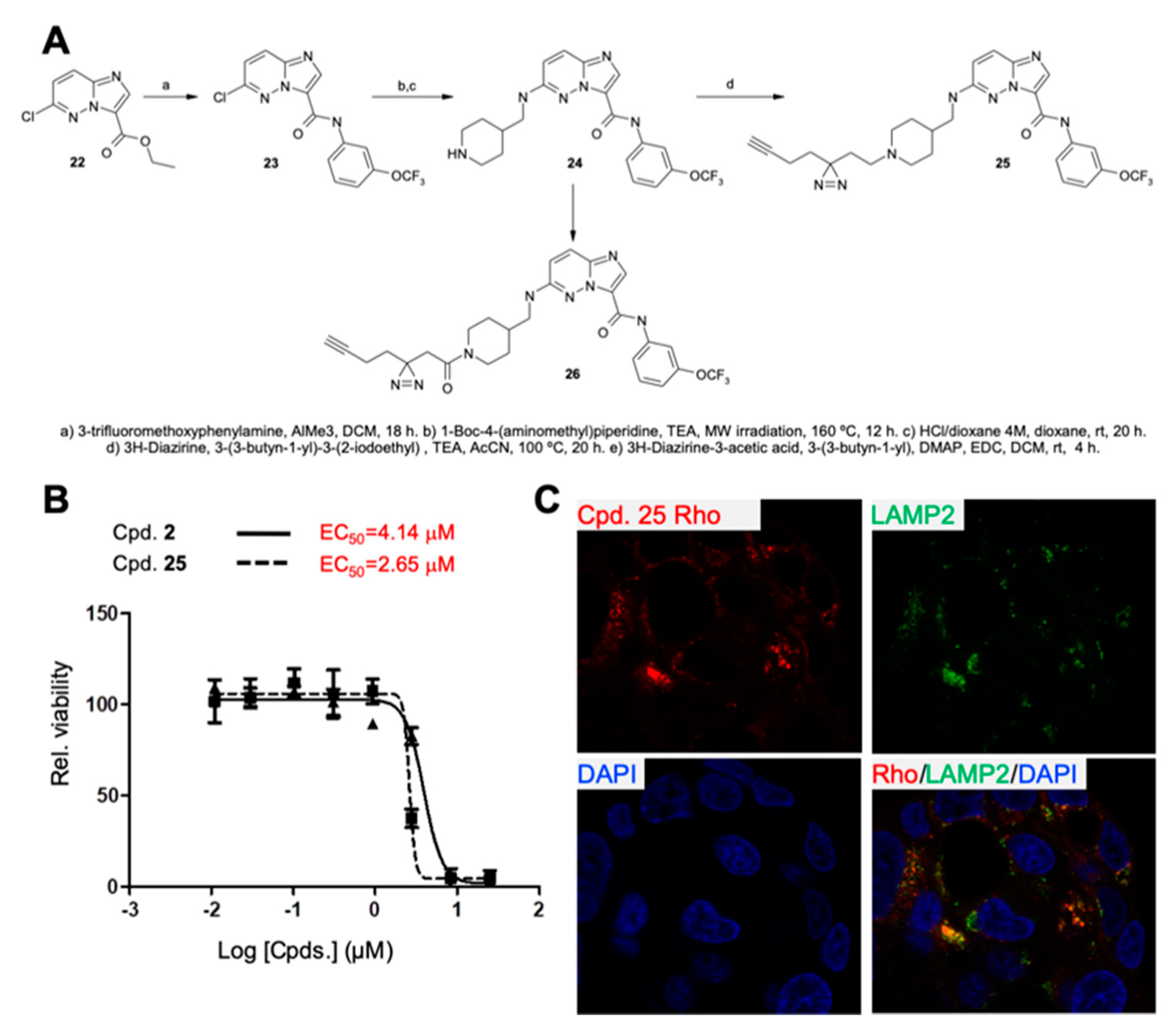
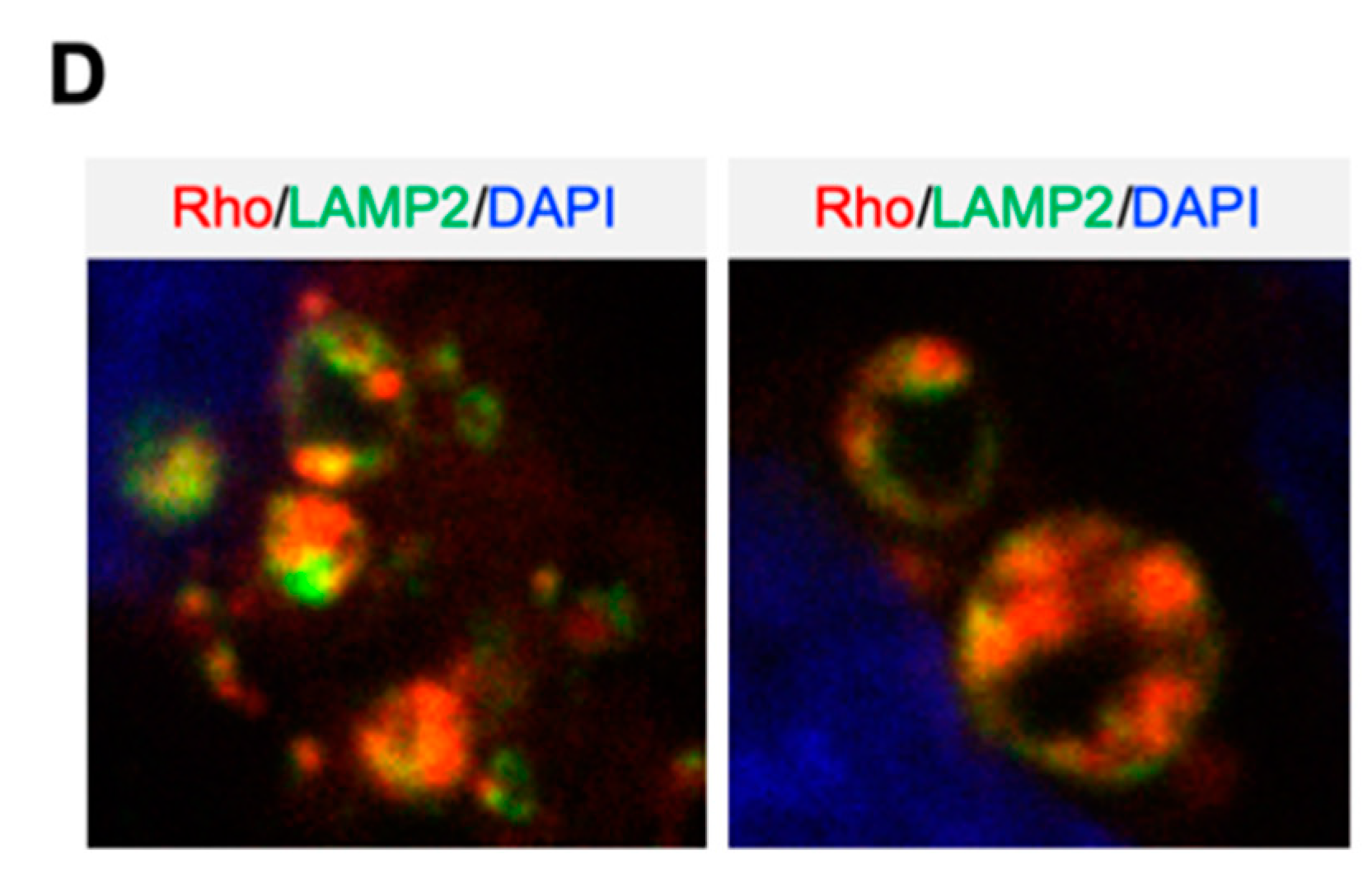
2.3. Characterization and Development of Chemical Probes to Study the Mechanism of Action of Series 1 Compounds
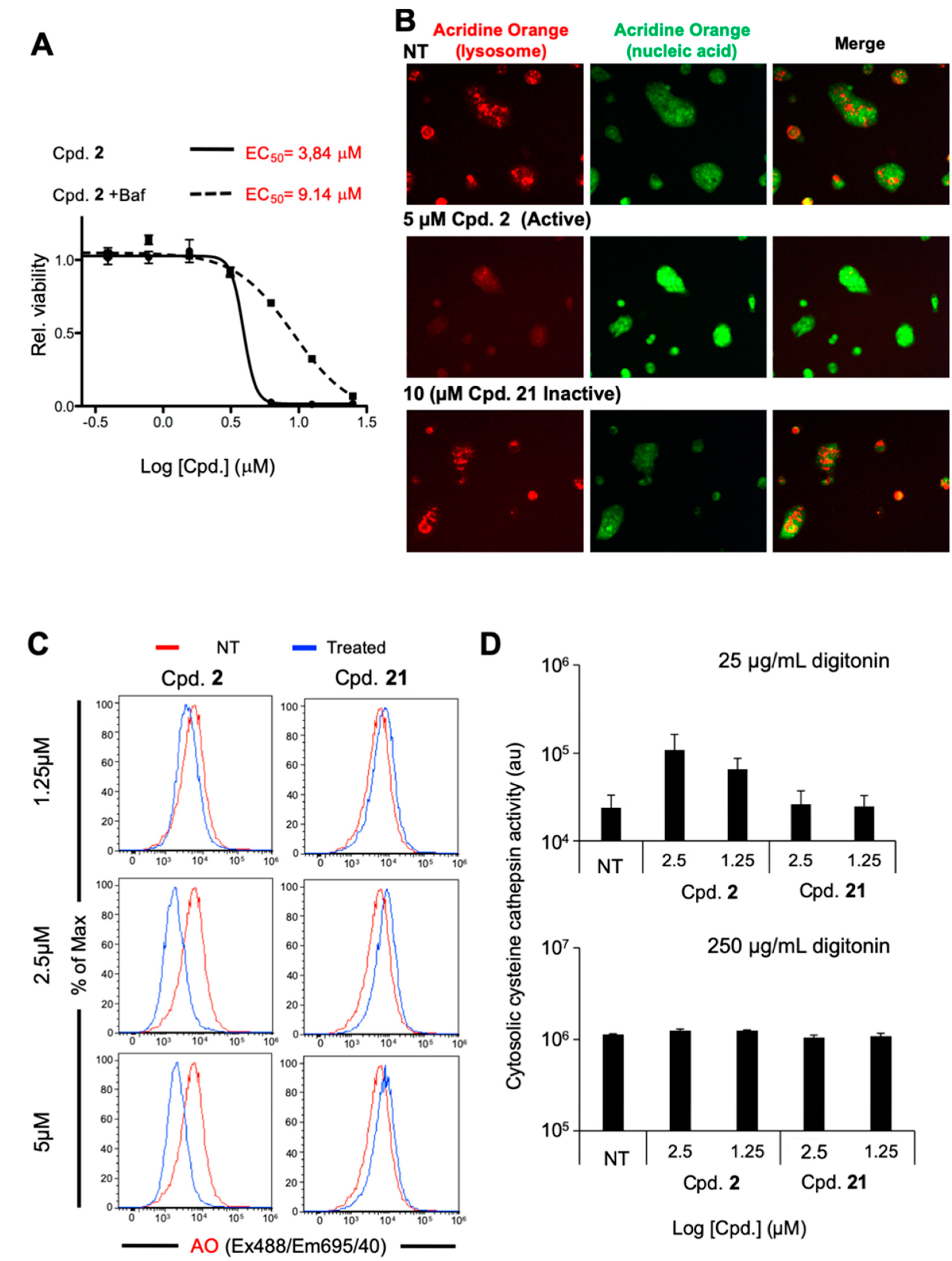
2.4. Compound 2 Induces Lysosomal Membrane Permeabilization
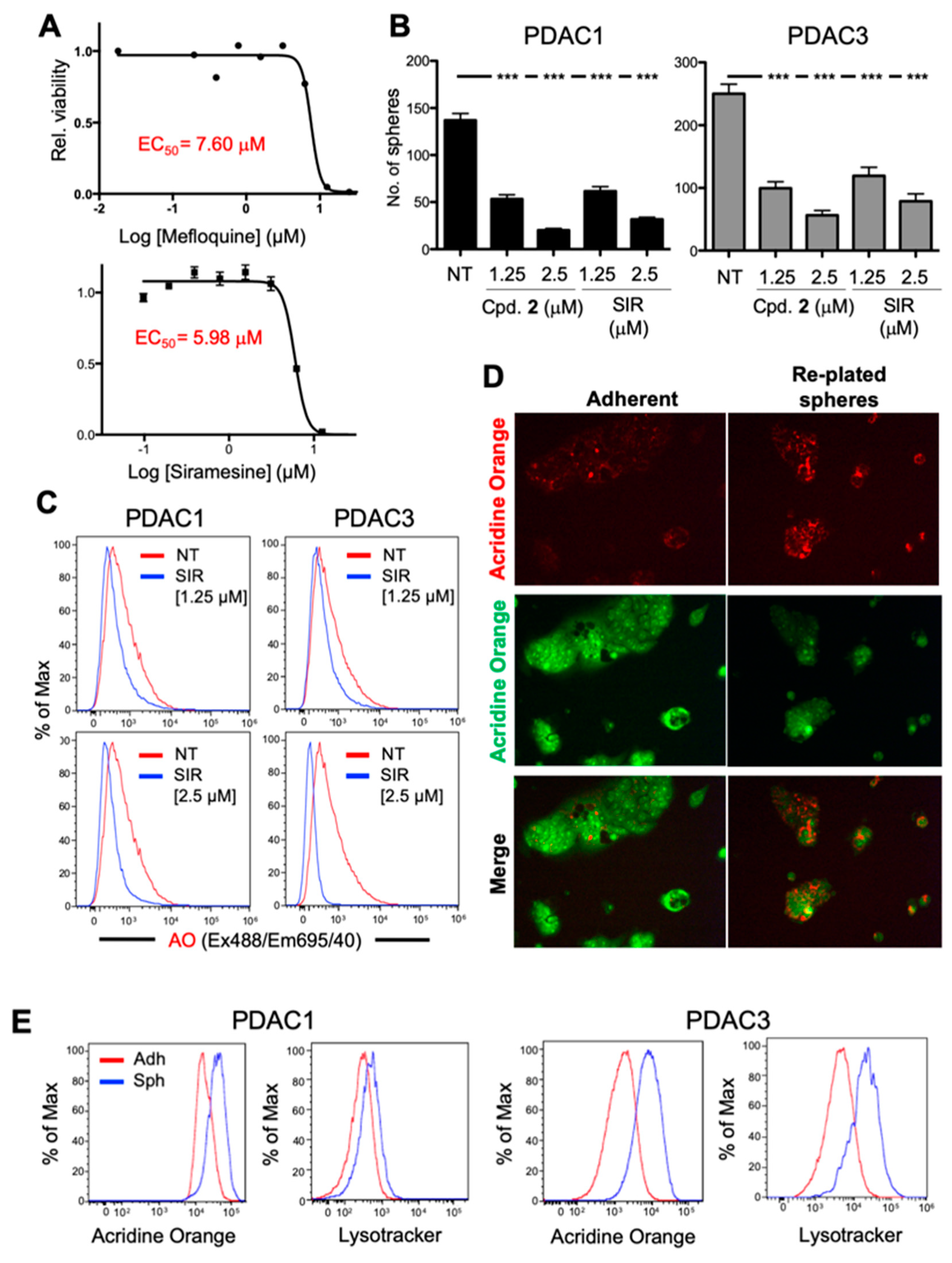
2.5. Broad Spectrum of LMP-Inducing Agents Target CSCs
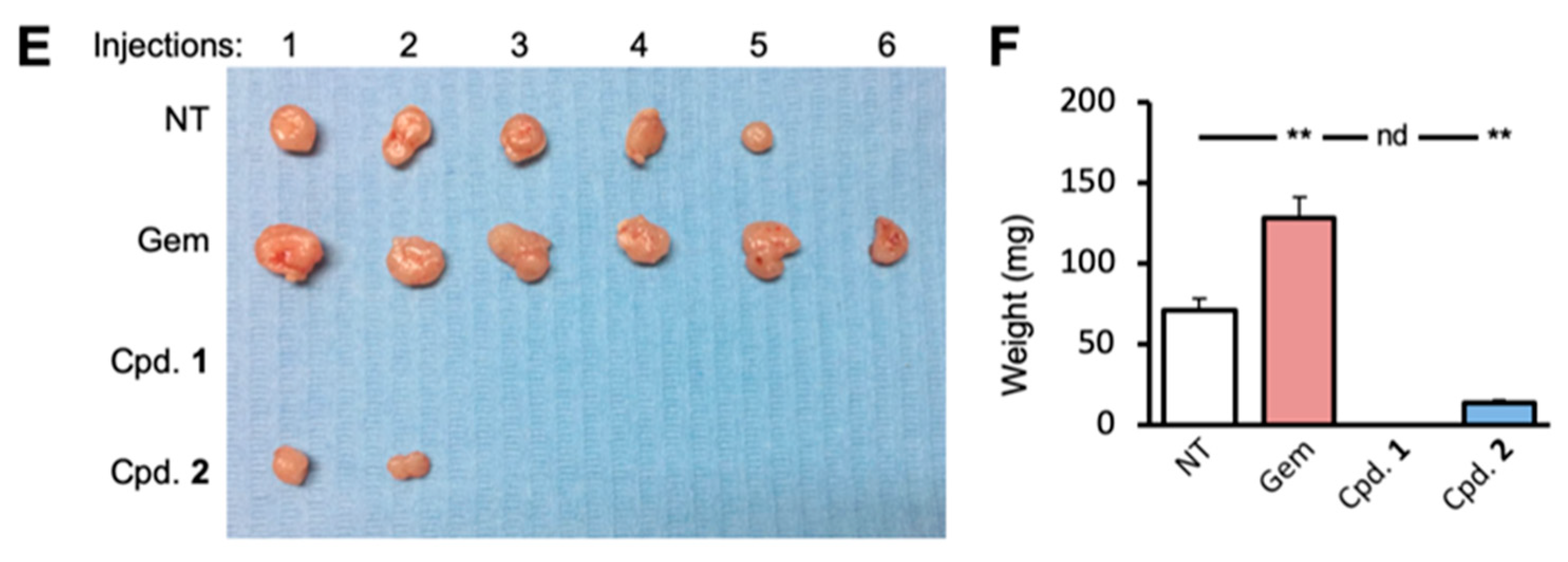
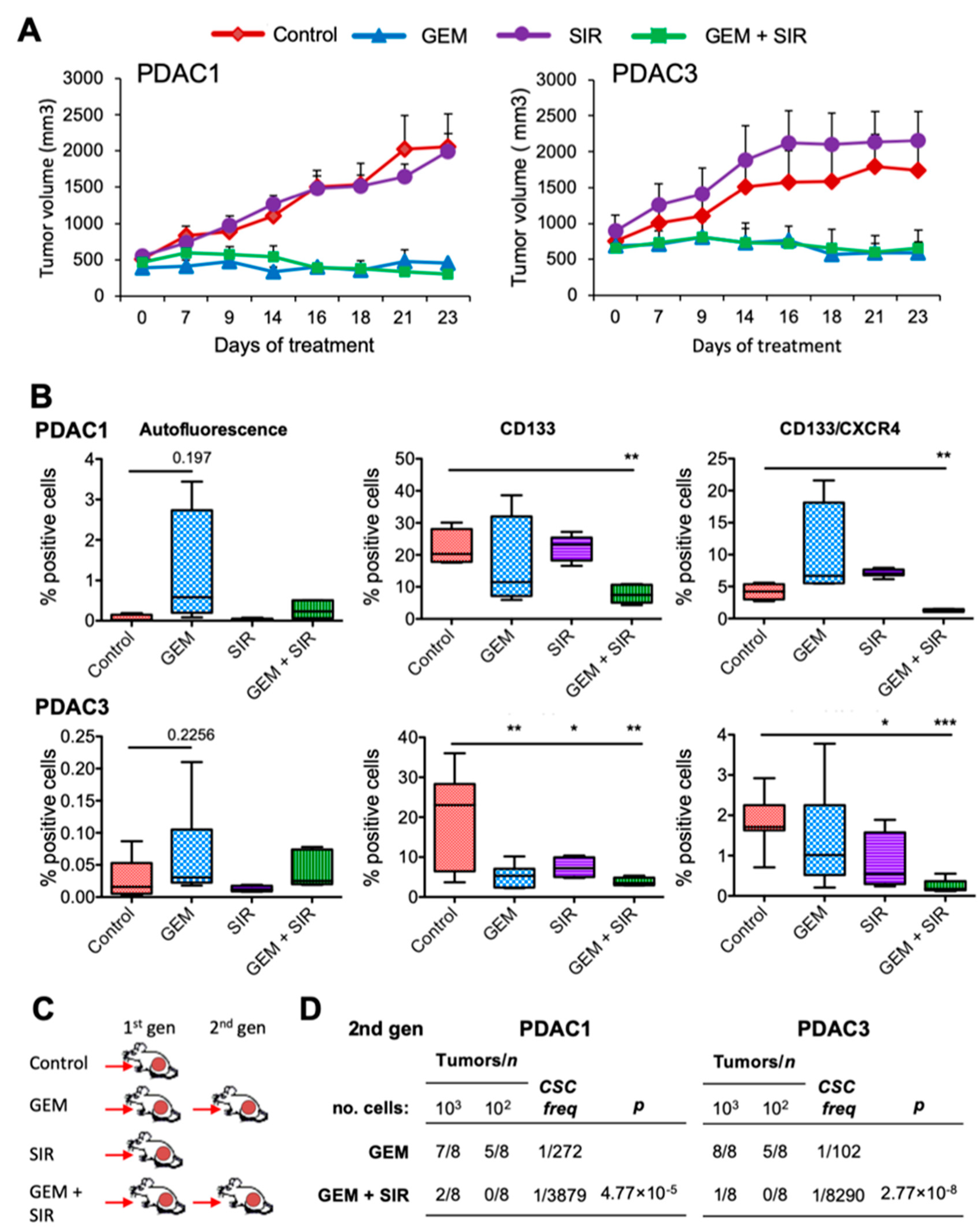
2.6. LMP Inhibits Pancreatic CSC-Mediated Tumorigenesis and Disease Recurrence In Vivo
3. Discussion
4. Materials and Methods
4.1. CNIO-640 Chemical Library
4.2. Murine Embryonic Stem Cells
4.3. Primary Human Pancreatic Cancer Cells
4.4. Sphere Formation Assay
4.5. Drug Screening and Cell Viability Assays
4.6. Flow Cytometry
4.7. In Vivo Assays
4.8. Pharmacokinetic Studies (PK)
4.9. KinomeScan
4.10. Synthesis of Affinity Chemical Probes
4.11. Kinase Assays
4.12. Immunofluorescence with Click Chemistry Probes
4.13. Acridine Orange Lysosomal Integrity Assay
4.14. Measurement of Cytosolic Cathepsin Activity
4.15. RNA Sequencing
4.16. Statistical Analyses
4.17. Data Availability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Da Silva-Diz, V.; Lorenzo-Sanz, L.; Bernat-Peguera, A.; Lopez-Cerda, M.; Munoz, P. Cancer cell plasticity: Impact on tumor progression and therapy response. Semin. Cancer Biol. 2018, 53, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Hermann, P.C.; Sainz, B., Jr. Pancreatic cancer stem cells: A state or an entity? Semin. Cancer Biol. 2018, 53, 223–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valle, S.; Martin-Hijano, L.; Alcala, S.; Alonso-Nocelo, M.; Sainz, B., Jr. The Ever-Evolving Concept of the Cancer Stem Cell in Pancreatic Cancer. Cancers 2018, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinbichler, T.B.; Dudas, J.; Skvortsov, S.; Ganswindt, U.; Riechelmann, H.; Skvortsova, I.I. Therapy resistance mediated by cancer stem cells. Semin. Cancer Biol. 2018, 53, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Saygin, C.; Matei, D.; Majeti, R.; Reizes, O.; Lathia, J.D. Targeting Cancer Stemness in the Clinic: From Hype to Hope. Cell Stem Cell 2019, 24, 25–40. [Google Scholar] [CrossRef] [Green Version]
- Valent, P.; Bonnet, D.; De Maria, R.; Lapidot, T.; Copland, M.; Melo, J.V.; Chomienne, C.; Ishikawa, F.; Schuringa, J.J.; Stassi, G.; et al. Cancer stem cell definitions and terminology: The devil is in the details. Nat. Rev. Cancer 2012, 12, 767–775. [Google Scholar] [CrossRef]
- Somervaille, T.C.; Matheny, C.J.; Spencer, G.J.; Iwasaki, M.; Rinn, J.L.; Witten, D.M.; Chang, H.Y.; Shurtleff, S.A.; Downing, J.R.; Cleary, M.L. Hierarchical maintenance of MLL myeloid leukemia stem cells employs a transcriptional program shared with embryonic rather than adult stem cells. Cell Stem Cell 2009, 4, 129–140. [Google Scholar] [CrossRef] [Green Version]
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; Weinberg, R.A. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008, 40, 499–507. [Google Scholar] [CrossRef]
- Boya, P.; Kroemer, G. Lysosomal membrane permeabilization in cell death. Oncogene 2008, 27, 6434–6451. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Gomez-Sintes, R.; Boya, P. Lysosomal membrane permeabilization and cell death. Traffic 2018, 19, 918–931. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Kim, S.O.; Han, J. Susceptibility of lysosomes to rupture is a determinant for plasma membrane disruption in tumor necrosis factor alpha-induced cell death. Mol. Cell Biol. 2003, 23, 665–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domagala, A.; Fidyt, K.; Bobrowicz, M.; Stachura, J.; Szczygiel, K.; Firczuk, M. Typical and Atypical Inducers of Lysosomal Cell Death: A Promising Anticancer Strategy. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, Y.; Sameni, M.; Sloane, B.F. Malignant transformation alters intracellular trafficking of lysosomal cathepsin D in human breast epithelial cells. Pathol. Oncol. Res. 1998, 4, 283–296. [Google Scholar] [CrossRef]
- Kallunki, T.; Olsen, O.D.; Jaattela, M. Cancer-associated lysosomal changes: Friends or foes? Oncogene 2013, 32, 1995–2004. [Google Scholar] [CrossRef] [Green Version]
- Fehrenbacher, N.; Bastholm, L.; Kirkegaard-Sorensen, T.; Rafn, B.; Bottzauw, T.; Nielsen, C.; Weber, E.; Shirasawa, S.; Kallunki, T.; Jaattela, M. Sensitization to the lysosomal cell death pathway by oncogene-induced down-regulation of lysosome-associated membrane proteins 1 and 2. Cancer Res. 2008, 68, 6623–6633. [Google Scholar] [CrossRef] [Green Version]
- Piao, S.; Amaravadi, R.K. Targeting the lysosome in cancer. Ann. N. Y. Acad. Sci. 2016, 1371, 45–54. [Google Scholar] [CrossRef] [Green Version]
- Serrano-Puebla, A.; Boya, P. Lysosomal membrane permeabilization as a cell death mechanism in cancer cells. Biochem. Soc. Trans. 2018, 46, 207–215. [Google Scholar] [CrossRef]
- Sukhai, M.A.; Prabha, S.; Hurren, R.; Rutledge, A.C.; Lee, A.Y.; Sriskanthadevan, S.; Sun, H.; Wang, X.; Skrtic, M.; Seneviratne, A.; et al. Lysosomal disruption preferentially targets acute myeloid leukemia cells and progenitors. J. Clin. Invest 2013, 123, 315–328. [Google Scholar] [CrossRef]
- Jensen, S.S.; Petterson, S.A.; Halle, B.; Aaberg-Jessen, C.; Kristensen, B.W. Effects of the lysosomal destabilizing drug siramesine on glioblastoma in vitro and in vivo. BMC Cancer 2017, 17, 178. [Google Scholar] [CrossRef] [Green Version]
- Xia, P.; Xu, X.Y. PI3K/Akt/mTOR signaling pathway in cancer stem cells: From basic research to clinical application. Am. J. Cancer Re.s 2015, 5, 1602–1609. [Google Scholar]
- Yu, J.S.; Cui, W. Proliferation, survival and metabolism: The role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development 2016, 143, 3050–3060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendiburu-Elicabe, M.; Gil-Ranedo, J.; Izquierdo, M. Efficacy of rapamycin against glioblastoma cancer stem cells. Clin. Transl. Oncol. 2014, 16, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Greenberger, S.; Yuan, S.; Walsh, L.A.; Boscolo, E.; Kang, K.T.; Matthews, B.; Mulliken, J.B.; Bischoff, J. Rapamycin suppresses self-renewal and vasculogenic potential of stem cells isolated from infantile hemangioma. J. Invest Dermatol. 2011, 131, 2467–2476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Liu, Y.; Lu, J.; Zhang, P.; Wang, Y.; Xu, Y.; Wang, Z.; Mao, J.H.; Wei, G. Rapamycin inhibits FBXW7 loss-induced epithelial-mesenchymal transition and cancer stem cell-like characteristics in colorectal cancer cells. Biochem. Biophys. Res. Commun. 2013, 434, 352–356. [Google Scholar] [CrossRef] [Green Version]
- Hambardzumyan, D.; Becher, O.J.; Rosenblum, M.K.; Pandolfi, P.P.; Manova-Todorova, K.; Holland, E.C. PI3K pathway regulates survival of cancer stem cells residing in the perivascular niche following radiation in medulloblastoma in vivo. Genes Dev. 2008, 22, 436–448. [Google Scholar] [CrossRef] [Green Version]
- Miranda-Lorenzo, I.; Dorado, J.; Lonardo, E.; Alcala, S.; Serrano, A.G.; Clausell-Tormos, J.; Cioffi, M.; Megias, D.; Zagorac, S.; Balic, A.; et al. Intracellular autofluorescence: A biomarker for epithelial cancer stem cells. Nat. Methods 2014, 11, 1161–1169. [Google Scholar] [CrossRef]
- Sainz, B., Jr.; Alcala, S.; Garcia, E.; Sanchez-Ripoll, Y.; Azevedo, M.M.; Cioffi, M.; Tatari, M.; Miranda-Lorenzo, I.; Hidalgo, M.; Gomez-Lopez, G.; et al. Microenvironmental hCAP-18/LL-37 promotes pancreatic ductal adenocarcinoma by activating its cancer stem cell compartment. Gut 2015, 64, 1921–1935. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Hao, P.; Li, L.; Tan, C.Y.; Cheng, X.; Chen, G.Y.; Sze, S.K.; Shen, H.M.; Yao, S.Q. Design and synthesis of minimalist terminal alkyne-containing diazirine photo-crosslinkers and their incorporation into kinase inhibitors for cell- and tissue-based proteome profiling. Angew. Chem. Int. Ed Engl. 2013, 52, 8551–8556. [Google Scholar] [CrossRef]
- Nadanaciva, S.; Lu, S.; Gebhard, D.F.; Jessen, B.A.; Pennie, W.D.; Will, Y. A high content screening assay for identifying lysosomotropic compounds. Toxicol. In Vitro 2011, 25, 715–723. [Google Scholar] [CrossRef]
- Salerno, M.; Avnet, S.; Bonuccelli, G.; Hosogi, S.; Granchi, D.; Baldini, N. Impairment of lysosomal activity as a therapeutic modality targeting cancer stem cells of embryonal rhabdomyosarcoma cell line RD. PLoS ONE 2014, 9, e110340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mai, T.T.; Hamai, A.; Hienzsch, A.; Caneque, T.; Muller, S.; Wicinski, J.; Cabaud, O.; Leroy, C.; David, A.; Acevedo, V.; et al. Salinomycin kills cancer stem cells by sequestering iron in lysosomes. Nat. Chem. 2017, 9, 1025–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sancho, P.; Burgos-Ramos, E.; Tavera, A.; Bou Kheir, T.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Grana, O.; et al. MYC/PGC-1alpha Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 2015, 22, 590–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, N.H.; Olsen, O.D.; Groth-Pedersen, L.; Ellegaard, A.M.; Bilgin, M.; Redmer, S.; Ostenfeld, M.S.; Ulanet, D.; Dovmark, T.H.; Lonborg, A.; et al. Transformation-associated changes in sphingolipid metabolism sensitize cells to lysosomal cell death induced by inhibitors of acid sphingomyelinase. Cancer Cell 2013, 24, 379–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heading, C. Siramesine H Lundbeck. Curr. Opin. Invest. Dr. 2001, 2, 266–270. [Google Scholar]
- Werbowetski-Ogilvie, T.E.; Bosse, M.; Stewart, M.; Schnerch, A.; Ramos-Mejia, V.; Rouleau, A.; Wynder, T.; Smith, M.J.; Dingwall, S.; Carter, T.; et al. Characterization of human embryonic stem cells with features of neoplastic progression. Nat. Biotechnol. 2009, 27, 91–97. [Google Scholar] [CrossRef]
- Sachlos, E.; Risueno, R.M.; Laronde, S.; Shapovalova, Z.; Lee, J.H.; Russell, J.; Malig, M.; McNicol, J.D.; Fiebig-Comyn, A.; Graham, M.; et al. Identification of drugs including a dopamine receptor antagonist that selectively target cancer stem cells. Cell 2012, 149, 1284–1297. [Google Scholar] [CrossRef] [Green Version]
- Oltulu, F.; Kocaturk, D.C.; Adali, Y.; Ozdil, B.; Acikgoz, E.; Gurel, C.; Karabay Yavasoglu, N.U.; Aktug, H. Autophagy and mTOR pathways in mouse embryonic stem cell, lung cancer and somatic fibroblast cell lines. J. Cell Biochem. 2019, 120, 18066–18076. [Google Scholar] [CrossRef]
- Kopras, E.; Potluri, V.; Bermudez, M.L.; Williams, K.; Belcher, S.; Kasper, S. Actions of endocrine-disrupting chemicals on stem/progenitor cells during development and disease. Endocr. Relat. Cancer 2014, 21, T1–T12. [Google Scholar] [CrossRef]
- Cioffi, M.; Trabulo, S.M.; Sanchez-Ripoll, Y.; Miranda-Lorenzo, I.; Lonardo, E.; Dorado, J.; Reis Vieira, C.; Ramirez, J.C.; Hidalgo, M.; Aicher, A.; et al. The miR-17-92 cluster counteracts quiescence and chemoresistance in a distinct subpopulation of pancreatic cancer stem cells. Gut 2015, 54, 1936–1948. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, C.; Arnt, J.; Costall, B.; Kelly, M.E.; Meier, E.; Naylor, R.J.; Perregaard, J. The selective sigma2-ligand Lu 28-179 has potent anxiolytic-like effects in rodents. J. Pharmacol. Exp. Ther. 1997, 283, 1323–1332. [Google Scholar] [PubMed]
- Ostenfeld, M.S.; Fehrenbacher, N.; Hoyer-Hansen, M.; Thomsen, C.; Farkas, T.; Jaattela, M. Effective tumor cell death by sigma-2 receptor ligand siramesine involves lysosomal leakage and oxidative stress. Cancer Res. 2005, 65, 8975–8983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostenfeld, M.S.; Hoyer-Hansen, M.; Bastholm, L.; Fehrenbacher, N.; Olsen, O.D.; Groth-Pedersen, L.; Puustinen, P.; Kirkegaard-Sorensen, T.; Nylandsted, J.; Farkas, T.; et al. Anti-cancer agent siramesine is a lysosomotropic detergent that induces cytoprotective autophagosome accumulation. Autophagy 2008, 4, 487–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harinasuta, T.; Bunnag, D.; Wernsdorfer, W.H. A phase II clinical trial of mefloquine in patients with chloroquine-resistant falciparum malaria in Thailand. Bull. World Health Organ. 1983, 61, 299–305. [Google Scholar]
- Groth-Pedersen, L.; Ostenfeld, M.S.; Hoyer-Hansen, M.; Nylandsted, J.; Jaattela, M. Vincristine induces dramatic lysosomal changes and sensitizes cancer cells to lysosome-destabilizing siramesine. Cancer Res. 2007, 67, 2217–2225. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Sainz, B., Jr.; Martin, B.; Tatari, M.; Heeschen, C.; Guerra, S. ISG15 is a critical microenvironmental factor for pancreatic cancer stem cells. Cancer Res. 2014, 74, 7309–7320. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Gonzalez, S.; Rodriguez-Aristegui, S.; Gomez de la Oliva, C.A.; Hernandez, A.I.; Gonzalez Cantalapiedra, E.; Varela, C.; Garcia, A.B.; Rabal, O.; Oyarzabal, J.; Bischoff, J.R.; et al. Discovery of novel triazolo[4,3-b]pyridazin-3-yl-quinoline derivatives as PIM inhibitors. Eur. J. Med. Chem. 2019, 168, 87–109. [Google Scholar] [CrossRef]
- Foghsgaard, L.; Wissing, D.; Mauch, D.; Lademann, U.; Bastholm, L.; Boes, M.; Elling, F.; Leist, M.; Jaattela, M. Cathepsin B acts as a dominant execution protease in tumor cell apoptosis induced by tumor necrosis factor. J. Cell Biol. 2001, 153, 999–1010. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Smyth, G.K. ELDA: Extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J. Immunol. Methods 2009, 347, 70–78. [Google Scholar] [CrossRef] [PubMed]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cash, T.P.; Alcalá, S.; Rico-Ferreira, M.d.R.; Hernández-Encinas, E.; García, J.; Albarrán, M.I.; Valle, S.; Muñoz, J.; Martínez-González, S.; Blanco-Aparicio, C.; et al. Induction of Lysosome Membrane Permeabilization as a Therapeutic Strategy to Target Pancreatic Cancer Stem Cells. Cancers 2020, 12, 1790. https://doi.org/10.3390/cancers12071790
Cash TP, Alcalá S, Rico-Ferreira MdR, Hernández-Encinas E, García J, Albarrán MI, Valle S, Muñoz J, Martínez-González S, Blanco-Aparicio C, et al. Induction of Lysosome Membrane Permeabilization as a Therapeutic Strategy to Target Pancreatic Cancer Stem Cells. Cancers. 2020; 12(7):1790. https://doi.org/10.3390/cancers12071790
Chicago/Turabian StyleCash, Timothy P., Sonia Alcalá, María del Rosario Rico-Ferreira, Elena Hernández-Encinas, Jennifer García, María Isabel Albarrán, Sandra Valle, Javier Muñoz, Sonia Martínez-González, Carmen Blanco-Aparicio, and et al. 2020. "Induction of Lysosome Membrane Permeabilization as a Therapeutic Strategy to Target Pancreatic Cancer Stem Cells" Cancers 12, no. 7: 1790. https://doi.org/10.3390/cancers12071790
APA StyleCash, T. P., Alcalá, S., Rico-Ferreira, M. d. R., Hernández-Encinas, E., García, J., Albarrán, M. I., Valle, S., Muñoz, J., Martínez-González, S., Blanco-Aparicio, C., Pastor, J., Serrano, M., & Sainz, B., Jr. (2020). Induction of Lysosome Membrane Permeabilization as a Therapeutic Strategy to Target Pancreatic Cancer Stem Cells. Cancers, 12(7), 1790. https://doi.org/10.3390/cancers12071790