Towards Clinical Implementation of Adeno-Associated Virus (AAV) Vectors for Cancer Gene Therapy: Current Status and Future Perspectives


Abstract
:1. Introduction
2. Adeno-Associated Virus and AAV Vector Technology
2.1. From Virus to Vectors—A Brief Introduction into the AAV Vector System
2.2. AAV Vector-Mediated Transduction
2.3. In Vivo Gene Therapy with First Generation AAV Vectors—Blueprints for Cancer Gene Therapy
2.3.1. Liver-Directed Gene Therapy
2.3.2. CNS-Directed Gene Therapy
2.4. Optimizing the AAV Vector System for Cancer Gene Therapy
2.4.1. Focus on the AAV Vector Genome—Achieving Transgene Expression in Cells of the Tumor Microenvironment
2.4.2. Focus on the AAV Vector Genome—Achieving Specificity by Transcriptional and Post-Transcriptional Targeting
2.4.3. Focus on the AAV Capsid—Achieving Specificity by Transductional Targeting
3. Use of AAV Vectors for Cancer Gene Therapy in Preclinical Models
3.1. AAV Vector-Mediated Targeting Tumor Cell Proliferation and Tumor Cell Death
3.1.1. Cytotoxic Killing of Tumor Cells: Suicide Gene Transfer
3.1.2. Inducing Tumor Cell Death: Apoptosis and Beyond
3.2. AAV Vector-Mediated Targeting of Tumor Angiogenesis
3.2.1. Overexpression of Anti-Angiogenic Factors
3.2.2. Strategies to Block the VEGF Pathway
3.3. Use of AAV Vectors for Immunotherapy of Cancer
3.3.1. AAV Vector-Mediated Modification of Immune Cells: CAR-T Cells and Beyond
3.3.2. AAV-Based Cancer Vaccination Approaches
3.3.3. AAV-Vector Mediated Delivery of Cytokines—Modulating the Immunosuppressive Microenvironment
3.3.4. Other AAV Vector-Based Approaches for Cancer Immunotherapy
4. Future Perspectives and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puzanov, I.; Milhem, M.M.; Minor, D.; Hamid, O.; Li, A.; Chen, L.; Chastain, M.; Gorski, K.S.; Anderson, A.; Chou, J.; et al. Talimogene Laherparepvec in Combination with Ipilimumab in Previously Untreated, Unresectable Stage IIIB-IV Melanoma. J. Clin. Oncol. 2016, 34, 2619–2626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keeler, A.M.; Flotte, T.R. Recombinant Adeno-Associated Virus Gene Therapy in Light of Luxturna (and Zolgensma and Glybera): Where Are We, and How Did We Get Here? Annu. Rev. Virol. 2019, 6, 601–621. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef]
- Hastie, E.; Samulski, R.J. Adeno-associated virus at 50: A golden anniversary of discovery, research, and gene therapy success--a personal perspective. Hum. Gene Ther. 2015, 26, 257–265. [Google Scholar] [CrossRef] [Green Version]
- Gao, G.; Vandenberghe, L.H.; Alvira, M.R.; Lu, Y.; Calcedo, R.; Zhou, X.; Wilson, J.M. Clades of Adeno-associated viruses are widely disseminated in human tissues. J. Virol. 2004, 78, 6381–6388. [Google Scholar] [CrossRef] [Green Version]
- Gao, G.P.; Alvira, M.R.; Wang, L.; Calcedo, R.; Johnston, J.; Wilson, J.M. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc. Natl. Acad. Sci. USA 2002, 99, 11854–11859. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.H.; Hallwirth, C.V.; Westerman, M.; Hetherington, N.A.; Tseng, Y.S.; Cecchini, S.; Virag, T.; Ziegler, M.L.; Rogozin, I.B.; Koonin, E.V.; et al. Germline viral “fossils” guide in silico reconstruction of a mid-Cenozoic era marsupial adeno-associated virus. Sci. Rep. 2016, 6, 28965. [Google Scholar] [CrossRef]
- Büning, H.; Srivastava, A. Capsid Modifications for Targeting and Improving the Efficacy of AAV Vectors. Mol. Ther. Methods Clin. Dev. 2019, 12, 248–265. [Google Scholar] [CrossRef]
- Santiago-Ortiz, J.L.; Schaffer, D.V. Adeno-associated virus (AAV) vectors in cancer gene therapy. J. Control. Release 2016, 240, 287–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotterman, M.A.; Schaffer, D.V. Engineering adeno-associated viruses for clinical gene therapy. Nat. Rev. Genet. 2014, 15, 445–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Recchia, A.; Mavilio, F. Site-specific integration by the adeno-associated virus rep protein. Curr. Gene Ther. 2011, 11, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Nygaard, S.; Barzel, A.; Haft, A.; Major, A.; Finegold, M.; Kay, M.A.; Grompe, M. A universal system to select gene-modified hepatocytes in vivo. Sci. Transl. Med. 2016, 8, 342ra79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagedorn, C.; Schnodt-Fuchs, M.; Boehme, P.; Abdelrazik, H.; Lipps, H.J.; Büning, H. S/MAR Element Facilitates Episomal Long-Term Persistence of Adeno-Associated Virus Vector Genomes in Proliferating Cells. Hum. Gene Ther. 2017, 28, 1169–1179. [Google Scholar] [CrossRef]
- Penaud-Budloo, M.; Francois, A.; Clement, N.; Ayuso, E. Pharmacology of Recombinant Adeno-associated Virus Production. Mol. Ther. Methods Clin. Dev. 2018, 8, 166–180. [Google Scholar] [CrossRef] [Green Version]
- Rabinowitz, J.E.; Rolling, F.; Li, C.; Conrath, H.; Xiao, W.; Xiao, X.; Samulski, R.J. Cross-packaging of a single adeno-associated virus (AAV) type 2 vector genome into multiple AAV serotypes enables transduction with broad specificity. J. Virol. 2002, 76, 791–801. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.Y.; Halder, S.; Agbandje-McKenna, M. Parvovirus glycan interactions. Curr. Opin. Virol. 2014, 7, 108–118. [Google Scholar] [CrossRef] [Green Version]
- Sonntag, F.; Bleker, S.; Leuchs, B.; Fischer, R.; Kleinschmidt, J.A. Adeno-associated virus type 2 capsids with externalized VP1/VP2 trafficking domains are generated prior to passage through the cytoplasm and are maintained until uncoating occurs in the nucleus. J. Virol. 2006, 80, 11040–11054. [Google Scholar] [CrossRef] [Green Version]
- Nonnenmacher, M.; Weber, T. Intracellular transport of recombinant adeno-associated virus vectors. Gene Ther. 2012, 19, 649–658. [Google Scholar] [CrossRef]
- Liu, X.; Huang, H.; Gao, Y.; Zhou, L.; Yang, J.; Li, X.; Li, Y.; Zhao, H.; Su, S.; Ke, C.; et al. Visualization of gene therapy with a liver cancer-targeted adeno-associated virus 3 vector. J. Cancer 2020, 11, 2192–2200. [Google Scholar] [CrossRef] [PubMed]
- Dhungel, B.; Jayachandran, A.; Layton, C.J.; Steel, J.C. Seek and destroy: Targeted adeno-associated viruses for gene delivery to hepatocellular carcinoma. Drug Deliv. 2017, 24, 289–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Presutti, D.; Ceccarelli, M.; Micheli, L.; Papoff, G.; Santini, S.; Samperna, S.; Lalli, C.; Zentilin, L.; Ruberti, G.; Tirone, F. Tis21-gene therapy inhibits medulloblastoma growth in a murine allograft model. PLoS ONE 2018, 13, e0194206. [Google Scholar] [CrossRef] [Green Version]
- GuhaSarkar, D.; Neiswender, J.; Su, Q.; Gao, G.; Sena-Esteves, M. Intracranial AAV-IFN-beta gene therapy eliminates invasive xenograft glioblastoma and improves survival in orthotopic syngeneic murine model. Mol. Oncol. 2017, 11, 180–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kattenhorn, L.M.; Tipper, C.H.; Stoica, L.; Geraghty, D.S.; Wright, T.L.; Clark, K.R.; Wadsworth, S.C. Adeno-Associated Virus Gene Therapy for Liver Disease. Hum. Gene Ther. 2016, 27, 947–961. [Google Scholar] [CrossRef] [PubMed]
- Manno, C.S.; Pierce, G.F.; Arruda, V.R.; Glader, B.; Ragni, M.; Rasko, J.J.; Ozelo, M.C.; Hoots, K.; Blatt, P.; Konkle, B.; et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 2006, 12, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Nathwani, A.C.; Tuddenham, E.G.; Rangarajan, S.; Rosales, C.; McIntosh, J.; Linch, D.C.; Chowdary, P.; Riddell, A.; Pie, A.J.; Harrington, C.; et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N. Engl. J. Med. 2011, 365, 2357–2365. [Google Scholar] [CrossRef]
- Nathwani, A.C.; Reiss, U.M.; Tuddenham, E.G.; Rosales, C.; Chowdary, P.; McIntosh, J.; Della Peruta, M.; Lheriteau, E.; Patel, N.; Raj, D.; et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N. Engl. J. Med. 2014, 371, 1994–2004. [Google Scholar] [CrossRef] [Green Version]
- Baruteau, J.; Waddington, S.N.; Alexander, I.E.; Gissen, P. Gene therapy for monogenic liver diseases: Clinical successes, current challenges and future prospects. J. Inherit. Metab. Dis. 2017, 40, 497–517. [Google Scholar] [CrossRef]
- Deverman, B.E.; Ravina, B.M.; Bankiewicz, K.S.; Paul, S.M.; Sah, D.W.Y. Gene therapy for neurological disorders: Progress and prospects. Nat. Rev. Drug Discov. 2018, 17, 641–659. [Google Scholar] [CrossRef]
- Mendell, J.R.; Al-Zaidy, S.; Shell, R.; Arnold, W.D.; Rodino-Klapac, L.R.; Prior, T.W.; Lowes, L.; Alfano, L.; Berry, K.; Church, K.; et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Albright, B.H.; Storey, C.M.; Murlidharan, G.; Castellanos Rivera, R.M.; Berry, G.E.; Madigan, V.J.; Asokan, A. Mapping the Structural Determinants Required for AAVrh.10 Transport across the Blood-Brain Barrier. Mol. Ther. 2018, 26, 510–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Li, S.; Wang, H.; Guo, Y.; Gessler, D.J.; Cao, C.; Su, Q.; Kramer, J.; Zhong, L.; Ahmed, S.S.; et al. Global CNS transduction of adult mice by intravenously delivered rAAVrh.8 and rAAVrh.10 and nonhuman primates by rAAVrh.10. Mol. Ther. 2014, 22, 1299–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hacker, U.T.; Wingenfeld, L.; Kofler, D.M.; Schuhmann, N.K.; Lutz, S.; Herold, T.; King, S.B.; Gerner, F.M.; Perabo, L.; Rabinowitz, J.; et al. Adeno-associated virus serotypes 1 to 5 mediated tumor cell directed gene transfer and improvement of transduction efficiency. J. Gene Med. 2005, 7, 1429–1438. [Google Scholar] [CrossRef]
- Cervelli, T.; Palacios, J.A.; Zentilin, L.; Mano, M.; Schwartz, R.A.; Weitzman, M.D.; Giacca, M. Processing of recombinant AAV genomes occurs in specific nuclear structures that overlap with foci of DNA-damage-response proteins. J. Cell Sci. 2008, 121, 349–357. [Google Scholar] [CrossRef] [Green Version]
- McCarty, D.M.; Monahan, P.E.; Samulski, R.J. Self-complementary recombinant adeno-associated virus (scAAV) vectors promote efficient transduction independently of DNA synthesis. Gene Ther. 2001, 8, 1248–1254. [Google Scholar] [CrossRef] [Green Version]
- McCarty, D.M. Self-complementary AAV vectors; advances and applications. Mol. Ther. 2008, 16, 1648–1656. [Google Scholar] [CrossRef]
- Aldrich, W.A.; Ren, C.; White, A.F.; Zhou, S.Z.; Kumar, S.; Jenkins, C.B.; Shaw, D.R.; Strong, T.V.; Triozzi, P.L.; Ponnazhagan, S. Enhanced transduction of mouse bone marrow-derived dendritic cells by repetitive infection with self-complementary adeno-associated virus 6 combined with immunostimulatory ligands. Gene Ther. 2006, 13, 29–39. [Google Scholar] [CrossRef]
- Veron, P.; Boutin, S.; Martin, S.; Chaperot, L.; Plumas, J.; Davoust, J.; Masurier, C. Highly efficient transduction of human plasmacytoid dendritic cells without phenotypic and functional maturation. J. Transl. Med. 2009, 7, 10. [Google Scholar] [CrossRef] [Green Version]
- Javan, B.; Shahbazi, M. Hypoxia-inducible tumour-specific promoters as a dual-targeting transcriptional regulation system for cancer gene therapy. Ecancermedicalscience 2017, 11, 751. [Google Scholar] [CrossRef]
- Yuan, X.; Larsson, C.; Xu, D. Mechanisms underlying the activation of TERT transcription and telomerase activity in human cancer: Old actors and new players. Oncogene 2019, 38, 6172–6183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinagre, J.; Almeida, A.; Populo, H.; Batista, R.; Lyra, J.; Pinto, V.; Coelho, R.; Celestino, R.; Prazeres, H.; Lima, L.; et al. Frequency of TERT promoter mutations in human cancers. Nat. Commun. 2013, 4, 2185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogg, R.T.; Garcia, J.A.; Gerard, R.D. Adenoviral targeting of gene expression to tumors. Cancer Gene Ther. 2010, 17, 375–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noyan, F.; Diez, I.A.; Hapke, M.; Klein, C.; Dewey, R.A. Induced transgene expression for the treatment of solid tumors by hematopoietic stem cell-based gene therapy. Cancer Gene Ther. 2012, 19, 352–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, R.K.; Dubeau, L.; Kleiner, H.; Parr, T.; Nichols, P.; Ko, B.; Dong, D.; Ko, H.; Mao, C.; DiGiovanni, J.; et al. Cancer-inducible transgene expression by the Grp94 promoter: Spontaneous activation in tumors of various origins and cancer-associated macrophages. Cancer Res. 2002, 62, 7207–7212. [Google Scholar] [PubMed]
- Sarcar, S.; Tulalamba, W.; Rincon, M.Y.; Tipanee, J.; Pham, H.Q.; Evens, H.; Boon, D.; Samara-Kuko, E.; Keyaerts, M.; Loperfido, M.; et al. Next-generation muscle-directed gene therapy by in silico vector design. Nat. Commun. 2019, 10, 492. [Google Scholar] [CrossRef]
- Nair, N.; Rincon, M.Y.; Evens, H.; Sarcar, S.; Dastidar, S.; Samara-Kuko, E.; Ghandeharian, O.; Man Viecelli, H.; Thony, B.; De Bleser, P.; et al. Computationally designed liver-specific transcriptional modules and hyperactive factor IX improve hepatic gene therapy. Blood 2014, 123, 3195–3199. [Google Scholar] [CrossRef] [Green Version]
- Geisler, A.; Fechner, H. MicroRNA-regulated viral vectors for gene therapy. World J. Exp. Med. 2016, 6, 37–54. [Google Scholar] [CrossRef]
- Chang, T.C.; Yu, D.; Lee, Y.S.; Wentzel, E.A.; Arking, D.E.; West, K.M.; Dang, C.V.; Thomas-Tikhonenko, A.; Mendell, J.T. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat. Genet. 2008, 40, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Della Peruta, M.; Badar, A.; Rosales, C.; Chokshi, S.; Kia, A.; Nathwani, D.; Galante, E.; Yan, R.; Arstad, E.; Davidoff, A.M.; et al. Preferential targeting of disseminated liver tumors using a recombinant adeno-associated viral vector. Hum. Gene Ther. 2015, 26, 94–103. [Google Scholar] [CrossRef] [Green Version]
- Brown, B.D.; Naldini, L. Exploiting and antagonizing microRNA regulation for therapeutic and experimental applications. Nat. Rev. Genet. 2009, 10, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Dhungel, B.; Ramlogan-Steel, C.A.; Steel, J.C. MicroRNA-Regulated Gene Delivery Systems for Research and Therapeutic Purposes. Molecules 2018, 23, 1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartlett, J.S.; Kleinschmidt, J.; Boucher, R.C.; Samulski, R.J. Targeted adeno-associated virus vector transduction of nonpermissive cells mediated by a bispecific F(ab’gamma)2 antibody. Nat. Biotechnol. 1999, 17, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Ponnazhagan, S.; Mahendra, G.; Kumar, S.; Thompson, J.A.; Castillas, M., Jr. Conjugate-based targeting of recombinant adeno-associated virus type 2 vectors by using avidin-linked ligands. J. Virol. 2002, 76, 12900–12907. [Google Scholar] [CrossRef] [Green Version]
- Mével, M.; Bouzelha, M.; Leray, A.; Pacouret, S.; Guilbaud, M.; Penaud-Budloo, M.; Alvarez-Dorta, D.; Dubreil, L.; Gouin, S.G.; Combal, J.P.; et al. Chemical modification of the adeno-associated virus capsid to improve gene delivery. Chem. Sci. 2020, 11, 1122–1131. [Google Scholar] [CrossRef] [Green Version]
- Munch, R.C.; Muth, A.; Muik, A.; Friedel, T.; Schmatz, J.; Dreier, B.; Trkola, A.; Pluckthun, A.; Büning, H.; Buchholz, C.J. Off-target-free gene delivery by affinity-purified receptor-targeted viral vectors. Nat. Commun. 2015, 6, 6246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichhoff, A.M.; Borner, K.; Albrecht, B.; Schafer, W.; Baum, N.; Haag, F.; Korbelin, J.; Trepel, M.; Braren, I.; Grimm, D.; et al. Nanobody-Enhanced Targeting of AAV Gene Therapy Vectors. Mol. Ther. Methods Clin. Dev. 2019, 15, 211–220. [Google Scholar] [CrossRef] [Green Version]
- Girod, A.; Ried, M.; Wobus, C.; Lahm, H.; Leike, K.; Kleinschmidt, J.; Deleage, G.; Hallek, M. Genetic capsid modifications allow efficient re-targeting of adeno-associated virus type 2. Nat. Med. 1999, 5, 1052–1056. [Google Scholar] [CrossRef]
- Shi, W.; Bartlett, J.S. RGD inclusion in VP3 provides adeno-associated virus type 2 (AAV2)-based vectors with a heparan sulfate-independent cell entry mechanism. Mol. Ther. 2003, 7, 515–525. [Google Scholar] [CrossRef]
- Boucas, J.; Lux, K.; Huber, A.; Schievenbusch, S.; Von Freyend, M.J.; Perabo, L.; Quadt-Humme, S.; Odenthal, M.; Hallek, M.; Büning, H. Engineering adeno-associated virus serotype 2-based targeting vectors using a new insertion site-position 453-and single point mutations. J. Gene Med. 2009, 11, 1103–1113. [Google Scholar] [CrossRef]
- Stachler, M.D.; Bartlett, J.S. Mosaic vectors comprised of modified AAV1 capsid proteins for efficient vector purification and targeting to vascular endothelial cells. Gene Ther. 2006, 13, 926–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Büning, H.; Huber, A.; Zhang, L.; Meumann, N.; Hacker, U. Engineering the AAV capsid to optimize vector-host-interactions. Curr. Opin. Pharmacol. 2015, 24, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Büning, H.; Ried, M.U.; Perabo, L.; Gerner, F.M.; Huttner, N.A.; Enssle, J.; Hallek, M. Receptor targeting of adeno-associated virus vectors. Gene Ther. 2003, 10, 1142–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perabo, L.; Büning, H.; Kofler, D.M.; Ried, M.U.; Girod, A.; Wendtner, C.M.; Enssle, J.; Hallek, M. In vitro selection of viral vectors with modified tropism: The adeno-associated virus display. Mol. Ther. 2003, 8, 151–157. [Google Scholar] [CrossRef]
- Michelfelder, S.; Kohlschutter, J.; Skorupa, A.; Pfennings, S.; Muller, O.; Kleinschmidt, J.A.; Trepel, M. Successful expansion but not complete restriction of tropism of adeno-associated virus by in vivo biopanning of random virus display peptide libraries. PLoS ONE 2009, 4, e5122. [Google Scholar] [CrossRef]
- Grifman, M.; Trepel, M.; Speece, P.; Gilbert, L.B.; Arap, W.; Pasqualini, R.; Weitzman, M.D. Incorporation of Tumor-Targeting Peptides into Recombinant Adeno-associated Virus Capsids. Mol. Ther. 2001, 3, 964–975. [Google Scholar] [CrossRef] [PubMed]
- Konkalmatt, P.R.; Deng, D.; Thomas, S.; Wu, M.T.; Logsdon, C.D.; French, B.A.; Kelly, K.A. Plectin-1 Targeted AAV Vector for the Molecular Imaging of Pancreatic Cancer. Front. Oncol. 2013, 3, 84. [Google Scholar] [CrossRef] [Green Version]
- Sayroo, R.; Nolasco, D.; Yin, Z.; Colon-Cortes, Y.; Pandya, M.; Ling, C.; Aslanidi, G. Development of novel AAV serotype 6 based vectors with selective tropism for human cancer cells. Gene Ther. 2015, 23, 18–25. [Google Scholar] [CrossRef]
- Marsch, S.; Huber, A.; Hallek, M.; Büning, H.; Perabo, L. A novel directed evolution method to enhance cell-type specificity of adeno-associated virus vectors. Comb. Chem. High. Throughput Screen 2010, 13, 807–812. [Google Scholar] [CrossRef]
- Waterkamp, D.A.; Muller, O.J.; Ying, Y.; Trepel, M.; Kleinschmidt, J.A. Isolation of targeted AAV2 vectors from novel virus display libraries. J. Gene Med. 2006, 8, 1307–1319. [Google Scholar] [CrossRef]
- Michelfelder, S.; Lee, M.K.; deLima-Hahn, E.; Wilmes, T.; Kaul, F.; Muller, O.; Kleinschmidt, J.A.; Trepel, M. Vectors selected from adeno-associated viral display peptide libraries for leukemia cell-targeted cytotoxic gene therapy. Exp. Hematol. 2007, 35, 1766–1776. [Google Scholar] [CrossRef] [PubMed]
- Stiefelhagen, M.; Sellner, L.; Kleinschmidt, J.A.; Jauch, A.; Laufs, S.; Wenz, F.; Zeller, W.J.; Fruehauf, S.; Veldwijk, M.R. Application of a haematopoetic progenitor cell-targeted adeno-associated viral (AAV) vector established by selection of an AAV random peptide library on a leukaemia cell line. Genet. Vaccines Ther. 2008, 6, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sellner, L.; Stiefelhagen, M.; Kleinschmidt, J.A.; Laufs, S.; Wenz, F.; Fruehauf, S.; Zeller, W.J.; Veldwijk, M.R. Generation of efficient human blood progenitor-targeted recombinant adeno-associated viral vectors (AAV) by applying an AAV random peptide library on primary human hematopoietic progenitor cells. Exp. Hematol. 2008, 36, 957–964. [Google Scholar] [CrossRef] [PubMed]
- Naumer, M.; Popa-Wagner, R.; Kleinschmidt, J.A. Impact of capsid modifications by selected peptide ligands on recombinant adeno-associated virus serotype 2-mediated gene transduction. J. Gen. Virol. 2012, 93, 2131–2141. [Google Scholar] [CrossRef]
- Michelfelder, S.; Varadi, K.; Raupp, C.; Hunger, A.; Korbelin, J.; Pahrmann, C.; Schrepfer, S.; Muller, O.J.; Kleinschmidt, J.A.; Trepel, M. Peptide ligands incorporated into the threefold spike capsid domain to re-direct gene transduction of AAV8 and AAV9 in vivo. PLoS ONE 2011, 6, e23101. [Google Scholar] [CrossRef] [Green Version]
- Naumer, M.; Ying, Y.; Michelfelder, S.; Reuter, A.; Trepel, M.; Muller, O.J.; Kleinschmidt, J.A. Development and validation of novel AAV2 random libraries displaying peptides of diverse lengths and at diverse capsid positions. Hum. Gene Ther. 2012, 23, 492–507. [Google Scholar] [CrossRef]
- Nicklin, S.A.; Buening, H.; Dishart, K.L.; de Alwis, M.; Girod, A.; Hacker, U.; Thrasher, A.J.; Ali, R.R.; Hallek, M.; Baker, A.H. Efficient and selective AAV2-mediated gene transfer directed to human vascular endothelial cells. Mol. Ther. 2001, 4, 174–181. [Google Scholar] [CrossRef]
- White, S.J.; Nicklin, S.A.; Büning, H.; Brosnan, M.J.; Leike, K.; Papadakis, E.D.; Hallek, M.; Baker, A.H. Targeted gene delivery to vascular tissue in vivo by tropism-modified adeno-associated virus vectors. Circulation 2004, 109, 513–519. [Google Scholar] [CrossRef]
- Work, L.M.; Büning, H.; Hunt, E.; Nicklin, S.A.; Denby, L.; Britton, N.; Leike, K.; Odenthal, M.; Drebber, U.; Hallek, M.; et al. Vascular bed-targeted in vivo gene delivery using tropism-modified adeno-associated viruses. Mol. Ther. 2006, 13, 683–693. [Google Scholar] [CrossRef]
- White, K.; Büning, H.; Kritz, A.; Janicki, H.; McVey, J.; Perabo, L.; Murphy, G.; Odenthal, M.; Work, L.M.; Hallek, M.; et al. Engineering adeno-associated virus 2 vectors for targeted gene delivery to atherosclerotic lesions. Gene Ther. 2008, 15, 443–451. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Rossi, A.; Lange, L.; Meumann, N.; Koitzsch, U.; Christie, K.; Nesbit, M.A.; Moore, C.B.T.; Hacker, U.T.; Morgan, M.; et al. Capsid Engineering Overcomes Barriers Toward Adeno-Associated Virus Vector-Mediated Transduction of Endothelial Cells. Hum. Gene Ther. 2019, 30, 1284–1296. [Google Scholar] [CrossRef]
- Muller, O.J.; Kaul, F.; Weitzman, M.D.; Pasqualini, R.; Arap, W.; Kleinschmidt, J.A.; Trepel, M. Random peptide libraries displayed on adeno-associated virus to select for targeted gene therapy vectors. Nat. Biotechnol. 2003, 21, 1040–1046. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Dupaty, L.; Aillot, L.; Zhang, L.; Gallien, C.; Hallek, M.; Odenthal, M.; Adriouch, S.; Salvetti, A.; Büning, H. Vector uncoating limits adeno-associated viral vector-mediated transduction of human dendritic cells and vector immunogenicity. Sci. Rep. 2019, 9, 3631. [Google Scholar] [CrossRef] [PubMed]
- Adachi, K.; Nakai, H. A New Recombinant Adeno-Associated Virus (Aav)-Based Random Peptide Display Library System: Infection-Defective Aav1.9-3 as a Novel Detargeted Platform for Vector Evolution. Gene Ther. Regul. 2010, 5, 31–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sallach, J.; Di Pasquale, G.; Larcher, F.; Niehoff, N.; Rubsam, M.; Huber, A.; Chiorini, J.; Almarza, D.; Eming, S.A.; Ulus, H.; et al. Tropism-modified AAV vectors overcome barriers to successful cutaneous therapy. Mol. Ther. 2014, 22, 929–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ried, M.U.; Girod, A.; Leike, K.; Büning, H.; Hallek, M. Adeno-associated virus capsids displaying immunoglobulin-binding domains permit antibody-mediated vector retargeting to specific cell surface receptors. J. Virol. 2002, 76, 4559–4566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stachler, M.D.; Chen, I.; Ting, A.Y.; Bartlett, J.S. Site-specific modification of AAV vector particles with biophysical probes and targeting ligands using biotin ligase. Mol. Ther. 2008, 16, 1467–1473. [Google Scholar] [CrossRef]
- Liu, Y.; Fang, Y.; Zhou, Y.; Zandi, E.; Lee, C.L.; Joo, K.I.; Wang, P. Site-specific modification of adeno-associated viruses via a genetically engineered aldehyde tag. Small 2013, 9, 421–429. [Google Scholar] [CrossRef]
- Li, Z.B.; Zeng, Z.J.; Chen, Q.; Luo, S.Q.; Hu, W.X. Recombinant AAV-mediated HSVtk gene transfer with direct intratumoral injections and Tet-On regulation for implanted human breast cancer. BMC Cancer 2006, 6, 66. [Google Scholar] [CrossRef] [Green Version]
- Kanazawa, T.; Mizukami, H.; Okada, T.; Hanazono, Y.; Kume, A.; Nishino, H.; Takeuchi, K.; Kitamura, K.; Ichimura, K.; Ozawa, K. Suicide gene therapy using AAV-HSVtk/ganciclovir in combination with irradiation results in regression of human head and neck cancer xenografts in nude mice. Gene Ther. 2003, 10, 51–58. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.G.; Luo, R.Q.; Zhou, X.; Han, R.F.; Zeng, G.W. Potent antitumor activity of the combination of HSV-TK and endostatin by adeno-associated virus vector for bladder cancer in vivo. Clin. Lab. 2013, 59, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Lu, R.; Ding, R.; Kan, Y.W. Adeno-associated viral-mediated gene transfer to hepatoma: Thymidine kinase/interleukin 2 is more effective in tumor killing in non-ganciclovir (GCV)-treated than in GCV-treated animals. Mol. Ther. 2000, 1, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Lu, R.; Chang, J.C.; Kan, Y.W. Tissue-specific expression of herpes simplex virus thymidine kinase gene delivered by adeno-associated virus inhibits the growth of human hepatocellular carcinoma in athymic mice. Proc. Natl. Acad. Sci. USA 1997, 94, 13891–13896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, N.; Cheemadan, S.; Saxena, H.; Bammidi, S.; Jayandharan, G.R. MicroRNA-based recombinant AAV vector assembly improves efficiency of suicide gene transfer in a murine model of lymphoma. Cancer Med. 2020, 9, 3188–3201. [Google Scholar] [CrossRef] [Green Version]
- Mohr, A.; Henderson, G.; Dudus, L.; Herr, I.; Kuerschner, T.; Debatin, K.M.; Weiher, H.; Fisher, K.J.; Zwacka, R.M. AAV-encoded expression of TRAIL in experimental human colorectal cancer leads to tumor regression. Gene Ther. 2004, 11, 534–543. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; Liu, Y.; Liu, S.; Kung, H.F.; Sun, X.; Zheng, D.; Xu, R. Recombinant adeno-associated virus-mediated TRAIL gene therapy suppresses liver metastatic tumors. Int. J. Cancer 2005, 116, 314–321. [Google Scholar] [CrossRef]
- Ma, H.; Liu, Y.; Liu, S.; Xu, R.; Zheng, D. Oral adeno-associated virus-sTRAIL gene therapy suppresses human hepatocellular carcinoma growth in mice. Hepatology 2005, 42, 1355–1363. [Google Scholar] [CrossRef]
- Shi, J.; Zheng, D.; Liu, Y.; Sham, M.H.; Tam, P.; Farzaneh, F.; Xu, R. Overexpression of soluble TRAIL induces apoptosis in human lung adenocarcinoma and inhibits growth of tumor xenografts in nude mice. Cancer Res. 2005, 65, 1687–1692. [Google Scholar] [CrossRef] [Green Version]
- Yoo, J.; Choi, S.; Hwang, K.S.; Cho, W.K.; Jung, C.R.; Kwon, S.T.; Im, D.S. Adeno-associated virus-mediated gene transfer of a secreted form of TRAIL inhibits tumor growth and occurrence in an experimental tumor model. J. Gene Med. 2006, 8, 163–174. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, F.; Cai, H.; Wu, Y.; He, G.; Tan, W.S. The efficacy of combination therapy using adeno-associated virus-TRAIL targeting to telomerase activity and cisplatin in a mice model of hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2010, 136, 1827–1837. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, F.; Cai, H.; Zhong, S.; Liu, X.; Tan, W.S. Potent antitumor effect of TRAIL mediated by a novel adeno-associated viral vector targeting to telomerase activity for human hepatocellular carcinoma. J. Gene Med. 2008, 10, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Crommentuijn, M.H.; Kantar, R.; Noske, D.P.; Vandertop, W.P.; Badr, C.E.; Wurdinger, T.; Maguire, C.A.; Tannous, B.A. Systemically administered AAV9-sTRAIL combats invasive glioblastoma in a patient-derived orthotopic xenograft model. Mol. Ther. Oncol. 2016, 3, 16017. [Google Scholar] [CrossRef] [PubMed]
- Xue, Z.; Sun, P.H.; Zhu, L.M.; Jiang, S.H.; Qiao, M.M.; Chi, A.L.; Tu, S.P. Adeno-associated virus-mediated survivin mutant Thr34Ala cooperates with oxaliplatin to inhibit tumor growth and angiogenesis in colon cancer. Oncol. Rep. 2011, 25, 1039–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, S.C.; Feng, S.; Wang, P.J.; Cui, L.; Qu, J.G.; Zhang, J.X. Overexpression of Survivin mutant Thr34Ala induces apoptosis and inhibits gastric cancer growth. Neoplasma 2015, 62, 81–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.J.; Lin, M.C.; Bai, Y.X.; Jing, D.D.; Wong, B.C.; Han, S.W.; Lin, J.; Xu, B.; Huang, C.F.; Kung, H.F. Ectopic expression of a COOH-terminal fragment of the human telomerase reverse transcriptase leads to telomere dysfunction and reduction of growth and tumorigenicity in HeLa cells. Cancer Res. 2002, 62, 3226–3232. [Google Scholar] [PubMed]
- Gao, Y.; Ng, S.S.; Chau, D.H.; Yao, H.; Yang, C.; Man, K.; Huang, P.T.; Huang, C.; Huang, J.J.; Kung, H.F.; et al. Development of recombinant adeno-associated virus and adenovirus cocktail system for efficient hTERTC27 polypeptide-mediated cancer gene therapy. Cancer Gene Ther. 2008, 15, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhang, C.; Le, Z.; Zeng, S.; Pan, C.; Shi, J.; Wang, J.; Zhao, X. Telomerase reverse transcriptase interference synergistically promotes tumor necrosis factor related apoptosis inducing ligand induced oral squamous cell carcinoma apoptosis and suppresses proliferation in vitro and in vivo. Int. J. Mol. Med. 2018, 42, 1283–1294. [Google Scholar] [CrossRef] [Green Version]
- Bhere, D.; Tamura, K.; Wakimoto, H.; Choi, S.H.; Purow, B.; Debatisse, J.; Shah, K. microRNA-7 upregulates death receptor 5 and primes resistant brain tumors to caspase-mediated apoptosis. Neuro Oncol. 2018, 20, 215–224. [Google Scholar] [CrossRef] [Green Version]
- Meng, L.; Liu, C.; Lu, J.; Zhao, Q.; Deng, S.; Wang, G.; Qiao, J.; Zhang, C.; Zhen, L.; Lu, Y.; et al. Small RNA zippers lock miRNA molecules and block miRNA function in mammalian cells. Nat. Commun. 2017, 8, 13964. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Sun, J.; Guo, Y.; Zhang, P.; Liu, Y.; Zheng, D.; Shi, J. Combination of AAV-TRAIL with miR-221-Zip Therapeutic Strategy Overcomes the Resistance to TRAIL Induced Apoptosis in Liver Cancer. Theranostics 2017, 7, 3228–3242. [Google Scholar] [CrossRef]
- Bhere, D.; Arghiani, N.; Lechtich, E.R.; Yao, Y.; Alsaab, S.; Bei, F.; Matin, M.M.; Shah, K. Simultaneous downregulation of miR-21 and upregulation of miR-7 has anti-tumor efficacy. Sci. Rep. 2020, 10, 1779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrocca, F.; Altschuler, G.; Tan, S.M.; Mendillo, M.L.; Yan, H.; Jerry, D.J.; Kung, A.L.; Hide, W.; Ince, T.A.; Lieberman, J. A genome-wide siRNA screen identifies proteasome addiction as a vulnerability of basal-like triple-negative breast cancer cells. Cancer Cell 2013, 24, 182–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, C.; Silva, G.; Ribeiro, A.S.; Oliveira, M.; Garrido, M.; Bandeira, V.S.; Nascimento, A.; Coroadinha, A.S.; Peixoto, C.; Barbas, A.; et al. Evaluation of AAV-mediated delivery of shRNA to target basal-like breast cancer genetic vulnerabilities. J. Biotechnol. 2019, 300, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Godlewski, J.; Nowicki, M.O.; Bronisz, A.; Williams, S.; Otsuki, A.; Nuovo, G.; Raychaudhury, A.; Newton, H.B.; Chiocca, E.A.; Lawler, S. Targeting of the Bmi-1 oncogene/stem cell renewal factor by microRNA-128 inhibits glioma proliferation and self-renewal. Cancer Res. 2008, 68, 9125–9130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Guo, W.; Wang, X.; Liu, X.; Huang, M.; Gan, L.; Cheng, Y.; Li, J. Antitumor activity and inhibitory effects on cancer stem cell-like properties of Adeno-associated virus (AAV) -mediated Bmi-1 interference driven by Bmi-1 promoter for gastric cancer. Oncotarget 2016, 7, 22733–22745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhen, Z.; Yang, K.; Ye, L.; You, Z.; Chen, R.; Liu, Y. Decorin gene upregulation mediated by an adeno-associated virus vector increases intratumoral uptake of nab-paclitaxel in neuroblastoma via inhibition of stabilin-1. Investig. New Drugs 2017, 35, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef]
- Prabhakar, S.; Taherian, M.; Gianni, D.; Conlon, T.J.; Fulci, G.; Brockmann, J.; Stemmer-Rachamimov, A.; Sena-Esteves, M.; Breakefield, X.O.; Brenner, G.J. Regression of schwannomas induced by adeno-associated virus-mediated delivery of caspase-1. Hum. Gene Ther. 2013, 24, 152–162. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.G.; Abdelanabi, A.; Doha, M.; Brenner, G.J. Schwannoma gene therapy by adeno-associated virus delivery of the pore-forming protein Gasdermin-D. Cancer Gene Ther. 2019, 26, 259–267. [Google Scholar] [CrossRef]
- Ahmed, S.G.; Abdelnabi, A.; Maguire, C.A.; Doha, M.; Sagers, J.E.; Lewis, R.M.; Muzikansky, A.; Giovannini, M.; Stemmer-Rachamimov, A.; Stankovic, K.M.; et al. Gene therapy with apoptosis-associated speck-like protein, a newly described schwannoma tumor suppressor, inhibits schwannoma growth in vivo. Neuro Oncol. 2019, 21, 854–866. [Google Scholar] [CrossRef]
- Bai, R.Y.; Esposito, D.; Tam, A.J.; McCormick, F.; Riggins, G.J.; Wade Clapp, D.; Staedtke, V. Feasibility of using NF1-GRD and AAV for gene replacement therapy in NF1-associated tumors. Gene Ther. 2019, 26, 277–286. [Google Scholar] [CrossRef]
- Yoshiba, T.; Saga, Y.; Urabe, M.; Uchibori, R.; Matsubara, S.; Fujiwara, H.; Mizukami, H. CRISPR/Cas9-mediated cervical cancer treatment targeting human papillomavirus E6. Oncol. Lett. 2019, 17, 2197–2206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hjertner, O.; Hjorth-Hansen, H.; Borset, M.; Seidel, C.; Waage, A.; Sundan, A. Bone morphogenetic protein-4 inhibits proliferation and induces apoptosis of multiple myeloma cells. Blood 2001, 97, 516–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westhrin, M.; Holien, T.; Zahoor, M.; Moen, S.H.; Buene, G.; Stordal, B.; Hella, H.; Yuan, H.; de Bruijn, J.D.; Martens, A.; et al. Bone Morphogenetic Protein 4 Gene Therapy in Mice Inhibits Myeloma Tumor Growth, But Has a Negative Impact on Bone. JBMR Plus 2020, 4, e10247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cano, C.E.; Hamidi, T.; Sandi, M.J.; Iovanna, J.L. Nupr1: The Swiss-knife of cancer. J. Cell Physiol. 2011, 226, 1439–1443. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yin, Y.; Ma, J.; Sun, Y.; Zhou, R.; Cui, B.; Zhang, Y.; Yang, J.; Yan, X.; Liu, Z.; et al. Combination of AAVmediated NUPR1 knockdown and trifluoperazine induces premature senescence in human lung adenocarcinoma A549 cells in nude mice. Oncol. Rep. 2020, 43, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Wu, J.; Wang, D.; Wang, J. Cancer gene therapy by NF-kappaB-activated cancer cell specific expression of CRISPR/Cas9 targeting telomeres. Gene Ther. 2020. [Google Scholar] [CrossRef]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Discov. 2011, 10, 417–427. [Google Scholar] [CrossRef]
- Büning, H.; Hacker, U.T. Inhibitors of Angiogenesis. In Protein Targeting Compounds; Springer: Heidelberg/Berlin, Germany, 2015; pp. 261–285. [Google Scholar] [CrossRef]
- Qin, S.; Li, A.; Yi, M.; Yu, S.; Zhang, M.; Wu, K. Recent advances on anti-angiogenesis receptor tyrosine kinase inhibitors in cancer therapy. J. Hematol. Oncol. 2019, 12, 27. [Google Scholar] [CrossRef] [Green Version]
- Baluk, P.; Hashizume, H.; McDonald, D.M. Cellular abnormalities of blood vessels as targets in cancer. Curr. Opin. Genet. Dev. 2005, 15, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Campesato, L.F.; Merghoub, T. Antiangiogenic therapy and immune checkpoint blockade go hand in hand. Ann. Transl. Med. 2017, 5, 497. [Google Scholar] [CrossRef] [PubMed]
- Atkins, M.B.; Plimack, E.R.; Puzanov, I.; Fishman, M.N.; McDermott, D.F.; Cho, D.C.; Vaishampayan, U.; George, S.; Olencki, T.E.; Tarazi, J.C.; et al. Axitinib in combination with pembrolizumab in patients with advanced renal cell cancer: A non-randomised, open-label, dose-finding, and dose-expansion phase 1b trial. Lancet Oncol. 2018. [Google Scholar] [CrossRef]
- Sun, E.; Han, R.; Lu, B. Gene therapy of renal cancer using recombinant adeno-associated virus encoding human endostatin. Oncol. Lett. 2018, 16, 2789–2796. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.G.; Zhou, X.; Zeng, G.W.; Han, R.F. Suppression of bladder cancer growth in mice by adeno-associated virus vector-mediated endostatin expression. Tumor Biol. 2011, 32, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Li, X.P.; Li, C.Y.; Li, X.; Ding, Y.; Chan, L.L.; Yang, P.H.; Li, G.; Liu, X.; Lin, J.S.; Wang, J.; et al. Inhibition of human nasopharyngeal carcinoma growth and metastasis in mice by adenovirus-associated virus-mediated expression of human endostatin. Mol. Cancer Ther. 2006, 5, 1290–1298. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, I.V.; Ghebre, R.; Ramakrishnan, S. Adeno-associated virus-mediated delivery of a mutant endostatin suppresses ovarian carcinoma growth in mice. Gene Ther. 2005, 12, 30–38. [Google Scholar] [CrossRef] [Green Version]
- Noro, T.; Miyake, K.; Suzuki-Miyake, N.; Igarashi, T.; Uchida, E.; Misawa, T.; Yamazaki, Y.; Shimada, T. Adeno-associated viral vector-mediated expression of endostatin inhibits tumor growth and metastasis in an orthotropic pancreatic cancer model in hamsters. Cancer Res. 2004, 64, 7486–7490. [Google Scholar] [CrossRef] [Green Version]
- Shi, W.; Teschendorf, C.; Muzyczka, N.; Siemann, D.W. Adeno-associated virus-mediated gene transfer of endostatin inhibits angiogenesis and tumor growth in vivo. Cancer Gene Ther. 2002, 9, 513–521. [Google Scholar] [CrossRef]
- Lalani, A.S.; Chang, B.; Lin, J.; Case, S.S.; Luan, B.; Wu-Prior, W.W.; VanRoey, M.; Jooss, K. Anti-tumor efficacy of human angiostatin using liver-mediated adeno-associated virus gene therapy. Mol. Ther. 2004, 9, 56–66. [Google Scholar] [CrossRef]
- Ma, H.I.; Lin, S.Z.; Chiang, Y.H.; Li, J.; Chen, S.L.; Tsao, Y.P.; Xiao, X. Intratumoral gene therapy of malignant brain tumor in a rat model with angiostatin delivered by adeno-associated viral (AAV) vector. Gene Ther. 2002, 9, 2–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponnazhagan, S.; Mahendra, G.; Kumar, S.; Shaw, D.R.; Stockard, C.R.; Grizzle, W.E.; Meleth, S. Adeno-associated virus 2-mediated antiangiogenic cancer gene therapy: Long-term efficacy of a vector encoding angiostatin and endostatin over vectors encoding a single factor. Cancer Res. 2004, 64, 1781–1787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, I.V.; Devineni, S.; Ghebre, R.; Ghosh, G.; Joshi, H.P.; Jing, Y.; Truskinovsky, A.M.; Ramakrishnan, S. AAV-P125A-endostatin and paclitaxel treatment increases endoreduplication in endothelial cells and inhibits metastasis of breast cancer. Gene Ther. 2011, 18, 145–154. [Google Scholar] [CrossRef]
- Subramanian, I.V.; Bui Nguyen, T.M.; Truskinovsky, A.M.; Tolar, J.; Blazar, B.R.; Ramakrishnan, S. Adeno-associated virus-mediated delivery of a mutant endostatin in combination with carboplatin treatment inhibits orthotopic growth of ovarian cancer and improves long-term survival. Cancer Res. 2006, 66, 4319–4328. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.G.; Luo, R.Q.; Zhou, X.; Han, R.F.; Zeng, G.W. Suppression of bladder cancer growth by adeno-associated virus vector-mediated combination of HSV-TK and endostatin in vitro. Clin. Lab. 2013, 59, 1077–1089. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.; Duquette, M.; Liu, J.; Drapkin, R.; Lawler, J.; Petrik, J. Combined therapy with thrombospondin-1 type I repeats (3TSR) and chemotherapy induces regression and significantly improves survival in a preclinical model of advanced stage epithelial ovarian cancer. FASEB J. 2015, 29, 576–588. [Google Scholar] [CrossRef] [Green Version]
- Yu, D.L.; Stegelmeier, A.A.; Chow, N.; Rghei, A.D.; Matuszewska, K.; Lawler, J.; Bridle, B.W.; Petrik, J.J.; Wootton, S.K. AAV-mediated expression of 3TSR inhibits tumor and metastatic lesion development and extends survival in a murine model of epithelial ovarian carcinoma. Cancer Gene Ther. 2019. [Google Scholar] [CrossRef]
- Chen, X.; Chen, S.; Pei, N.; Mao, Y.; Wang, S.; Yan, R.; Bai, N.; Li, A.; Zhang, Y.; Du, H.; et al. AAV-Mediated angiotensin 1-7 overexpression inhibits tumor growth of lung cancer in vitro and in vivo. Oncotarget 2017, 8, 354–363. [Google Scholar] [CrossRef] [Green Version]
- Pei, N.; Wan, R.; Chen, X.; Li, A.; Zhang, Y.; Li, J.; Du, H.; Chen, B.; Wei, W.; Qi, Y.; et al. Angiotensin-(1-7) Decreases Cell Growth and Angiogenesis of Human Nasopharyngeal Carcinoma Xenografts. Mol. Cancer Ther. 2016, 15, 37–47. [Google Scholar] [CrossRef] [Green Version]
- Mao, Y.; Pei, N.; Chen, X.; Chen, H.; Yan, R.; Bai, N.; Li, A.; Li, J.; Zhang, Y.; Du, H.; et al. Angiotensin 1-7 Overexpression Mediated by a Capsid-optimized AAV8 Vector Leads to Significant Growth Inhibition of Hepatocellular Carcinoma In vivo. Int. J. Biol. Sci. 2018, 14, 57–68. [Google Scholar] [CrossRef] [Green Version]
- Shen, Z.; Yao, C.; Wang, Z.; Yue, L.; Fang, Z.; Yao, H.; Lin, F.; Zhao, H.; Sun, Y.J.; Bian, X.W.; et al. Vastatin, an Endogenous Antiangiogenesis Polypeptide That Is Lost in Hepatocellular Carcinoma, Effectively Inhibits Tumor Metastasis. Mol. Ther. 2016, 24, 1358–1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Browne, M.; Stellmach, V.; Cornwell, M.; Chung, C.; Doll, J.A.; Lee, E.J.; Jameson, J.L.; Reynolds, M.; Superina, R.A.; Abramson, L.P.; et al. Gene transfer of pigment epithelium-derived factor suppresses tumor growth and angiogenesis in a hepatoblastoma xenograft model. Pediatr. Res. 2006, 60, 282–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, S.S.; Shi, H.S.; Yin, T.; Li, Y.X.; Luo, S.T.; Wu, Q.J.; Lu, L.; Wei, Y.Q.; Yang, L. AAV-mediated gene transfer of human pigment epithelium-derived factor inhibits Lewis lung carcinoma growth in mice. Oncol. Rep. 2012, 27, 1142–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, S.S.; Wu, Q.J.; Gong, C.Y.; Luo, S.T.; Zhang, S.; Li, M.; Lu, L.; Wei, Y.Q.; Yang, L. Enhanced efficacy of combination therapy with adenoassociated virus-delivered pigment epithelium-derived factor and cisplatin in a mouse model of Lewis lung carcinoma. Mol. Med. Rep. 2014, 9, 2069–2076. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; He, S.; Wei, X.; Shao, B.; Luo, S.; Guo, F.; Zhang, H.; Wang, Y.; Gong, C.; Yang, L. Synergistic antitumor effect of recombinant adeno-associated virus-mediated pigment epithelium-derived factor with hyperthermia on solid tumor. Hum. Gene Ther. 2014, 25, 811–823. [Google Scholar] [CrossRef]
- Wu, Q.J.; Gong, C.Y.; Luo, S.T.; Zhang, D.M.; Zhang, S.; Shi, H.S.; Lu, L.; Yan, H.X.; He, S.S.; Li, D.D.; et al. AAV-mediated human PEDF inhibits tumor growth and metastasis in murine colorectal peritoneal carcinomatosis model. BMC Cancer 2012, 12, 129. [Google Scholar] [CrossRef] [Green Version]
- Ruan, Q.; Xi, L.; Boye, S.L.; Han, S.; Chen, Z.J.; Hauswirth, W.W.; Lewin, A.S.; Boulton, M.E.; Law, B.K.; Jiang, W.G.; et al. Development of an anti-angiogenic therapeutic model combining scAAV2-delivered siRNAs and noninvasive photoacoustic imaging of tumor vasculature development. Cancer Lett. 2013, 332, 120–129. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Hicks, M.J.; Kaminsky, S.M.; Moore, M.A.; Crystal, R.G.; Rafii, A. AAV-mediated persistent bevacizumab therapy suppresses tumor growth of ovarian cancer. Gynecol. Oncol. 2014, 135, 325–332. [Google Scholar] [CrossRef] [Green Version]
- Hicks, M.J.; Funato, K.; Wang, L.; Aronowitz, E.; Dyke, J.P.; Ballon, D.J.; Havlicek, D.F.; Frenk, E.Z.; De, B.P.; Chiuchiolo, M.J.; et al. Genetic modification of neurons to express bevacizumab for local anti-angiogenesis treatment of glioblastoma. Cancer Gene Ther. 2015, 22, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Luo, S.T.; Shi, H.S.; Li, M.; Zhang, H.L.; He, S.S.; Liu, Y.; Pan, Y.; Yang, L. AAV2-mediated gene transfer of VEGF-Trap with potent suppression of primary breast tumor growth and spontaneous pulmonary metastases by long-term expression. Oncol. Rep. 2012, 28, 1332–1338. [Google Scholar] [CrossRef]
- Zhao, S.; Zhang, Y.; Wang, L.; Yang, L.; Zou, L.; Gao, F. Adeno-associated virus 2 mediated gene transfer of vascular endothelial growth factor Trap: A new treatment option for glioma. Cancer Biol. Ther. 2019, 20, 65–72. [Google Scholar] [CrossRef]
- Li, J.; Zhu, P.; Wang, L.; Yang, L.; Zou, L.; Gao, F. Study of diffusion-weighted magnetic resonance imaging in the evaluation of the response to AAV2-VEGF-Trap neoadjuvant treatment in a triple-negative breast cancer animal model. Cancer Med. 2019, 8, 1594–1603. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Lalani, A.S.; Harding, T.C.; Gonzalez, M.; Wu, W.W.; Luan, B.; Tu, G.H.; Koprivnikar, K.; VanRoey, M.J.; He, Y.; et al. Inhibition of lymphogenous metastasis using adeno-associated virus-mediated gene transfer of a soluble VEGFR-3 decoy receptor. Cancer Res. 2005, 65, 6901–6909. [Google Scholar] [CrossRef] [Green Version]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, O.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef] [PubMed]
- Titov, A.; Valiullina, A.; Zmievskaya, E.; Zaikova, E.; Petukhov, A.; Miftakhova, R.; Bulatov, E.; Rizvanov, A. Advancing CAR T-Cell Therapy for Solid Tumors: Lessons Learned from Lymphoma Treatment. Cancers 2020, 12, 125. [Google Scholar] [CrossRef] [Green Version]
- Moco, P.D.; Aharony, N.; Kamen, A. Adeno-Associated Viral Vectors for Homology-Directed Generation of CAR-T Cells. Biotechnol. J. 2020, 15, e1900286. [Google Scholar] [CrossRef] [PubMed]
- Sather, B.D.; Romano Ibarra, G.S.; Sommer, K.; Curinga, G.; Hale, M.; Khan, I.F.; Singh, S.; Song, Y.; Gwiazda, K.; Sahni, J.; et al. Efficient modification of CCR5 in primary human hematopoietic cells using a megaTAL nuclease and AAV donor template. Sci. Transl. Med. 2015, 7, 307ra156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hale, M.; Lee, B.; Honaker, Y.; Leung, W.H.; Grier, A.E.; Jacobs, H.M.; Sommer, K.; Sahni, J.; Jackson, S.W.; Scharenberg, A.M.; et al. Homology-Directed Recombination for Enhanced Engineering of Chimeric Antigen Receptor T Cells. Mol. Ther. Methods Clin. Dev. 2017, 4, 192–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacLeod, D.T.; Antony, J.; Martin, A.J.; Moser, R.J.; Hekele, A.; Wetzel, K.J.; Brown, A.E.; Triggiano, M.A.; Hux, J.A.; Pham, C.D.; et al. Integration of a CD19 CAR into the TCR Alpha Chain Locus Streamlines Production of Allogeneic Gene-Edited CAR T Cells. Mol. Ther. 2017, 25, 949–961. [Google Scholar] [CrossRef] [Green Version]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gonen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef] [Green Version]
- Albers, J.J.; Ammon, T.; Gosmann, D.; Audehm, S.; Thoene, S.; Winter, C.; Secci, R.; Wolf, A.; Stelzl, A.; Steiger, K.; et al. Gene editing enables T-cell engineering to redirect antigen specificity for potent tumor rejection. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef] [PubMed]
- Wiebking, V.; Lee, C.M.; Mostrel, N.; Lahiri, P.; Bak, R.; Bao, G.; Roncarolo, M.G.; Bertaina, A.; Porteus, M.H. Genome editing of donor-derived T-cells to generate allogenic chimeric antigen receptor-modified T cells: Optimizing alphabeta T cell-depleted haploidentical hematopoietic stem cell transplantation. Haematologica 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, X.; Park, J.J.; Du, Y.; Kim, H.R.; Wang, G.; Errami, Y.; Chen, S. One-step generation of modular CAR-T cells with AAV-Cpf1. Nat. Methods 2019, 16, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Pomeroy, E.J.; Hunzeker, J.T.; Kluesner, M.G.; Lahr, W.S.; Smeester, B.A.; Crosby, M.R.; Lonetree, C.L.; Yamamoto, K.; Bendzick, L.; Miller, J.S.; et al. A Genetically Engineered Primary Human Natural Killer Cell Platform for Cancer Immunotherapy. Mol. Ther. 2020, 28, 52–63. [Google Scholar] [CrossRef]
- Chang, M.C.; Chen, Y.L.; Chiang, Y.C.; Chen, T.C.; Tang, Y.C.; Chen, C.A.; Sun, W.Z.; Cheng, W.F. Mesothelin-specific cell-based vaccine generates antigen-specific immunity and potent antitumor effects by combining with IL-12 immunomodulator. Gene Ther. 2016, 23, 38–49. [Google Scholar] [CrossRef]
- Chen, Y.L.; Chang, M.C.; Chiang, Y.C.; Lin, H.W.; Sun, N.Y.; Chen, C.A.; Sun, W.Z.; Cheng, W.F. Immuno-modulators enhance antigen-specific immunity and anti-tumor effects of mesothelin-specific chimeric DNA vaccine through promoting DC maturation. Cancer Lett. 2018, 425, 152–163. [Google Scholar] [CrossRef]
- Li, J.; Huang, S.; Zhou, Z.; Lin, W.; Chen, S.; Chen, M.; Ye, Y. Exosomes derived from rAAV/AFP-transfected dendritic cells elicit specific T cell-mediated immune responses against hepatocellular carcinoma. Cancer Manag. Res. 2018, 10, 4945–4957. [Google Scholar] [CrossRef] [Green Version]
- Mirandola, L.; Chiriva-Internati, M.; Bresalier, R.; Piccotti, L.; Grizzi, F.; Marincola, F.M. A novel method for efficient generation of antigen-specific effector T-cells using dendritic cells transduced with recombinant adeno-associated virus and p38 kinase blockade. J. Transl. Med. 2019, 17, 424. [Google Scholar] [CrossRef] [Green Version]
- Hensel, J.A.; Khattar, V.; Ashton, R.; Ponnazhagan, S. Recombinant AAV-CEA Tumor Vaccine in Combination with an Immune Adjuvant Breaks Tolerance and Provides Protective Immunity. Mol. Ther. Oncol. 2019, 12, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Krotova, K.; Day, A.; Aslanidi, G. An Engineered AAV6-Based Vaccine Induces High Cytolytic Anti-Tumor Activity by Directly Targeting DCs and Improves Ag Presentation. Mol. Ther. Oncol. 2019, 15, 166–177. [Google Scholar] [CrossRef] [Green Version]
- Pandya, M.; Britt, K.; Hoffman, B.; Ling, C.; Aslanidi, G.V. Reprogramming Immune Response with Capsid-Optimized AAV6 Vectors for Immunotherapy of Cancer. J. Immunother. 2015, 38, 292–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Liu, J.Q.; Shi, M.; Cheng, X.; Ding, M.; Zhang, J.C.; Davis, J.P.; Varikuti, S.; Satoskar, A.R.; Lu, L.; et al. IL-27 gene therapy induces depletion of Tregs and enhances the efficacy of cancer immunotherapy. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, A.; Ding, M.; Zhu, J.; Liu, J.Q.; Pan, X.; Ghoshal, K.; Bai, X.F. Intra-Tumoral Delivery of IL-27 Using Adeno-Associated Virus Stimulates Anti-tumor Immunity and Enhances the Efficacy of Immunotherapy. Front. Cell Dev. Biol. 2020, 8, 210. [Google Scholar] [CrossRef] [PubMed]
- Vasquez, M.; Paredes-Cervantes, V.; Aranda, F.; Ardaiz, N.; Gomar, C.; Berraondo, P. Antitumor effect of an adeno-associated virus expressing apolipoprotein A-1 fused to interferon alpha in an interferon alpha-resistant murine tumor model. Oncotarget 2017, 8, 5247–5255. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Ma, P.; Li, J.; Cui, X.; Song, W. Improvement of the cytotoxic T lymphocyte response against hepatocellular carcinoma by transduction of cancer cells with an adeno-associated virus carrying the interferon-gamma gene. Mol. Med. Rep. 2016, 13, 3197–3205. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Ma, P.; Li, J.; Song, W. Comparative analysis of cytotoxic T lymphocyte response induced by dendritic cells pulsed with recombinant adeno-associated virus carrying alpha-fetoprotein gene or cancer cell lysate. Mol. Med. Rep. 2015, 11, 3174–3180. [Google Scholar] [CrossRef] [Green Version]
- Xiao, R.; Mansour, A.G.; Huang, W.; Chrislip, L.A.; Wilkins, R.K.; Queen, N.J.; Youssef, Y.; Mao, H.C.; Caligiuri, M.A.; Cao, L. Adipocytes: A Novel Target for IL-15/IL-15Ralpha Cancer Gene Therapy. Mol. Ther. 2019, 27, 922–932. [Google Scholar] [CrossRef]
- Reul, J.; Frisch, J.; Engeland, C.E.; Thalheimer, F.B.; Hartmann, J.; Ungerechts, G.; Buchholz, C.J. Tumor-Specific Delivery of Immune Checkpoint Inhibitors by Engineered AAV Vectors. Front. Oncol. 2019, 9, 52. [Google Scholar] [CrossRef] [Green Version]
- Simoes, I.T.; Aranda, F.; Casado-Llombart, S.; Velasco-de Andres, M.; Catala, C.; Alvarez, P.; Consuegra-Fernandez, M.; Orta-Mascaro, M.; Merino, R.; Merino, J.; et al. Multifaceted effects of soluble human CD6 in experimental cancer models. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Snyder, A.G.; Hubbard, N.W.; Messmer, M.N.; Kofman, S.B.; Hagan, C.E.; Orozco, S.L.; Chiang, K.; Daniels, B.P.; Baker, D.; Oberst, A. Intratumoral activation of the necroptotic pathway components RIPK1 and RIPK3 potentiates antitumor immunity. Sci. Immunol. 2019, 4. [Google Scholar] [CrossRef]
- Ye, L.; Park, J.J.; Dong, M.B.; Yang, Q.; Chow, R.D.; Peng, L.; Du, Y.; Guo, J.; Dai, X.; Wang, G.; et al. In vivo CRISPR screening in CD8 T cells with AAV-Sleeping Beauty hybrid vectors identifies membrane targets for improving immunotherapy for glioblastoma. Nat. Biotechnol. 2019, 37, 1302–1313. [Google Scholar] [CrossRef] [PubMed]

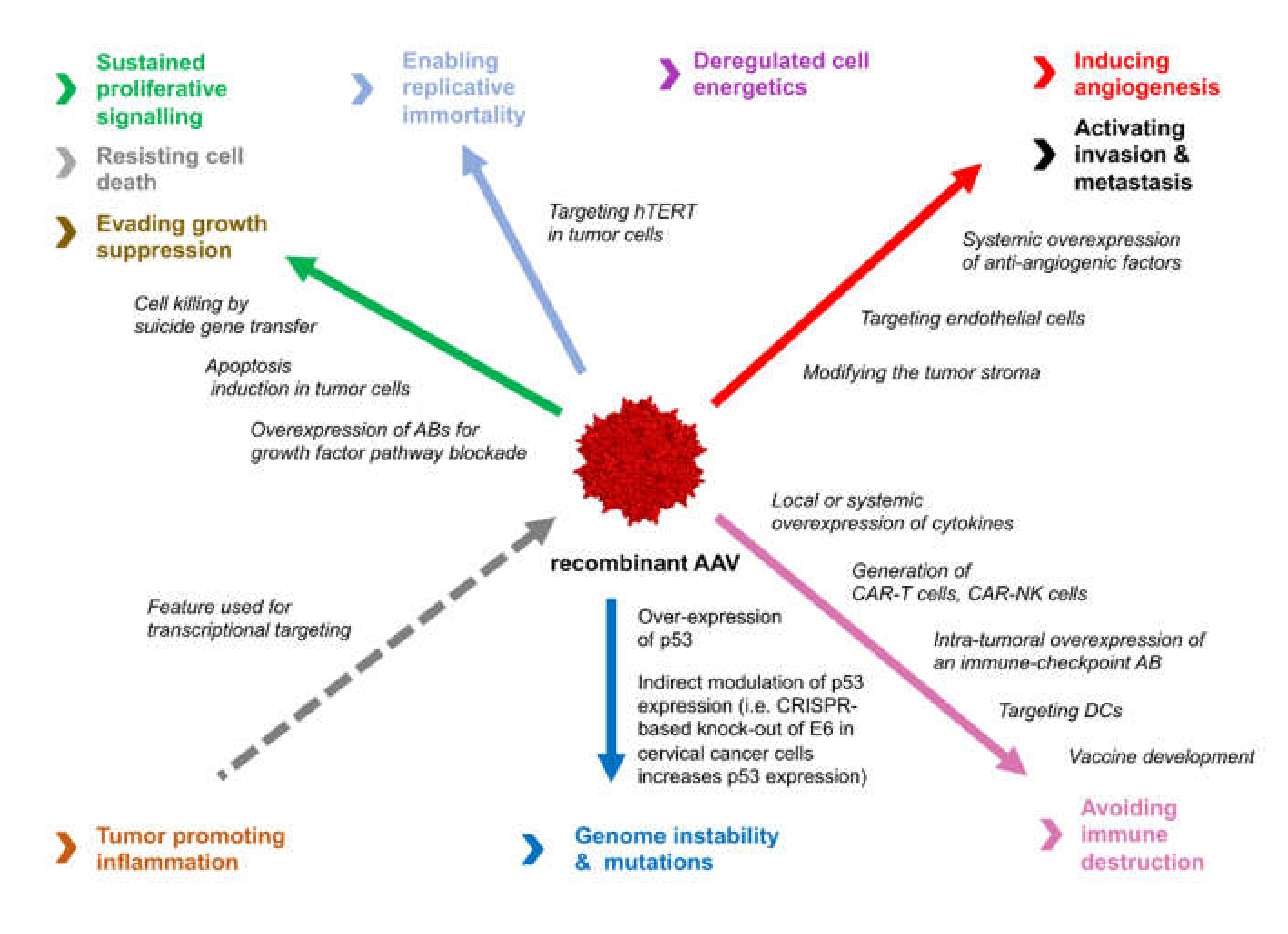

{kind=link}
{kind=link}
| Hallmark of Cancer. | Therapeutic Principle |
|---|---|
| Evading growth suppression | Cyclin-dependent kinase inhibitors: CDK4/6 inhibitors |
| Sustained proliferative signaling | Blocking growth factor receptor pathways (i.e., EGFR and others): monoclonal antibodies, tyrosine kinase inhibitors |
| Resisting cell death | BH3-mimetics that specifically bind to the hydrophobic groove of BCL-2: venetoclax |
| Enabling replicative immortality | Inhibition of telomerase activity, induction of senescence in tumor cells: a number of commonly used cancer therapeutics are involved in induction of senescence in cancer cells |
| Deregulating cellular energetics | Inhibition of aerobic glycolysis, inhibition of isocitrate dehydrogenase (IDH) and others: IDH1 and IDH2 inhibitors |
| Genome instability and mutation | PARP inhibition to facilitate synthetical lethality in homologous recombination deficient tumor cells: PARP inhibitors |
| Inducing angiogenesis | Inhibition of angiogenic pathways (i.e., VEGF, PlGF): monoclonal antibodies, fusion constructs, tyrosine kinase inhibitors |
| Activating invasion and metastasis | Targeting the epithelial- mesenchymal transition (EMT) program (e.g., HGF/cMET inhibition), anti-angiogenic treatment strategies |
| Avoiding immune destruction | Immune activation: immune checkpoint antibodies (i.e., anti-PD-1, anti-PD-L1, anti CLTA-4) |
| Tumor promoting inflammation | Selective anti-inflammatory drugs: not yet available clinically |
| Name | Target Cell Type | Serotype | Position | Insert | Reference |
|---|---|---|---|---|---|
| rRGD453ko | αν-integrin-positive tumor cells | AAV2 | I-453 | CDCRGDCFC | [60] |
| A584-RGD4C | αν-integrin-positive tumor cells | AAV2 | I-584 | CDCRGDCFC | [59] |
| AAV-ΔIV-NGR | CD13-positive tumor cells | AAV2 | I-585 | NGRAHA | [66] |
| AAV-PTP | Plectin-positive tumor cells | AAV2 | I-585 | KTLLPTP | [67] |
| AAV6-RGD | tumor cells | AAV6 | I-585 | RGD | [68] |
| AAV-I-587 | β1-integrin positive tumor cells | AAV2 | I-587 | QAGTFALRGDNPQG | [58] |
| AAV-588NGR | CD13-positive tumor cells | AAV2 | I-587 | NGRAHA | [66] |
| AAV-MO7A | tumor cells | AAV2 | I-587 | RGDAVGV | [64] |
| AAV-MO7T | tumor cells | AAV2 | I-587 | RGDTPTS | [64] |
| AAV-MecA | tumor cells | AAV2 | I-587 | GENQARS | [64] |
| AAV-MecB | tumor cells | AAV2 | I-587 | RSNAVVP | [64] |
| rRGD587 | αν-integrin positive tumor cells | AAV2 | I-587 | CDCRGDCFC | [60] |
| AAV-C4 | tumor cells | AAV2 | I-587 | PRGTNGP | [69] |
| AAV-D10 | tumor cells | AAV2 | I-587 | SRGATTT | [69] |
| A588-RGD4C | an integrin-positive tumor cell | AAV2 | I-588 | CDCRGDCFC | [59] |
| A588-RGD4CGLS | αν-integrin positive tumor cells | AAV2 | I-588 | CDCRGDCFC | [59] |
| AAV-VTAGRAP | tumor cells | AAV2 | I-588 | VTAGRAP | [70] |
| AAV-APVTRPA | tumor cells | AAV2 | I-588 | APVTRPA | [70] |
| AAV-DLSNLTR | tumor cells | AAV2 | I-588 | DLSNLTR | [70] |
| AAV-NQVGSWS | tumor cells | AAV2 | I-588 | NQVGSWS | [71] |
| AAV-EARVRPP | tumor cells | AAV2 | I-588 | EARVRPP | [72] |
| AAV-NSVSLYT | tumor cells (CML) | AAV2 | I-588 | NSVSLYT | [72] |
| AAV-LS1 | tumor cells (CML) CD34+ cells | AAV2 | I-588 | NDVRSAN | [73] |
| AAV-LS2 | tumor cells (CML) CD34+ cells | AAV2 | I-588 | NESRVLS | [73] |
| AAV-LS3 | tumor cells (CML) CD34+ cells | AAV2 | I-588 | NRTWEQQ | [73] |
| AAV-LS4 | tumor cells (CML) CD34+ cells | AAV2 | I-588 | NSVQSSW | [73] |
| AAV-RGDLGLS | tumor cells | AAV2 | I-588 | RGDLGLS | [65] |
| AAV-RGDMSRE | tumor cells | AAV2 | I-588 | RGDMSRE | [65] |
| AAV-ESGLSQS | tumor cells | AAV2 | I-588 | ESGLSQS | [65] |
| AAV-EYRDSSG | tumor cells | AAV2 | I-588 | EYRDSSG | [65] |
| AAV-DLGSARA | tumor cells | AAV2 | I-588 | DLGSARA | [65] |
| AAV-NDVRSAN | tumor cells | AAV2 | I-588 | NDVRSAN | [74] |
| AAV-GPQGKNS | tumor cells | AAV2 | I-588 | GPQGKNS | [74] |
| AAV1-RGD | tumor cells | AAV1 | I-590 | CDCRGDCFC | [61] |
| AAV8-ESGLSOS | tumor cells | AAV8 | I-590 | ESGLSQS | [65,75] |
| AAV-MNVRGDL | endothelial cells | AAV2 | I-453 | MNVRGDL | [76] |
| AAV-ENVRGDL | endothelial cells | AAV2 | I-453 | ENVRGDL | [76] |
| AAV-SIG | endothelial cells | AAV2 | I-587 | SIGYPLP | [77] |
| AAV-MTP | endothelial cells | AAV2 | I-587 | MTPFPTSNEANL | [78] |
| AAV-QPE | endothelial cells | AAV2 | I-587 | QPEHSST | [79] |
| AAV-VNT | endothelial cells | AAV2 | I-587 | VNTANST | [79] |
| AAV-CNH | endothelial cells | AAV2 | I-587 | CNHRYMQMC | [80] |
| AAV-CAP | endothelial cells | AAV2 | I-587 | CAPGPSKSG | [80] |
| AAV-VEC | endothelial cells | AAV2 | I-587 | VSSSTPR | [81] |
| AAV-NSSRDLG | endothelial cells | AAV2 | I-588 | NSSRDLG | [82] |
| AAV-NDVRAVS | endothelial cells | AAV2 | I-588 | NDVRAVS | [70,82] |
| AAV-NDVRSAN | endothelial cells | AAV2 | I-588 | NDVRSAN | [70] |
| AAV-DIIRA | endothelial cells | AAV2 | I-588 | DIIRA | [76] |
| AAV-SYENV | endothelial cells | AAV2 | I-588 | SYENVASRRPEG | [76] |
| AAV-PENSV | endothelial cells | AAV2 | I-588 | PENSVRRYGLEE | [76] |
| AAV-LSLAS | endothelial cells | AAV2 | I-588 | LSLASNRPTATS | [76] |
| AAV-NDVWN | endothelial cells | AAV2 | I-588 | NDVWNRDNSSKRGGTTEAS | [76] |
| AAV-NRTYS | endothelial cells | AAV2 | I-588 | NRTYSSTSNSTSRSEWDNS | [76] |
| AAV1-RGD | endothelial cells | AAV1 | I-590 | CDCRGDCFC | [61] |
| AAV-V | dendritic cells | AAV2 | I-587 | VSSTSPR | [83] |
| AAV-I | dendritic cells | AAV2 | I-587 | ISSSTAR | [83] |
| AAV1.9-3-SKAGRSP | fibroblasts | AAV1 (aa 445–568 from AAV9) | I-590 | SKAGRSP | [84] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hacker, U.T.; Bentler, M.; Kaniowska, D.; Morgan, M.; Büning, H. Towards Clinical Implementation of Adeno-Associated Virus (AAV) Vectors for Cancer Gene Therapy: Current Status and Future Perspectives. Cancers 2020, 12, 1889. https://doi.org/10.3390/cancers12071889
Hacker UT, Bentler M, Kaniowska D, Morgan M, Büning H. Towards Clinical Implementation of Adeno-Associated Virus (AAV) Vectors for Cancer Gene Therapy: Current Status and Future Perspectives. Cancers. 2020; 12(7):1889. https://doi.org/10.3390/cancers12071889
Chicago/Turabian StyleHacker, Ulrich T., Martin Bentler, Dorota Kaniowska, Michael Morgan, and Hildegard Büning. 2020. "Towards Clinical Implementation of Adeno-Associated Virus (AAV) Vectors for Cancer Gene Therapy: Current Status and Future Perspectives" Cancers 12, no. 7: 1889. https://doi.org/10.3390/cancers12071889
APA StyleHacker, U. T., Bentler, M., Kaniowska, D., Morgan, M., & Büning, H. (2020). Towards Clinical Implementation of Adeno-Associated Virus (AAV) Vectors for Cancer Gene Therapy: Current Status and Future Perspectives. Cancers, 12(7), 1889. https://doi.org/10.3390/cancers12071889