Increase of Antitumoral Effects of Cytokine-Induced Killer Cells by Antibody-Mediated Inhibition of MICA Shedding


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
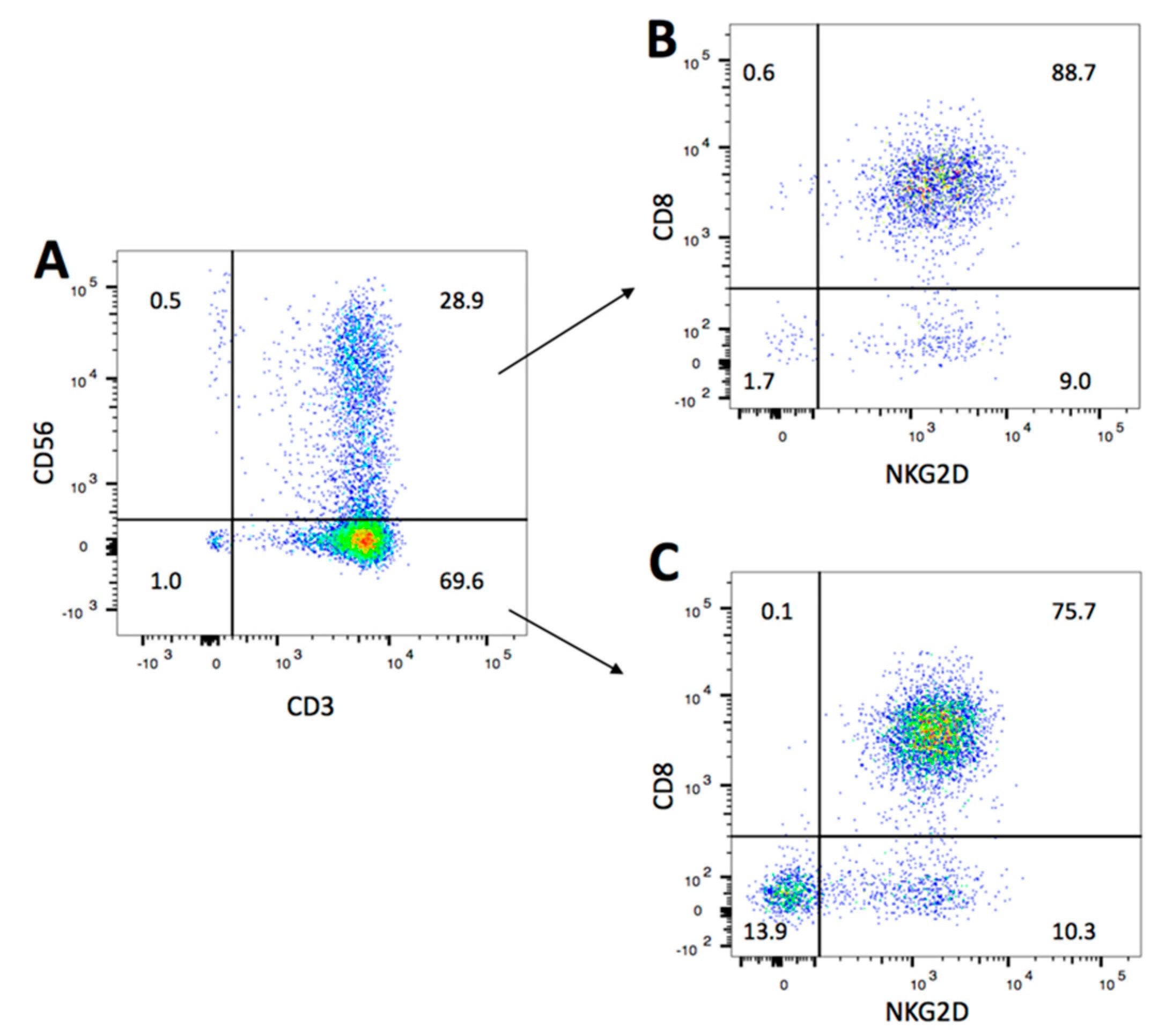
2.1. NKG2D Expression and Phenotype of CIK Cells
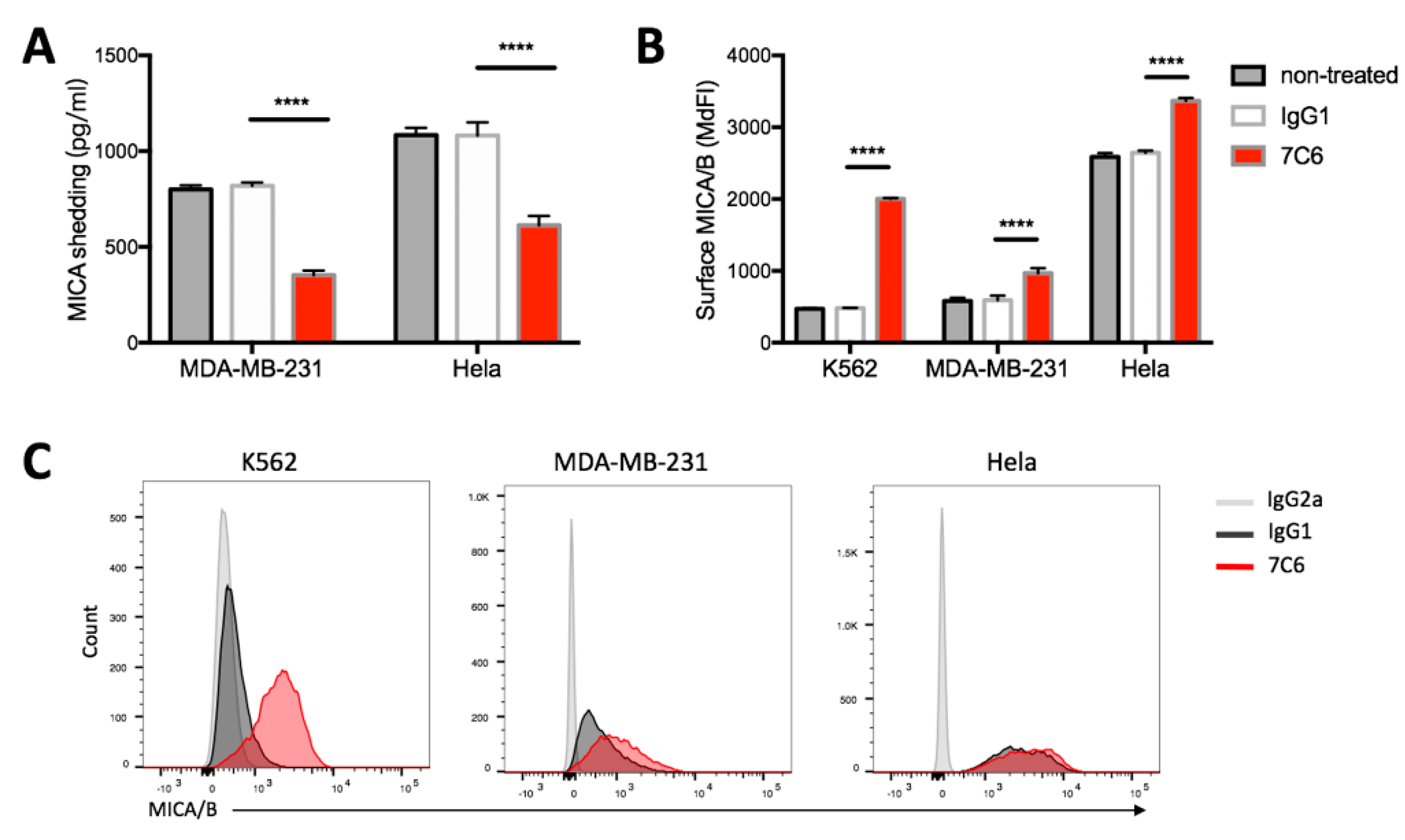
2.2. Inhibition of MICA Shedding and Stabilization of Surface MICA/B Expression on Tumor Cells by 7C6 Antibody
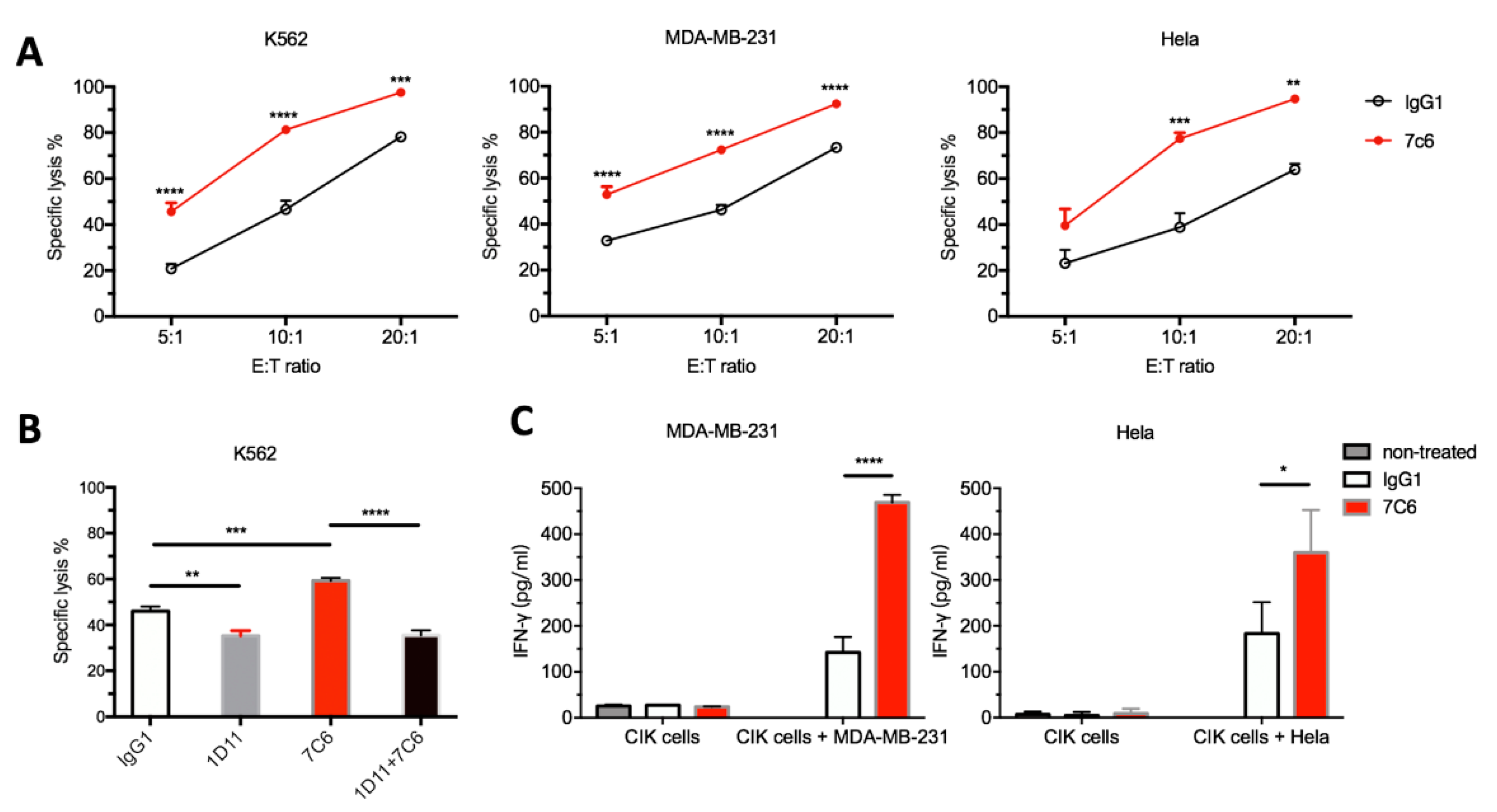
2.3. 7C6 mAb Enhances the In Vitro Antitumor Activity of CIK through the NKG2D-MICA/B Axis
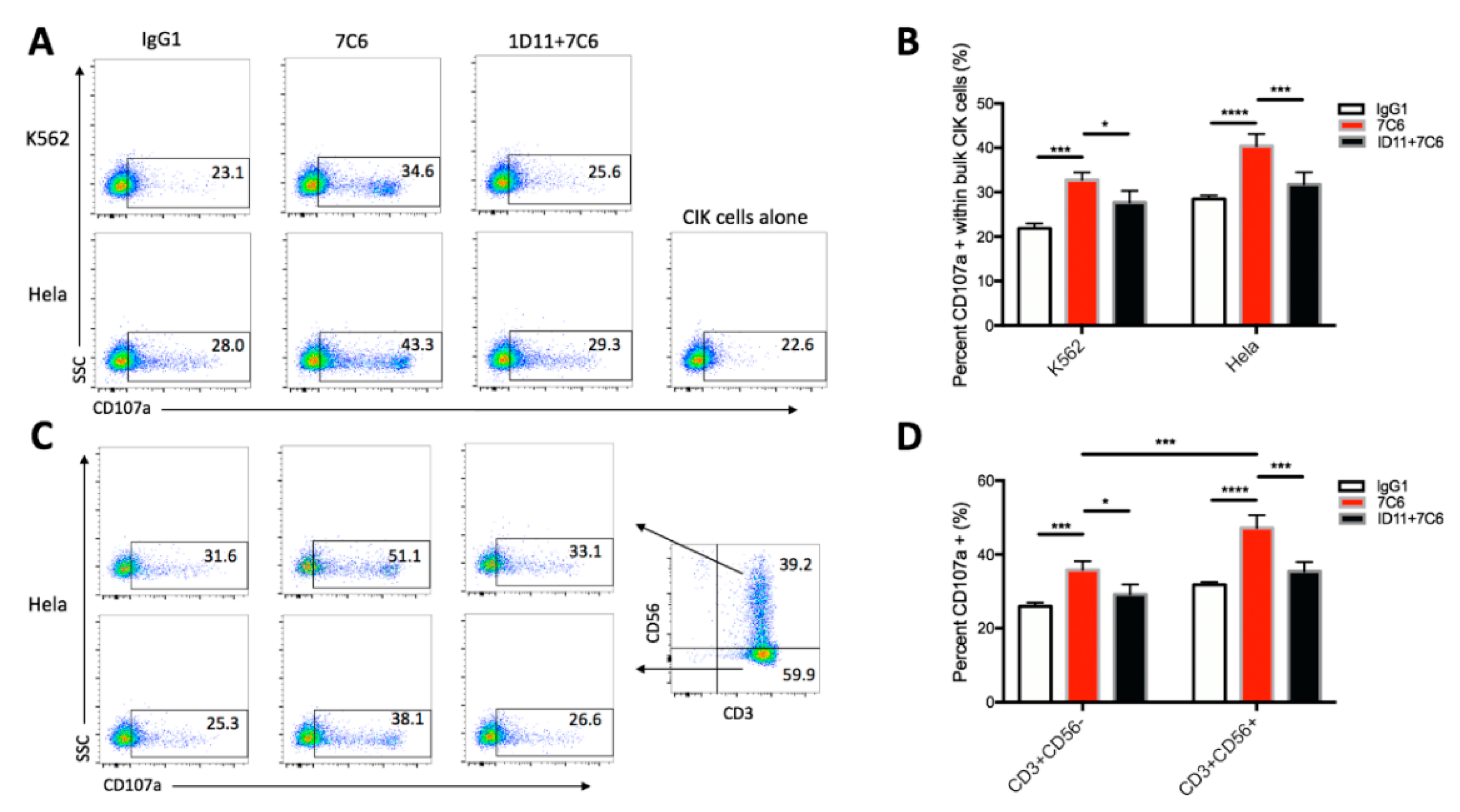
2.4. NKG2D-MICA/B Ligation Leads to the Triggering and Activation of both CD3+CD56+ NKT Cells and CD3+CD56− T Subset of CIK Cells
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Generation of CIK Cells
4.3. Antibody Reagents
4.4. Phenotypic Analysis
4.5. Surface Expression of MICA/B on Tumor Cells
4.6. FACS Cytotoxicity Assay
4.7. Degranulation Assay
4.8. ELISA Assay
4.8.1. IFN-γ Production by CIK Cells
4.8.2. MICA Shedding by Tumor Cells
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schmidt-Wolf, I.G.; Negrin, R.S.; Kiem, H.P.; Blume, K.G.; Weissman, I.L. Use of a SCID mouse/human lymphoma model to evaluate cytokine-induced killer cells with potent antitumor cell activity. J. Exp. Med. 1991, 174, 139–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmeel, F.C.; Schmeel, L.C.; Gast, S.M.; Schmidt-Wolf, I.G. Adoptive immunotherapy strategies with cytokine-induced killer (CIK) cells in the treatment of hematological malignancies. Int. J. Mol. Sci. 2014, 15, 14632–14648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mata-Molanes, J.J.; Sureda Gonzalez, M.; Valenzuela Jimenez, B.; Martinez Navarro, E.M.; Brugarolas Masllorens, A. Cancer Immunotherapy with Cytokine-Induced Killer Cells. Target Oncol. 2017, 12, 289–299. [Google Scholar] [CrossRef]
- Nwangwu, C.A.; Weiher, H.; Schmidt-Wolf, I.G.H. Increase of CIK cell efficacy by upregulating cell surface MICA and inhibition of NKG2D ligand shedding in multiple myeloma. Hematol. Oncol. 2017, 35, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Narayan, R.; Benjamin, J.E.; Shah, O.; Tian, L.; Tate, K.; Armstrong, R.; Xie, B.J.; Lowsky, R.; Laport, G.; Negrin, R.S.; et al. Donor-Derived Cytokine-Induced Killer Cell Infusion as Consolidation after Nonmyeloablative Allogeneic Transplantation for Myeloid Neoplasms. Biol. Blood Marrow Transpl. 2019, 25, 1293–1303. [Google Scholar] [CrossRef]
- Bremm, M.; Pfeffermann, L.M.; Cappel, C.; Katzki, V.; Erben, S.; Betz, S.; Quaiser, A.; Merker, M.; Bonig, H.; Schmidt, M.; et al. Improving Clinical Manufacturing of IL-15 Activated Cytokine-Induced Killer (CIK) Cells. Front. Immunol. 2019, 10, 1218. [Google Scholar] [CrossRef]
- Luah, Y.H.; Sundar Raj, K.; Koh, M.B.C.; Linn, Y.C. A novel simplified method of generating cytomegalovirus-specific cytokine-induced killer cells of high specificity and superior potency with GMP compliance. Clin. Immunol. 2019, 205, 83–92. [Google Scholar] [CrossRef]
- Schlegel, P.; Lang, A.M.; Matela, M.; Horrer, A.; Schilling, A.; Jochner, A.; Wiedenmann, M.; Seitz, C.; Doring, M.; Feuchtinger, T.; et al. Ex vivo expansion of autologous, donor-derived NK-, gammadeltaT-, and cytokine induced killer (CIK) cells post haploidentical hematopoietic stem cell transplantation results in increased antitumor activity. Bone Marrow Transplant 2019, 54, 727–732. [Google Scholar] [CrossRef]
- Schmidt-Wolf, I.G.; Lefterova, P.; Johnston, V.; Huhn, D.; Blume, K.G.; Negrin, R.S. Propagation of large numbers of T cells with natural killer cell markers. Br. J. Haematol. 1994, 87, 453–458. [Google Scholar] [CrossRef]
- Mehta, B.A.; Schmidt-Wolf, I.G.; Weissman, I.L.; Negrin, R.S. Two pathways of exocytosis of cytoplasmic granule contents and target cell killing by cytokine-induced CD3+ CD56+ killer cells. Blood 1995, 86, 3493–3499. [Google Scholar] [CrossRef] [Green Version]
- Pievani, A.; Borleri, G.; Pende, D.; Moretta, L.; Rambaldi, A.; Golay, J.; Introna, M. Dual-functional capability of CD3+CD56+ CIK cells, a T-cell subset that acquires NK function and retains TCR-mediated specific cytotoxicity. Blood 2011, 118, 3301–3310. [Google Scholar] [CrossRef] [Green Version]
- Verneris, M.R.; Karimi, M.; Baker, J.; Jayaswal, A.; Negrin, R.S. Role of NKG2D signaling in the cytotoxicity of activated and expanded CD8+ T cells. Blood 2004, 103, 3065–3072. [Google Scholar] [CrossRef] [Green Version]
- Cappuzzello, E.; Tosi, A.; Zanovello, P.; Sommaggio, R.; Rosato, A. Retargeting cytokine-induced killer cell activity by CD16 engagement with clinical-grade antibodies. Oncoimmunology 2016, 5, e1199311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cappel, C.; Huenecke, S.; Suemmerer, A.; Erben, S.; Rettinger, E.; Pfirrmann, V.; Heinze, A.; Zimmermann, O.; Klingebiel, T.; Ullrich, E.; et al. Cytotoxic potential of IL-15-activated cytokine-induced killer cells against human neuroblastoma cells. Pediatr Blood Cancer 2016, 63, 2230–2239. [Google Scholar] [CrossRef]
- Durrieu, L.; Lemieux, W.; Dieng, M.M.; Fontaine, F.; Duval, M.; Le Deist, F.; Haddad, E. Implication of different effector mechanisms by cord blood-derived and peripheral blood-derived cytokine-induced killer cells to kill precursor B acute lymphoblastic leukemia cell lines. Cytotherapy 2014, 16, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Bauer, S.; Groh, V.; Wu, J.; Steinle, A.; Phillips, J.H.; Lanier, L.L.; Spies, T. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 1999, 285, 727–729. [Google Scholar] [CrossRef]
- Groh, V.; Rhinehart, R.; Randolph-Habecker, J.; Topp, M.S.; Riddell, S.R.; Spies, T. Costimulation of CD8alphabeta T cells by NKG2D via engagement by MIC induced on virus-infected cells. Nat. Immunol. 2001, 2, 255–260. [Google Scholar] [CrossRef]
- Eagle, R.A.; Trowsdale, J. Promiscuity and the single receptor: NKG2D. Nat. Rev. Immunol. 2007, 7, 737–744. [Google Scholar] [CrossRef] [PubMed]
- El-Gazzar, A.; Groh, V.; Spies, T. Immunobiology and conflicting roles of the human NKG2D lymphocyte receptor and its ligands in cancer. J. Immunol. 2013, 191, 1509–1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhar, P.; Wu, J.D. NKG2D and its ligands in cancer. Curr. Opin. Immunol. 2018, 51, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, B.K.; Yim, D.; Chow, I.T.; Gonzalez, S.; Dai, Z.; Mann, H.H.; Strong, R.K.; Groh, V.; Spies, T. Disulphide-isomerase-enabled shedding of tumour-associated NKG2D ligands. Nature 2007, 447, 482–486. [Google Scholar] [CrossRef] [PubMed]
- Waldhauer, I.; Goehlsdorf, D.; Gieseke, F.; Weinschenk, T.; Wittenbrink, M.; Ludwig, A.; Stevanovic, S.; Rammensee, H.G.; Steinle, A. Tumor-associated MICA is shed by ADAM proteases. Cancer Res. 2008, 68, 6368–6376. [Google Scholar] [CrossRef] [Green Version]
- Boutet, P.; Aguera-Gonzalez, S.; Atkinson, S.; Pennington, C.J.; Edwards, D.R.; Murphy, G.; Reyburn, H.T.; Vales-Gomez, M. Cutting edge: The metalloproteinase ADAM17/TNF-alpha-converting enzyme regulates proteolytic shedding of the MHC class I-related chain B protein. J. Immunol. 2009, 182, 49–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Atteridge, C.L.; Wang, X.; Lundgren, A.D.; Wu, J.D. The membrane type matrix metalloproteinase MMP14 mediates constitutive shedding of MHC class I chain-related molecule A independent of A disintegrin and metalloproteinases. J. Immunol. 2010, 184, 3346–3350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrari de Andrade, L.; Tay, R.E.; Pan, D.; Luoma, A.M.; Ito, Y.; Badrinath, S.; Tsoucas, D.; Franz, B.; May, K.F., Jr.; Harvey, C.J.; et al. Antibody-mediated inhibition of MICA and MICB shedding promotes NK cell-driven tumor immunity. Science 2018, 359, 1537–1542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt-Wolf, I.G.; Lefterova, P.; Mehta, B.A.; Fernandez, L.P.; Huhn, D.; Blume, K.G.; Weissman, I.L.; Negrin, R.S. Phenotypic characterization and identification of effector cells involved in tumor cell recognition of cytokine-induced killer cells. Exp. Hematol. 1993, 21, 1673–1679. [Google Scholar]
- Linn, Y.C.; Lau, S.K.; Liu, B.H.; Ng, L.H.; Yong, H.X.; Hui, K.M. Characterization of the recognition and functional heterogeneity exhibited by cytokine-induced killer cell subsets against acute myeloid leukaemia target cell. Immunology 2009, 126, 423–435. [Google Scholar] [CrossRef]
- Cluxton, C.D.; Spillane, C.; O’Toole, S.A.; Sheils, O.; Gardiner, C.M.; O’Leary, J.J. Suppression of Natural Killer cell NKG2D and CD226 anti-tumour cascades by platelet cloaked cancer cells: Implications for the metastatic cascade. PLoS ONE 2019, 14, e0211538. [Google Scholar] [CrossRef] [Green Version]
- Betts, M.R.; Brenchley, J.M.; Price, D.A.; De Rosa, S.C.; Douek, D.C.; Roederer, M.; Koup, R.A. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J. Immunol. Methods 2003, 281, 65–78. [Google Scholar] [CrossRef]
- Alter, G.; Malenfant, J.M.; Altfeld, M. CD107a as a functional marker for the identification of natural killer cell activity. J Immunol. Methods 2004, 294, 15–22. [Google Scholar] [CrossRef]
- Merker, M.; Salzmann-Manrique, E.; Katzki, V.; Huenecke, S.; Bremm, M.; Bakhtiar, S.; Willasch, A.; Jarisch, A.; Soerensen, J.; Schulz, A.; et al. Clearance of Hematologic Malignancies by Allogeneic Cytokine-Induced Killer Cell or Donor Lymphocyte Infusions. Biol. Blood Marrow Transplant 2019, 25, 1281–1292. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, J.H.; Lim, Y.S.; Yeon, J.E.; Song, T.J.; Yu, S.J.; Gwak, G.Y.; Kim, K.M.; Kim, Y.J.; Lee, J.W.; et al. Sustained efficacy of adjuvant immunotherapy with cytokine-induced killer cells for hepatocellular carcinoma: An extended 5-year follow-up. Cancer Immunol. Immunother 2019, 68, 23–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, P.H.; Negrin, R.S. A novel population of expanded human CD3+CD56+ cells derived from T cells with potent in vivo antitumor activity in mice with severe combined immunodeficiency. J. Immunol. 1994, 153, 1687–1696. [Google Scholar] [PubMed]
- Nishimura, R.; Baker, J.; Beilhack, A.; Zeiser, R.; Olson, J.A.; Sega, E.I.; Karimi, M.; Negrin, R.S. In vivo trafficking and survival of cytokine-induced killer cells resulting in minimal GVHD with retention of antitumor activity. Blood 2008, 112, 2563–2574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrari de Andrade, L.; Kumar, S.; Luoma, A.M.; Ito, Y.; Alves da Silva, P.H.; Pan, D.; Pyrdol, J.W.; Yoon, C.H.; Wucherpfennig, K.W. Inhibition of MICA and MICB Shedding Elicits NK-Cell-Mediated Immunity against Tumors Resistant to Cytotoxic T Cells. Cancer Immunol. Res. 2020, 8, 769–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groh, V.; Wu, J.; Yee, C.; Spies, T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature 2002, 419, 734–738. [Google Scholar] [CrossRef]
- Raulet, D.H.; Gasser, S.; Gowen, B.G.; Deng, W.; Jung, H. Regulation of ligands for the NKG2D activating receptor. Annu. Rev. Immunol. 2013, 31, 413–441. [Google Scholar] [CrossRef] [Green Version]
- Deng, W.; Gowen, B.G.; Zhang, L.; Wang, L.; Lau, S.; Iannello, A.; Xu, J.; Rovis, T.L.; Xiong, N.; Raulet, D.H. Antitumor immunity. A shed NKG2D ligand that promotes natural killer cell activation and tumor rejection. Science 2015, 348, 136–139. [Google Scholar] [CrossRef] [Green Version]
- Clayton, A.; Mitchell, J.P.; Court, J.; Linnane, S.; Mason, M.D.; Tabi, Z. Human tumor-derived exosomes down-modulate NKG2D expression. J. Immunol. 2008, 180, 7249–7258. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.D.; Higgins, L.M.; Steinle, A.; Cosman, D.; Haugk, K.; Plymate, S.R. Prevalent expression of the immunostimulatory MHC class I chain-related molecule is counteracted by shedding in prostate cancer. J. Clin. Investig. 2004, 114, 560–568. [Google Scholar] [CrossRef]
- Bryceson, Y.T.; March, M.E.; Ljunggren, H.G.; Long, E.O. Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood 2006, 107, 159–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, C.; Bevers, J., 3rd; Cook, R.; Lombana, T.N.; Rajasekaran, K.; Matsumoto, M.; Spiess, C.; Kim, J.M.; Ye, Z. MICA immune complex formed with alpha 3 domain-specific antibody activates human NK cells in a Fc-dependent manner. J. Immunother. Cancer 2019, 7, 207. [Google Scholar] [CrossRef] [Green Version]
- Huergo-Zapico, L.; Acebes-Huerta, A.; Gonzalez-Rodriguez, A.P.; Contesti, J.; Gonzalez-Garcia, E.; Payer, A.R.; Villa-Alvarez, M.; Fernandez-Guizan, A.; Lopez-Soto, A.; Gonzalez, S. Expansion of NK cells and reduction of NKG2D expression in chronic lymphocytic leukemia. Correlation with progressive disease. PLoS ONE 2014, 9, e108326. [Google Scholar] [CrossRef] [PubMed]
- Konjevic, G.; Jurisic, V.; Jovic, V.; Vuletic, A.; Mirjacic Martinovic, K.; Radenkovic, S.; Spuzic, I. Investigation of NK cell function and their modulation in different malignancies. Immunol. Res. 2012, 52, 139–156. [Google Scholar] [CrossRef] [PubMed]
- Cholujova, D.; Jakubikova, J.; Kubes, M.; Arendacka, B.; Sapak, M.; Ihnatko, R.; Sedlak, J. Comparative study of four fluorescent probes for evaluation of natural killer cell cytotoxicity assays. Immunobiology 2008, 213, 629–640. [Google Scholar] [CrossRef]
- Lorenzo-Herrero, S.; Sordo-Bahamonde, C.; Gonzalez, S.; Lopez-Soto, A. CD107a Degranulation Assay to Evaluate Immune Cell Antitumor Activity. Methods Mol. Biol. 2019, 1884, 119–130. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, X.; Zhang, Y.; Li, Y.; Schmidt-Wolf, I.G.H. Increase of Antitumoral Effects of Cytokine-Induced Killer Cells by Antibody-Mediated Inhibition of MICA Shedding. Cancers 2020, 12, 1818. https://doi.org/10.3390/cancers12071818
Wu X, Zhang Y, Li Y, Schmidt-Wolf IGH. Increase of Antitumoral Effects of Cytokine-Induced Killer Cells by Antibody-Mediated Inhibition of MICA Shedding. Cancers. 2020; 12(7):1818. https://doi.org/10.3390/cancers12071818
Chicago/Turabian StyleWu, Xiaolong, Ying Zhang, Yutao Li, and Ingo G.H. Schmidt-Wolf. 2020. "Increase of Antitumoral Effects of Cytokine-Induced Killer Cells by Antibody-Mediated Inhibition of MICA Shedding" Cancers 12, no. 7: 1818. https://doi.org/10.3390/cancers12071818