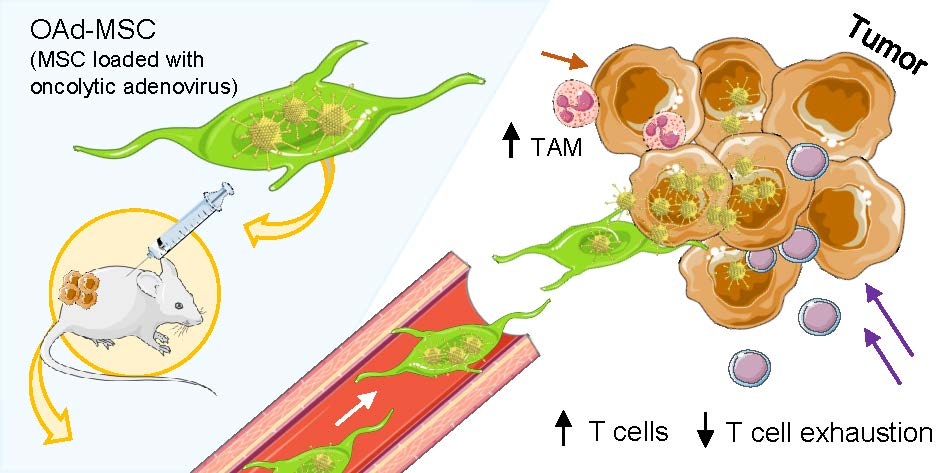
Cellular Virotherapy Increases Tumor-Infiltrating Lymphocytes (TIL) and Decreases their PD-1+ Subsets in Mouse Immunocompetent Models

 , and
, and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Oncolytic Adenovirus
2.3. Western Blot and NF-κB Activity
2.4. Animal Models
2.5. Flow Cytometry
2.6. Cytokine Array
2.7. Statistical Analysis
3. Results
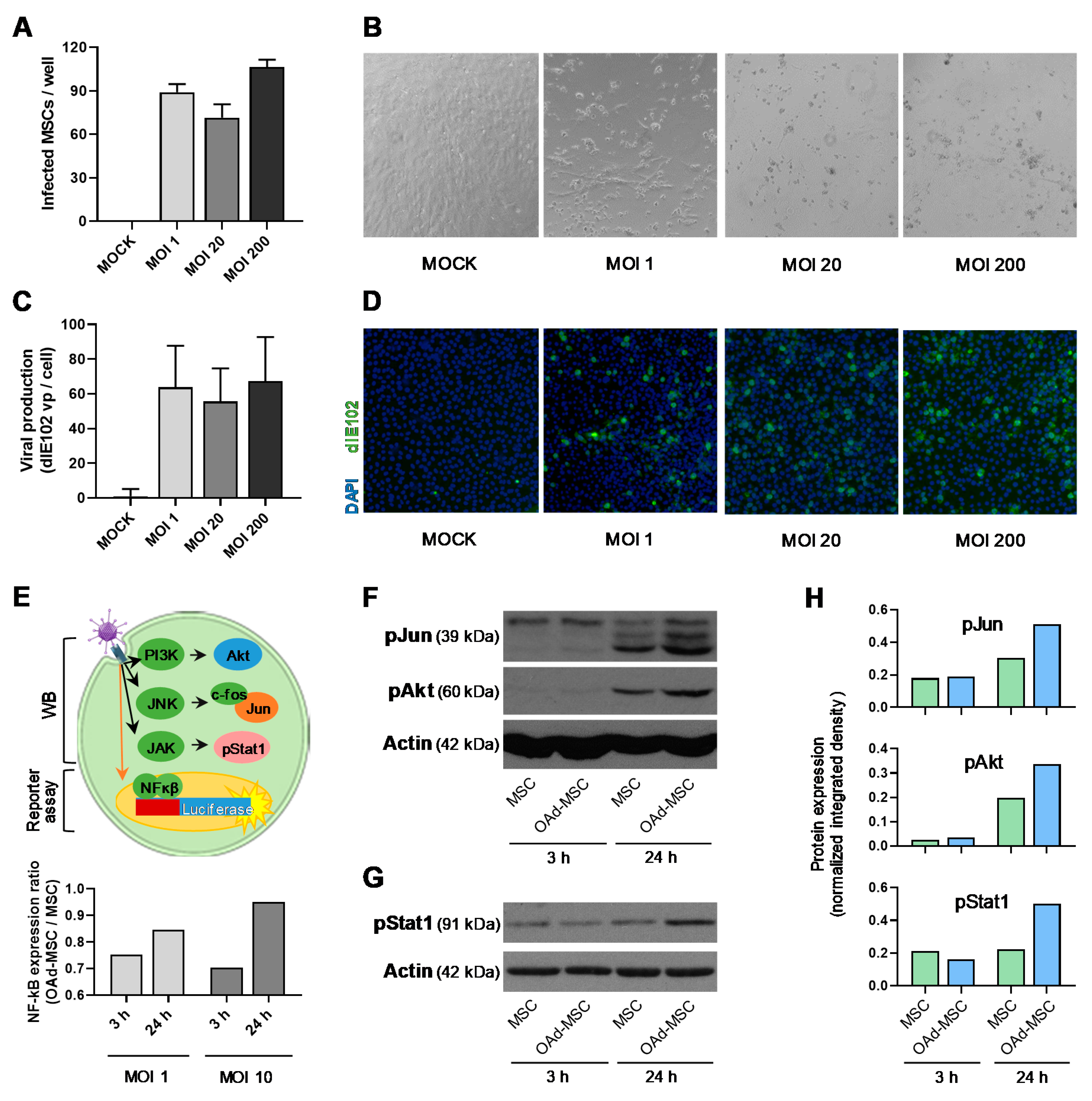
3.1. Mouse Oncolytic Adenovirus dlE102 Replicates in Mouse MSCs, and Does Not Activate Pro-Inflammatory Pathways
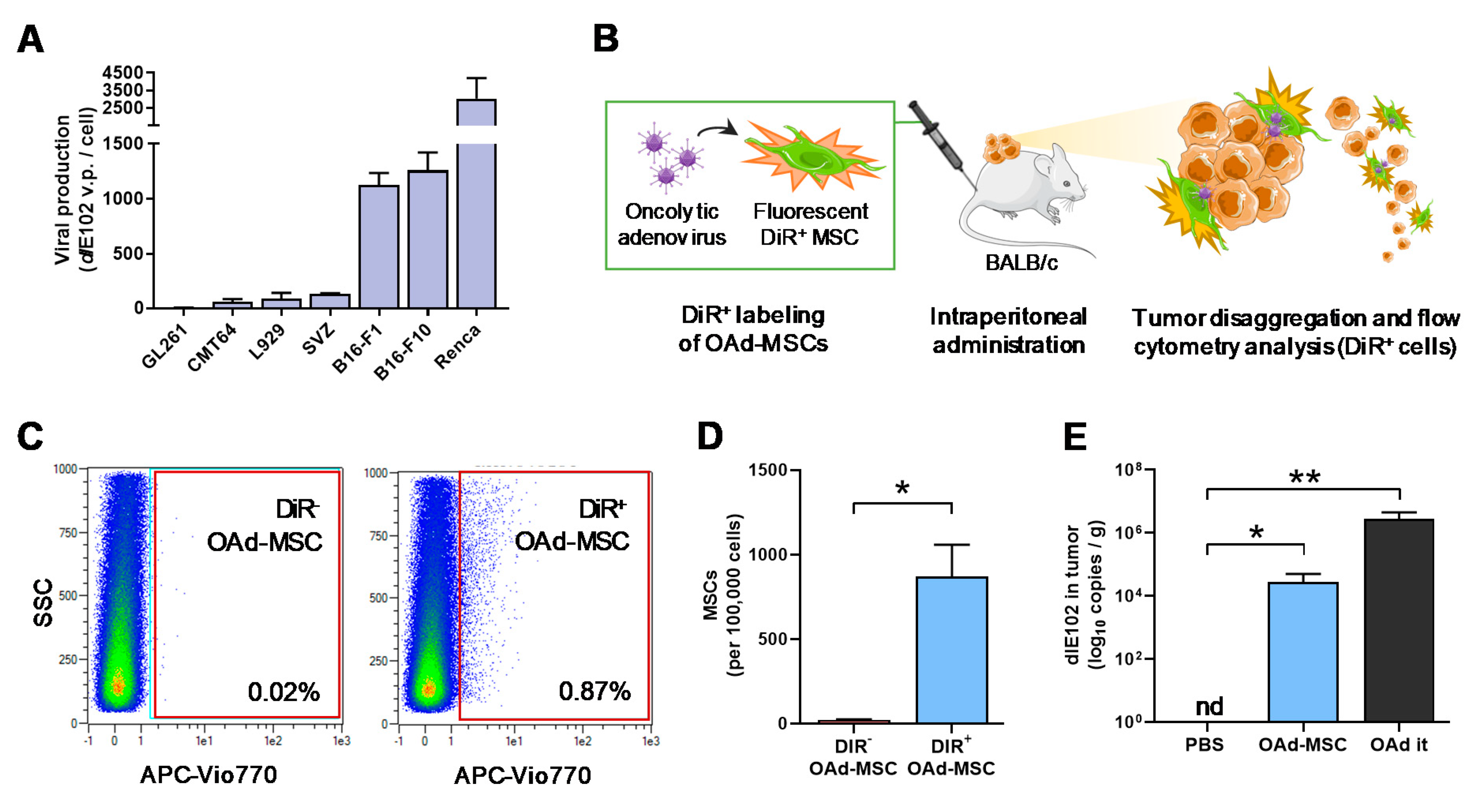
3.2. Mouse Oncolytic Adenovirus dlE102 Successfully Replicates in Mouse Tumor Cells
3.3. MSCs and Oncolytic Adenovirus dlE102 Are Detected in the Tumor Site after Systemic Administration of OAd-MSCs
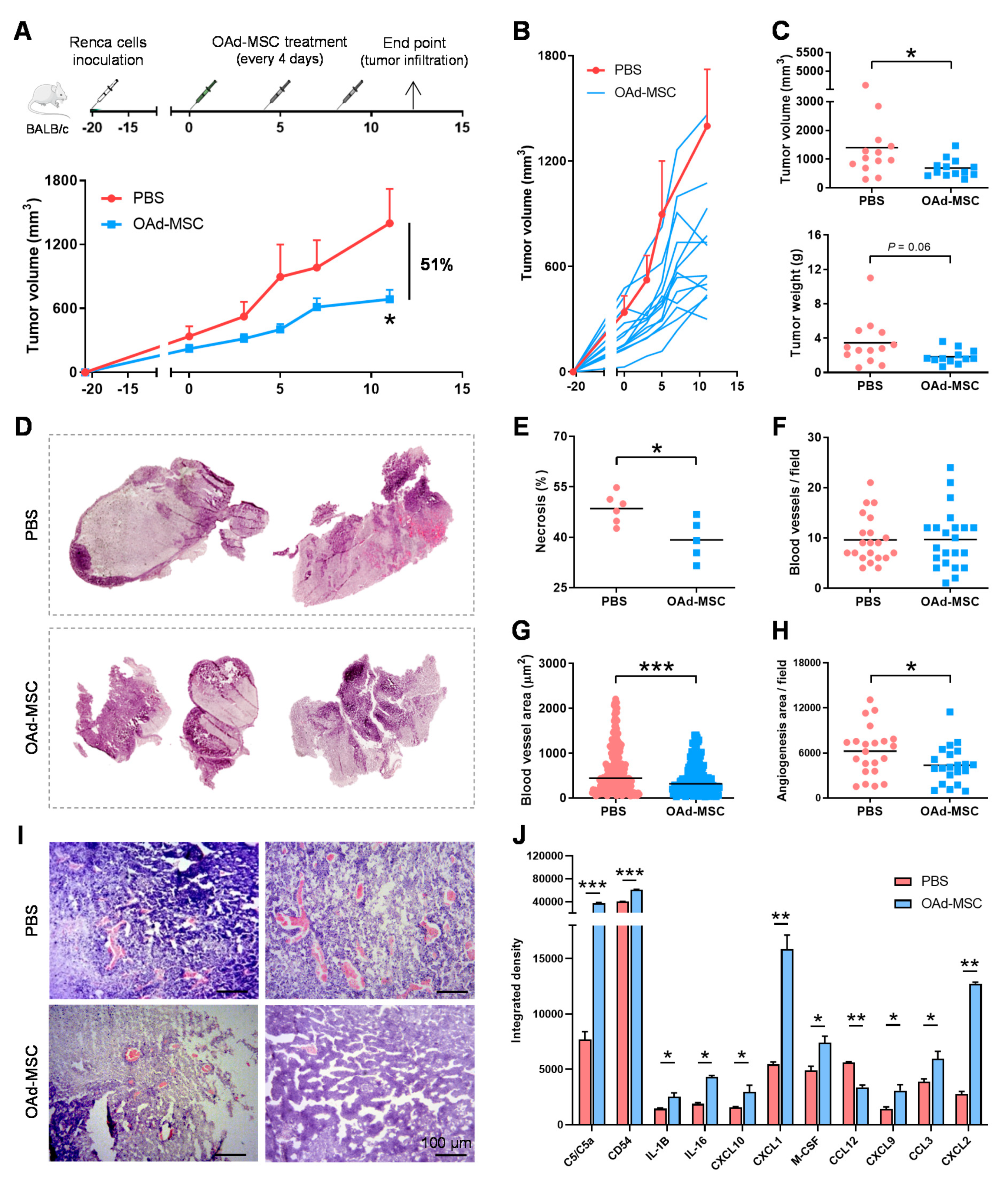
3.4. Treatment with OAd-MSCs Presents Significant Antitumor Efficacy and Induces a Pro-Inflammatory Profile in a Renal Cancer Model
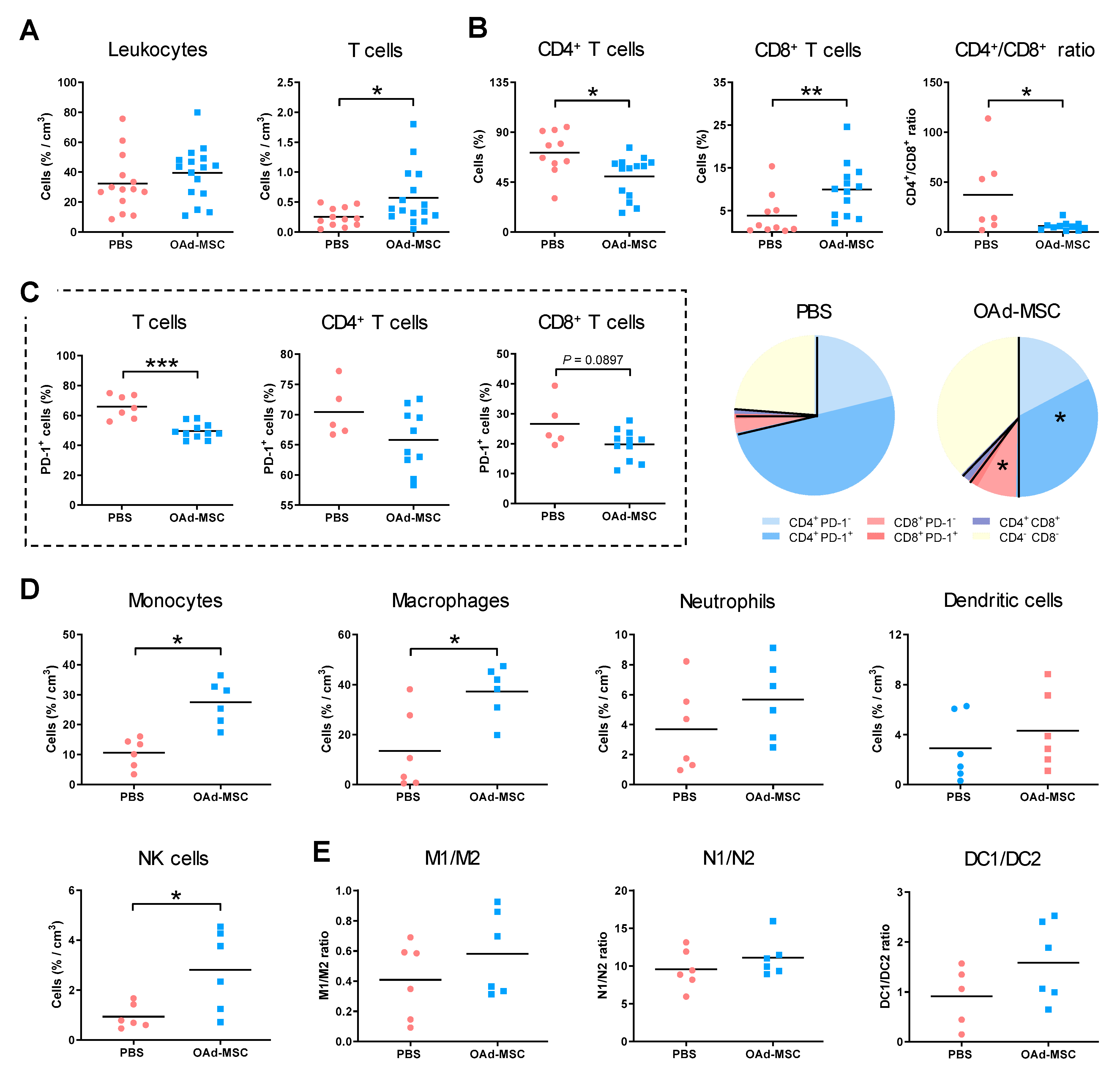
3.5. Treatment with OAd-MSCs Increases Tumor-Infiltrating Immune Cells in a Renal Cancer Model
3.6. Treatment with OAd-MSCs Presents Significant Antitumor Efficacy and Induces a Pro-Inflammatory Profile in a Melanoma Model
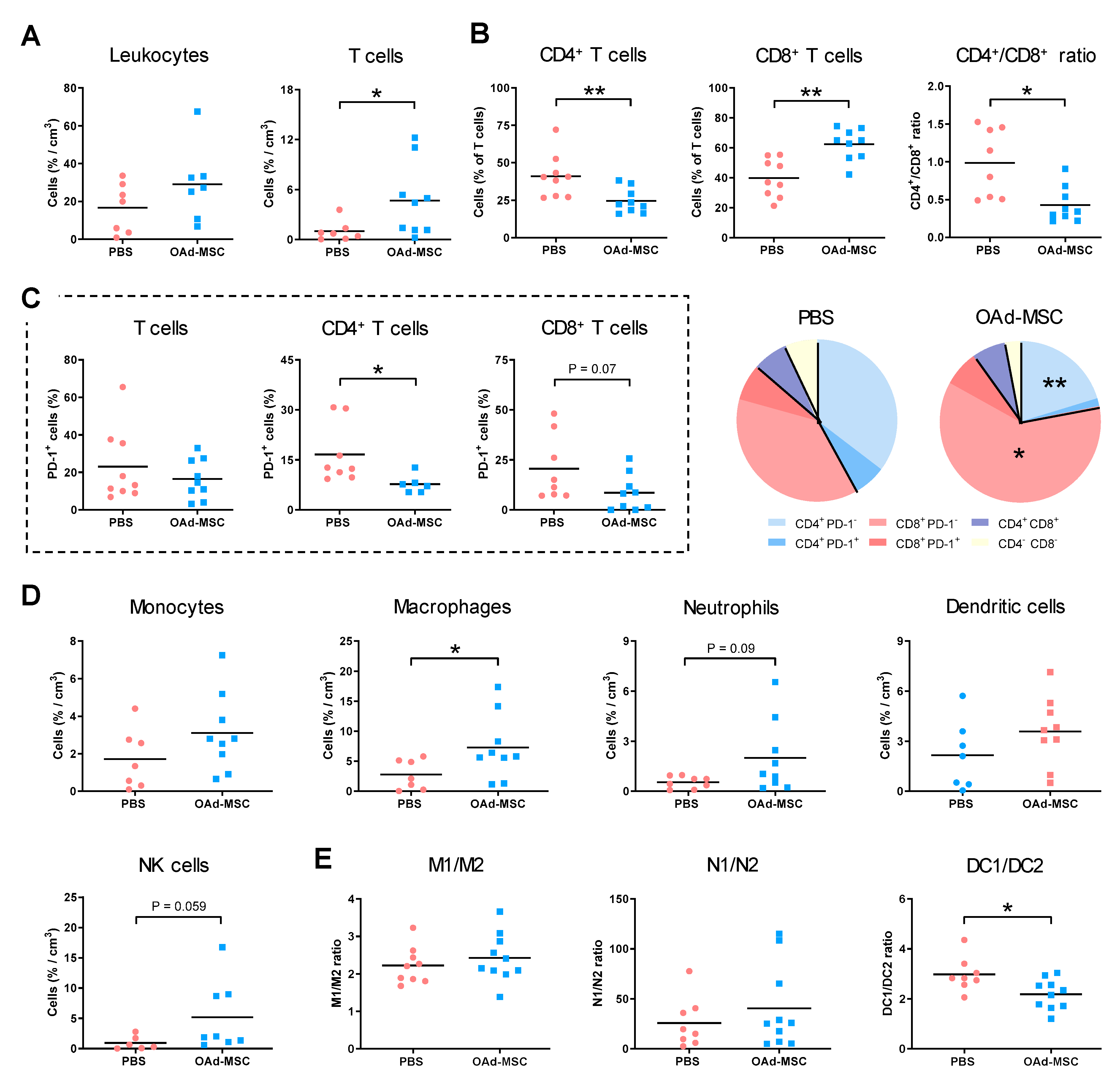
3.7. Treatment with OAd-MSCs Induces Changes in Tumor-Infiltrating Immune Cells in a Melanoma Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Russell, S.J.; Peng, K.W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, P.; Li, H.; Du, X.; Liu, M.; Huang, Q.; Wang, Y.; Wang, S. The Efficacy of Oncolytic Adenovirus Is Mediated by T-cell Responses against Virus and Tumor in Syrian Hamster Model. Clin. Cancer Res. 2017, 23, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Mahasa, K.J.; de Pillis, L.; Ouifki, R.; Eladdadi, A.; Maini, P.; Yoon, A.R.; Yun, C.O. Mesenchymal stem cells used as carrier cells of oncolytic adenovirus results in enhanced oncolytic virotherapy. Sci. Rep. 2020, 10, 425. [Google Scholar] [CrossRef] [PubMed]
- Kidd, S.; Spaeth, E.; Dembinski, J.L.; Dietrich, M.; Watson, K.; Klopp, A.; Battula, V.L.; Weil, M.; Andreeff, M.; Marini, F.C. Direct evidence of mesenchymal stem cell tropism for tumor and wounding microenvironments using in vivo bioluminescent imaging. Stem Cells 2009, 27, 2614–2623. [Google Scholar] [CrossRef] [PubMed]
- Hadrys, A.; Sochanik, A.; McFadden, G.; Jazowiecka-Rakus, J. Mesenchymal stem cells as carriers for systemic delivery of oncolytic viruses. Eur. J. Pharmacol. 2020, 874, 172991. [Google Scholar] [CrossRef]
- Ramirez, M.; Garcia-Castro, J.; Melen, G.J.; Gonzalez-Murillo, A.; Franco-Luzon, L. Patient-derived mesenchymal stem cells as delivery vehicles for oncolytic virotherapy: Novel state-of-the-art technology. Oncolytic Virother. 2015, 4, 149–155. [Google Scholar] [CrossRef]
- Yoon, A.R.; Hong, J.; Li, Y.; Shin, H.C.; Lee, H.; Kim, H.S.; Yun, C.O. Mesenchymal Stem Cell-Mediated Delivery of an Oncolytic Adenovirus Enhances Antitumor Efficacy in Hepatocellular Carcinoma. Cancer Res. 2019, 79, 4503–4514. [Google Scholar] [CrossRef]
- Barlabe, P.; Sostoa, J.; Fajardo, C.A.; Alemany, R.; Moreno, R. Enhanced antitumor efficacy of an oncolytic adenovirus armed with an EGFR-targeted BiTE using menstrual blood-derived mesenchymal stem cells as carriers. Cancer Gene Ther. 2020, 27, 383–388. [Google Scholar] [CrossRef]
- Moreno, R.; Fajardo, C.A.; Farrera-Sal, M.; Perise-Barrios, A.J.; Morales-Molina, A.; Al-Zaher, A.A.; García-Castro, J.; Alemany, R. Enhanced Antitumor Efficacy of Oncolytic Adenovirus-loaded Menstrual Blood-derived Mesenchymal Stem Cells in Combination with Peripheral Blood Mononuclear Cells. Mol. Cancer Ther. 2019, 18, 127–138. [Google Scholar] [CrossRef]
- Cascallo, M.; Alonso, M.M.; Rojas, J.J.; Perez-Gimenez, A.; Fueyo, J.; Alemany, R. Systemic toxicity-efficacy profile of ICOVIR-5, a potent and selective oncolytic adenovirus based on the pRB pathway. Mol. Ther. 2007, 15, 1607–1615. [Google Scholar] [CrossRef]
- Alonso, M.M.; Cascallo, M.; Gomez-Manzano, C.; Jiang, H.; Bekele, B.N.; Perez-Gimenez, A.; Lang, F.F.; Piao, Y.; Alemany, R.; Fueyo, J. ICOVIR-5 shows E2F1 addiction and potent antiglioma effect in vivo. Cancer Res. 2007, 67, 8255–8263. [Google Scholar] [CrossRef]
- Garcia-Castro, J.; Alemany, R.; Cascallo, M.; Martinez-Quintanilla, J.; Arriero Mdel, M.; Lassaletta, A.; Madero, L.; Ramírez, M. Treatment of metastatic neuroblastoma with systemic oncolytic virotherapy delivered by autologous mesenchymal stem cells: An exploratory study. Cancer Gene Ther. 2010, 17, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Melen, G.J.; Franco-Luzon, L.; Ruano, D.; Gonzalez-Murillo, A.; Alfranca, A.; Casco, F.; Lassaletta, Á.; Alonso, M.; Madero, L.; Alemany, R. Influence of carrier cells on the clinical outcome of children with neuroblastoma treated with high dose of oncolytic adenovirus delivered in mesenchymal stem cells. Cancer Lett. 2016, 371, 161–170. [Google Scholar] [CrossRef]
- Ramirez, M.; Ruano, D.; Moreno, L.; Lassaletta, Á.; Sirvent, F.J.B.; Andión, M.; Hernández, C.; González-Murillo, Á.; Melen, G. First-in-child trial of celyvir (autologous mesenchymal stem cells carrying the oncolytic virus ICOVIR-5) in patients with relapsed and refractory pediatric solid tumors. J. Clin. Oncol. 2018, 36, 10543. [Google Scholar] [CrossRef]
- Laborda, E.; Puig-Saus, C.; Rodriguez-Garcia, A.; Moreno, R.; Cascallo, M.; Pastor, J.; Alemany, R. A pRb-responsive, RGD-modified, and hyaluronidase-armed canine oncolytic adenovirus for application in veterinary oncology. Mol. Ther. 2014, 22, 986–998. [Google Scholar] [CrossRef] [PubMed]
- Cejalvo, T.; Perise-Barrios, A.J.; Del Portillo, I.; Laborda, E.; Rodriguez-Milla, M.A.; Cubillo, I.; Vázquez, F.; Sardón, D.; Ramírez, M.; Alemany, R.; et al. Remission of Spontaneous Canine Tumors after Systemic Cellular Viroimmunotherapy. Cancer Res. 2018, 78, 4891–4901. [Google Scholar] [CrossRef] [PubMed]
- Gooden, M.J.; de Bock, G.H.; Leffers, N.; Daemen, T.; Nijman, H.W. The prognostic influence of tumour-infiltrating lymphocytes in cancer: A systematic review with meta-analysis. Br. J. Cancer 2011, 105, 93–103. [Google Scholar] [CrossRef]
- Badalamenti, G.; Fanale, D.; Incorvaia, L.; Barraco, N.; Listi, A.; Maragliano, R.; Vincenzi, B.; Calò, V.; Iovanna, J.L.; Bazan, V.; et al. Role of tumor-infiltrating lymphocytes in patients with solid tumors: Can a drop dig a stone? Cell. Immunol. 2019, 343, 103753. [Google Scholar] [CrossRef]
- Rincon, E.; Cejalvo, T.; Kanojia, D.; Alfranca, A.; Rodriguez-Milla, M.A.; Gil Hoyos, R.A.; Han, Y.; Zhang, L.; Alemany, R.; Lesniak, M.S.; et al. Mesenchymal stem cell carriers enhance antitumor efficacy of oncolytic adenoviruses in an immunocompetent mouse model. Oncotarget 2017, 8, 45415–45431. [Google Scholar] [CrossRef]
- Morales-Molina, A.; Gambera, S.; Cejalvo, T.; Moreno, R.; Rodriguez-Milla, M.A.; Perise-Barrios, A.J.; García-Castro, J. Antitumor virotherapy using syngeneic or allogeneic mesenchymal stem cell carriers induces systemic immune response and intratumoral leukocyte infiltration in mice. Cancer Immunol. Immunother. 2018, 67, 1589–1602. [Google Scholar] [CrossRef]
- Filley, A.C.; Dey, M. Immune System, Friend or Foe of Oncolytic Virotherapy? Front. Oncol. 2017, 7, 106. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.; Ying, B.; Ball, A.O.; Beard, C.W.; Spindler, K.R. Interaction of mouse adenovirus type 1 early region 1A protein with cellular proteins pRb and p107. Virology 1996, 224, 184–197. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Robinson, M.; Li, B.; Ge, Y.; Ko, D.; Yendluri, S.; Harding, T.; VanRoev, M.; Spindler, K.R.; Jooss, K. Novel immunocompetent murine tumor model for evaluation of conditionally replication-competent (oncolytic) murine adenoviral vectors. J. Virol. 2009, 83, 3450–3462. [Google Scholar] [CrossRef] [PubMed]
- Mosna, F.; Sensebe, L.; Krampera, M. Human bone marrow and adipose tissue mesenchymal stem cells: A user’s guide. Stem Cells Dev. 2010, 19, 1449–1470. [Google Scholar] [CrossRef] [PubMed]
- Gargini, R.; Segura-Collar, B.; Herránz, B.; García-Escudero, V.; Romero-Bravo, A.; Núñez, F.P.; García-Pérez, D.; Gutiérrez-Guamán, J.; Ayuso-Sacido, A.; Seoane, J.; et al. The IDH-TAU-EGFR triad defines the neovascular landscape of diffuse gliomas. Sci. Transl. Med. 2020, 12, eaax1501. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.A.; Kwok, L.W.; Porter, E.L.; Payne, J.G.; McElroy, G.S.; Ohle, S.J.; Greenhill, S.R.; Blahna, M.T.; Yamamoto, K.; Jean, J.C.; et al. Lentiviral delivery of RNAi for in vivo lineage-specific modulation of gene expression in mouse lung macrophages. Mol. Ther. 2013, 21, 825–833. [Google Scholar] [CrossRef]
- Rodríguez-Milla, M.Á.; Morales-Molina, A.; Perisé-Barrios, A.J.; Cejalvo, T.; García-Castro, J. AKT and JUN are differentially activated in mesenchymal stem cells after infection with human and canine oncolytic adenoviruses. Cancer Gene Ther. 2020. [Google Scholar] [CrossRef]
- Hallden, G.; Hill, R.; Wang, Y.; Anand, A.; Liu, T.C.; Lemoine, N.R.; Francis, J.; Hawkins, L.; Kim, D. Novel immunocompetent murine tumor models for the assessment of replication-competent oncolytic adenovirus efficacy. Mol. Ther. 2003, 8, 412–424. [Google Scholar] [CrossRef]
- Rahman, M.M.; McFadden, G. Modulation of NF-kappaB signalling by microbial pathogens. Nat. Rev. Microbiol. 2011, 9, 291–306. [Google Scholar] [CrossRef]
- Franco-Luzon, L.; Gonzalez-Murillo, A.; Alcantara-Sanchez, C.; Garcia-Garcia, L.; Tabasi, M.; Huertas, A.L.; Chesler, L.; Ramírez, M. Systemic oncolytic adenovirus delivered in mesenchymal carrier cells modulate tumor infiltrating immune cells and tumor microenvironment in mice with neuroblastoma. Oncotarget 2020, 11, 347–361. [Google Scholar] [CrossRef]
- Hendrickx, R.; Stichling, N.; Koelen, J.; Kuryk, L.; Lipiec, A.; Greber, U.F. Innate immunity to adenovirus. Hum. Gene Ther. 2014, 25, 265–284. [Google Scholar] [CrossRef] [PubMed]
- Molloy, C.T.; Adkins, L.J.; Griffin, C.; Singer, K.; Weinberg, J.B. Mouse adenovirus type 1 infection of adipose tissue. Virus Res. 2018, 244, 90–98. [Google Scholar] [CrossRef]
- Benko, M.; Harrach, B. Molecular evolution of adenoviruses. Curr. Top. Microbiol. Immunol. 2003, 272, 3–35. [Google Scholar] [PubMed]
- Lenaerts, L.; Daelemans, D.; Geukens, N.; De Clercq, E.; Naesens, L. Mouse adenovirus type 1 attachment is not mediated by the coxsackie-adenovirus receptor. FEBS Lett. 2006, 580, 3937–3942. [Google Scholar] [CrossRef] [PubMed]
- Quon, H.; Hasbini, A.; Cougnard, J.; Djafari, L.; Lacroix, C.; Abdulkarim, B. Assessment of tumor angiogenesis as a prognostic factor of survival in patients with oligodendroglioma. J. Neurooncol. 2010, 96, 277–285. [Google Scholar] [CrossRef]
- Matsuda, Y.; Hagio, M.; Ishiwata, T. Nestin: A novel angiogenesis marker and possible target for tumor angiogenesis. World J. Gastroenterol. 2013, 19, 42–48. [Google Scholar] [CrossRef]
- Zhang, L.; Zha, Z.; Qu, W.; Zhao, H.; Yuan, J.; Feng, Y.; Wu, B. Tumor necrosis as a prognostic variable for the clinical outcome in patients with renal cell carcinoma: A systematic review and meta-analysis. BMC Cancer 2018, 18, 870. [Google Scholar] [CrossRef]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef]
- Watanabe, H.; Numata, K.; Ito, T.; Takagi, K.; Matsukawa, A. Innate immune response in Th1- and Th2-dominant mouse strains. Shock 2004, 22, 460–466. [Google Scholar] [CrossRef]
- Straetemans, T.; Berrevoets, C.; Coccoris, M.; Treffers-Westerlaken, E.; Wijers, R.; Cole, D.K.; Dardalhon, V.; Sewell, A.K.; Taylor, N.; Verweij, J.; et al. Recurrence of melanoma following T cell treatment: Continued antigen expression in a tumor that evades T cell recruitment. Mol. Ther. 2015, 23, 396–406. [Google Scholar] [CrossRef]
- Basingab, F.S.; Ahmadi, M.; Morgan, D.J. IFNgamma-Dependent Interactions between ICAM-1 and LFA-1 Counteract Prostaglandin E2-Mediated Inhibition of Antitumor CTL Responses. Cancer Immunol. Res. 2016, 4, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Chaves, K.C.; Peron, J.P.; Chammas, R.; Turaca, L.T.; Pesquero, J.B.; Braga, M.S.; Foguer, K.; Schor, N.; Bellini, M.H. Endostatin gene therapy stimulates upregulation of ICAM-1 and VCAM-1 in a metastatic renal cell carcinoma model. Cancer Gene Ther. 2012, 19, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Raulet, D.H.; Moretta, A.; Caligiuri, M.A.; Zitvogel, L.; Lanier, L.L.; Yokoyama, W.M.; Ugolini, S. Innate or adaptive immunity? The example of natural killer cells. Science 2011, 331, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef] [PubMed]
- Kollmann, D.; Schweiger, T.; Schwarz, S.; Ignatova, D.; Chang, Y.T.; Lewik, G.; Schoppmann, S.F.; Hoetzenecker, W.; Klepetko, W.; Guenova, E.; et al. PD1-positive tumor-infiltrating lymphocytes are associated with poor clinical outcome after pulmonary metastasectomy for colorectal cancer. Oncoimmunology 2017, 6, e1331194. [Google Scholar] [CrossRef]
- Granier, C.; Dariane, C.; Combe, P.; Verkarre, V.; Urien, S.; Badoual, C.; Roussel, H.; Mandavit, M.; Ravel, P.; Sibony, M.; et al. Tim-3 Expression on Tumor-Infiltrating PD-1(+)CD8(+) T Cells Correlates with Poor Clinical Outcome in Renal Cell Carcinoma. Cancer Res. 2017, 77, 1075–1082. [Google Scholar] [CrossRef]
- Wang, R.; Shi, F.; Zhao, L.; Zhao, Y.; Wu, G.; Song, Q.K. High expression of E-cadherin and Ki-67 associated with functional/dysfunctional phenotypes of tumor-infiltrating lymphocytes among Chinese patients with operable breast cancer. J. Int. Med. Res. 2018, 46, 5219–5227. [Google Scholar] [CrossRef]
- Saito, H.; Shimizu, S.; Kono, Y.; Murakami, Y.; Shishido, Y.; Miyatani, K.; Matsunaga, T.; Fukumoto, Y.; Ashida, K. Fujiwara, Y. PD-1 Expression on Circulating CD8(+) T-Cells as a Prognostic Marker for Patients With Gastric Cancer. Anticancer Res. 2019, 39, 443–448. [Google Scholar] [CrossRef]
- Penaloza-MacMaster, P.; Provine, N.M.; Ra, J.; Borducchi, E.N.; McNally, A.; Simmons, N.L.; Iampietro, M.J.; Barouch, D.H. Alternative serotype adenovirus vaccine vectors elicit memory T cells with enhanced anamnestic capacity compared to Ad5 vectors. J. Virol. 2013, 87, 1373–1384. [Google Scholar] [CrossRef][Green Version]
- Penaloza-MacMaster, P.; Alayo, Q.A.; Ra, J.; Provine, N.M.; Larocca, R.; Lee, B.; Barouch, D.H. Inhibitory receptor expression on memory CD8 T cells following Ad vector immunization. Vaccine 2016, 34, 4955–4963. [Google Scholar] [CrossRef]
- Feist, M.; Zhu, Z.; Dai, E.; Ma, C.; Liu, Z.; Giehl, E.; Ravindranathan, R.; Kowalsky, S.J.; Obermajer, N.; Kammula, U.D.; et al. Oncolytic virus promotes tumor-reactive infiltrating lymphocytes for adoptive cell therapy. Cancer Gene Ther. 2020. [Google Scholar] [CrossRef] [PubMed]
- Haanen, J. Converting Cold into Hot Tumors by Combining Immunotherapies. Cell 2017, 170, 1055–1056. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morales-Molina, A.; Rodríguez-Milla, M.Á.; Gimenez-Sanchez, A.; Perisé-Barrios, A.J.; García-Castro, J. Cellular Virotherapy Increases Tumor-Infiltrating Lymphocytes (TIL) and Decreases their PD-1+ Subsets in Mouse Immunocompetent Models. Cancers 2020, 12, 1920. https://doi.org/10.3390/cancers12071920
Morales-Molina A, Rodríguez-Milla MÁ, Gimenez-Sanchez A, Perisé-Barrios AJ, García-Castro J. Cellular Virotherapy Increases Tumor-Infiltrating Lymphocytes (TIL) and Decreases their PD-1+ Subsets in Mouse Immunocompetent Models. Cancers. 2020; 12(7):1920. https://doi.org/10.3390/cancers12071920
Chicago/Turabian StyleMorales-Molina, Alvaro, Miguel Ángel Rodríguez-Milla, Alicia Gimenez-Sanchez, Ana Judith Perisé-Barrios, and Javier García-Castro. 2020. "Cellular Virotherapy Increases Tumor-Infiltrating Lymphocytes (TIL) and Decreases their PD-1+ Subsets in Mouse Immunocompetent Models" Cancers 12, no. 7: 1920. https://doi.org/10.3390/cancers12071920
APA StyleMorales-Molina, A., Rodríguez-Milla, M. Á., Gimenez-Sanchez, A., Perisé-Barrios, A. J., & García-Castro, J. (2020). Cellular Virotherapy Increases Tumor-Infiltrating Lymphocytes (TIL) and Decreases their PD-1+ Subsets in Mouse Immunocompetent Models. Cancers, 12(7), 1920. https://doi.org/10.3390/cancers12071920