SRC-Family Kinases in Acute Myeloid Leukaemia and Mastocytosis



Abstract
1. Introduction
2. SFKs in Acute Myeloid Leukemia (AML)
2.1. Expression of SFKs in AML
2.2. SFKs Are Activated in AML
2.3. Lessons from SFK Inhibitors in AML
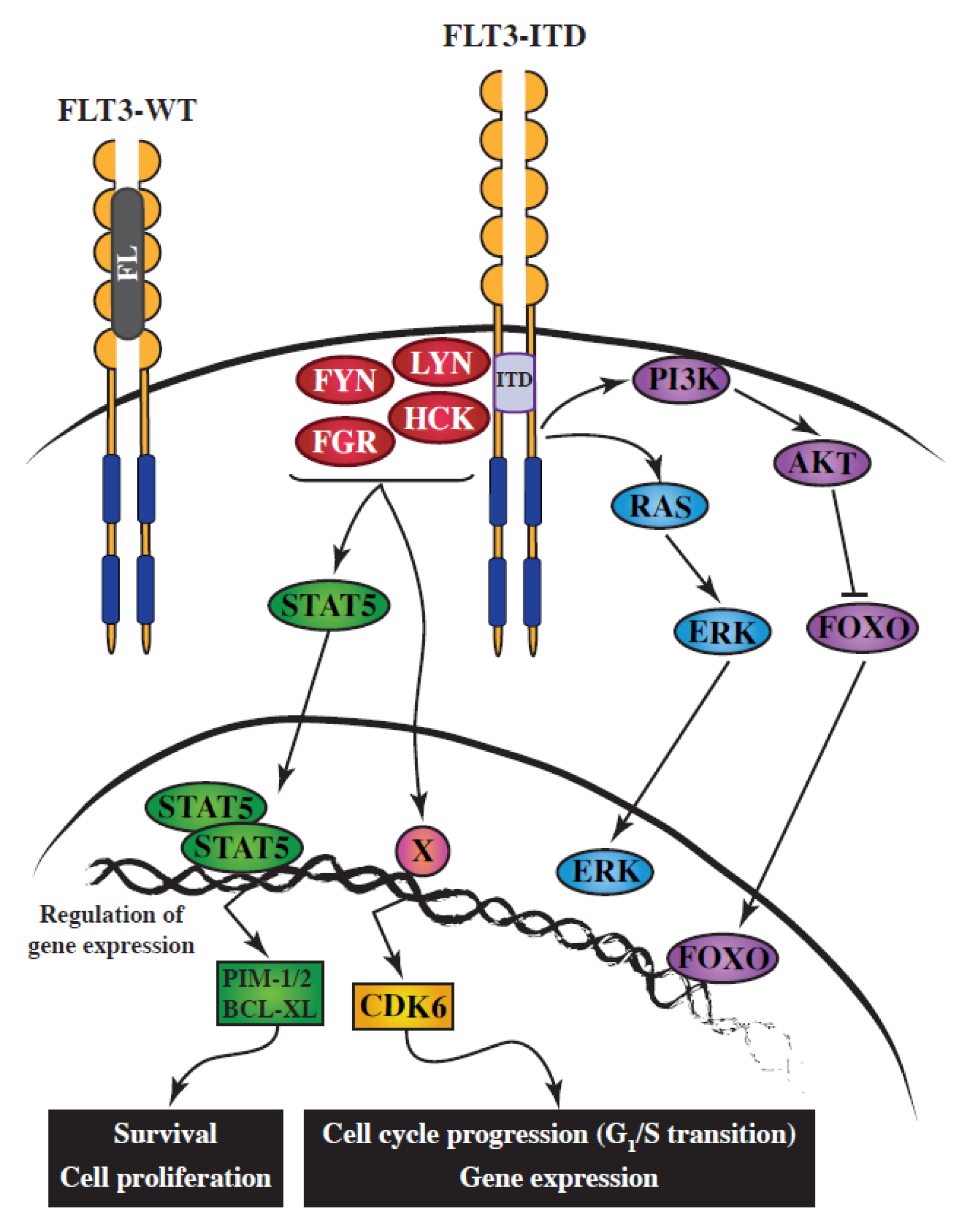
2.4. SFKs and FLT3-ITD in AML
2.5. SFKs and Oncogenic KIT in AML
3. SFKs in Mastocytosis
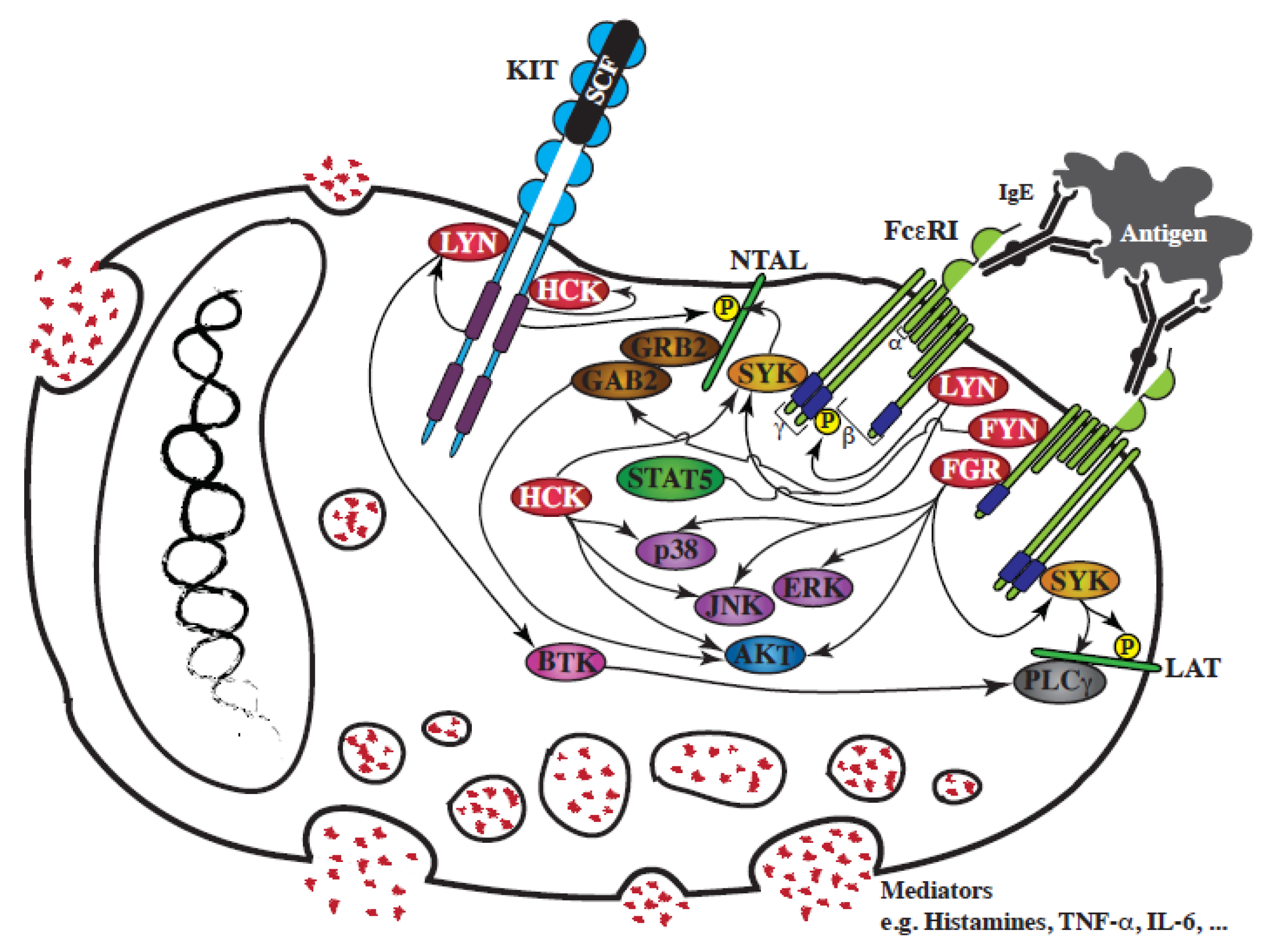
3.1. SFKs and KIT Signaling
3.2. SFKs and FcεRI Signaling
3.3. SFK Inhibitors in Mastocytosis
4. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hatakeyama, S.; Iwabuchi, K.; Ato, M.; Iwabuchi, C.; Kajino, K.; Takami, K.; Katoh, M.; Ogasawara, K.; Good, R.A.; Onoe, K. Fgr expression restricted to subpopulation of monocyte/macrophage lineage in resting conditions is induced in various hematopoietic cells after activation or transformation. Microbiol. Immunol. 1996, 40, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Ingley, E. Functions of the Lyn tyrosine kinase in health and disease. Cell Commun. Signal. 2012, 10, 21. [Google Scholar] [CrossRef]
- Quintrell, N.; Lebo, R.; Varmus, H.; Bishop, J.M.; Pettenati, M.J.; Le Beau, M.M.; Diaz, M.O.; Rowley, J.D. Identification of a human gene (HCK) that encodes a protein-tyrosine kinase and is expressed in hemopoietic cells. Mol. Cell Biol. 1987, 7, 2267–2275. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, S.F.; Marth, J.D.; Lewis, D.B.; Perlmutter, R.M. Novel protein-tyrosine kinase gene (HCK) preferentially expressed in cells of hematopoietic origin. Mol. Cell Biol. 1987, 7, 2276–2285. [Google Scholar] [CrossRef] [PubMed]
- Dymecki, S.M.; Niederhuber, J.E.; Desiderio, S.V. Specific expression of a tyrosine kinase gene, blk, in B lymphoid cells. Science 1990, 247, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Straus, D.B.; Weiss, A. Genetic evidence for the involvement of the lck tyrosine kinase in signal transduction through the T cell antigen receptor. Cell 1992, 70, 585–593. [Google Scholar] [CrossRef]
- Ingley, E. Src family kinases: Regulation of their activities, levels and identification of new pathways. Biochim. Biophys. Acta 2008, 1784, 56–65. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacol. Res. 2015, 94, 9–25. [Google Scholar] [CrossRef]
- Parsons, S.J.; Parsons, J.T. Src family kinases, key regulators of signal transduction. Oncogene 2004, 23, 7906–7909. [Google Scholar] [CrossRef]
- Gleixner, K.V.; Mayerhofer, M.; Cerny-Reiterer, S.; Hormann, G.; Rix, U.; Bennett, K.L.; Hadzijusufovic, E.; Meyer, R.A.; Pickl, W.F.; Gotlib, J.; et al. KIT-D816V-independent oncogenic signaling in neoplastic cells in systemic mastocytosis: Role of LYN and BTK activation and disruption by dasatinib and bosutinib. Blood 2011, 118, 1885–1898. [Google Scholar] [CrossRef]
- Loriaux, M.M.; Levine, R.L.; Tyner, J.W.; Frohling, S.; Scholl, C.; Stoffregen, E.P.; Wernig, G.; Erickson, H.; Eide, C.A.; Berger, R.; et al. High-throughput sequence analysis of the tyrosine kinome in acute myeloid leukemia. Blood 2008, 111, 4788–4796. [Google Scholar] [CrossRef] [PubMed]
- Tomasson, M.H.; Xiang, Z.; Walgren, R.; Zhao, Y.; Kasai, Y.; Miner, T.; Ries, R.E.; Lubman, O.; Fremont, D.H.; McLellan, M.D.; et al. Somatic mutations and germline sequence variants in the expressed tyrosine kinase genes of patients with de novo acute myeloid leukemia. Blood 2008, 111, 4797–4808. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Takeuchi, M.; Takeda, Y.; Sakai, S.; Abe, D.; Ohwada, C.; Sakaida, E.; Shimizu, N.; Saito, Y.; Miyagi, S.; et al. Identification of a novel TEL-Lyn fusion gene in primary myelofibrosis. Leukemia 2010, 24, 197–200. [Google Scholar] [CrossRef]
- Ma, E.S.K.; Wan, T.S.K.; Au, C.H.; Ho, D.N.; Ma, S.Y.; Ng, M.H.L.; Chan, T.L. Next-generation sequencing and molecular cytogenetic characterization of ETV6-LYN fusion due to chromosomes 1, 8 and 12 rearrangement in acute myeloid leukemia. Cancer Genet. 2017, 218–219, 15–19. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dohner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Buchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef]
- Daver, N.; Schlenk, R.F.; Russell, N.H.; Levis, M.J. Targeting FLT3 mutations in AML: Review of current knowledge and evidence. Leukemia 2019, 33, 299–312. [Google Scholar] [CrossRef]
- Kiyoi, H.; Towatari, M.; Yokota, S.; Hamaguchi, M.; Ohno, R.; Saito, H.; Naoe, T. Internal tandem duplication of the FLT3 gene is a novel modality of elongation mutation which causes constitutive activation of the product. Leukemia 1998, 12, 1333–1337. [Google Scholar] [CrossRef]
- Voisset, E.; Lopez, S.; Chaix, A.; Georges, C.; Hanssens, K.; Prebet, T.; Dubreuil, P.; De Sepulveda, P. FES kinases are required for oncogenic FLT3 signaling. Leukemia 2010, 24, 721–728. [Google Scholar] [CrossRef]
- Puissant, A.; Fenouille, N.; Alexe, G.; Pikman, Y.; Bassil, C.F.; Mehta, S.; Du, J.; Kazi, J.U.; Luciano, F.; Ronnstrand, L.; et al. SYK is a critical regulator of FLT3 in acute myeloid leukemia. Cancer Cell 2014, 25, 226–242. [Google Scholar] [CrossRef]
- Heiss, E.; Masson, K.; Sundberg, C.; Pedersen, M.; Sun, J.; Bengtsson, S.; Ronnstrand, L. Identification of Y589 and Y599 in the juxtamembrane domain of Flt3 as ligand-induced autophosphorylation sites involved in binding of Src family kinases and the protein tyrosine phosphatase SHP2. Blood 2006, 108, 1542–1550. [Google Scholar] [CrossRef]
- Mitina, O.; Warmuth, M.; Krause, G.; Hallek, M.; Obermeier, A. Src family tyrosine kinases phosphorylate Flt3 on juxtamembrane tyrosines and interfere with receptor maturation in a kinase-dependent manner. Ann. Hematol. 2007, 86, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Robinson, L.J.; Xue, J.; Corey, S.J. Src family tyrosine kinases are activated by Flt3 and are involved in the proliferative effects of leukemia-associated Flt3 mutations. Exp. Hematol. 2005, 33, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Kazi, J.U.; Ronnstrand, L. The role of SRC family kinases in FLT3 signaling. Int. J. Biochem. Cell Biol. 2019, 107, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Lennartsson, J.; Ronnstrand, L. Stem cell factor receptor/c-Kit: From basic science to clinical implications. Physiol. Rev. 2012, 92, 1619–1649. [Google Scholar] [CrossRef] [PubMed]
- Willman, C.L.; Stewart, C.C.; Longacre, T.L.; Head, D.R.; Habbersett, R.; Ziegler, S.F.; Perlmutter, R.M. Expression of the c-FGR and hck protein-tyrosine kinases in acute myeloid leukemic blasts is associated with early commitment and differentiation events in the monocytic and granulocytic lineages. Blood 1991, 77, 726–734. [Google Scholar] [CrossRef]
- Chougule, R.A.; Kazi, J.U.; Ronnstrand, L. FYN expression potentiates FLT3-ITD induced STAT5 signaling in acute myeloid leukemia. Oncotarget 2016, 7, 9964–9974. [Google Scholar] [CrossRef]
- Patel, R.K.; Weir, M.C.; Shen, K.; Snyder, D.; Cooper, V.S.; Smithgall, T.E. Expression of myeloid Src-family kinases is associated with poor prognosis in AML and influences Flt3-ITD kinase inhibitor acquired resistance. PLoS ONE 2019, 14, e0225887. [Google Scholar] [CrossRef]
- Dos Santos, C.; Demur, C.; Bardet, V.; Prade-Houdellier, N.; Payrastre, B.; Recher, C. A critical role for Lyn in acute myeloid leukemia. Blood 2008, 111, 2269–2279. [Google Scholar] [CrossRef]
- Dos Santos, C.; McDonald, T.; Ho, Y.W.; Liu, H.; Lin, A.; Forman, S.J.; Kuo, Y.H.; Bhatia, R. The Src and c-Kit kinase inhibitor dasatinib enhances p53-mediated targeting of human acute myeloid leukemia stem cells by chemotherapeutic agents. Blood 2013, 122, 1900–1913. [Google Scholar] [CrossRef]
- Lopez, S.; Voisset, E.; Tisserand, J.C.; Mosca, C.; Prebet, T.; Santamaria, D.; Dubreuil, P.; De Sepulveda, P. An essential pathway links FLT3-ITD, HCK and CDK6 in acute myeloid leukemia. Oncotarget 2016, 7, 51163–51173. [Google Scholar] [CrossRef]
- Weir, M.C.; Shu, S.T.; Patel, R.K.; Hellwig, S.; Chen, L.; Tan, L.; Gray, N.S.; Smithgall, T.E. Selective Inhibition of the Myeloid Src-Family Kinase Fgr Potently Suppresses AML Cell Growth in Vitro and in Vivo. ACS Chem. Biol. 2018, 13, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Kitamura, H.; Hijikata, A.; Tomizawa-Murasawa, M.; Tanaka, S.; Takagi, S.; Uchida, N.; Suzuki, N.; Sone, A.; Najima, Y.; et al. Identification of therapeutic targets for quiescent, chemotherapy-resistant human leukemia stem cells. Sci. Transl. Med. 2010, 2, 17ra9. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, Y.; Williams, A.H.; Estes, M.L.; Matsushita, N.; Boschelli, F.; Jove, R.; List, A.F. Src family kinases promote AML cell survival through activation of signal transducers and activators of transcription (STAT). Leuk. Res. 2008, 32, 893–903. [Google Scholar] [CrossRef]
- Guerrouahen, B.S.; Futami, M.; Vaklavas, C.; Kanerva, J.; Whichard, Z.L.; Nwawka, K.; Blanchard, E.G.; Lee, F.Y.; Robinson, L.J.; Arceci, R.; et al. Dasatinib inhibits the growth of molecularly heterogeneous myeloid leukemias. Clin. Cancer Res. 2010, 16, 1149–1158. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Hayakawa, F.; Miyata, Y.; Watamoto, K.; Emi, N.; Abe, A.; Kiyoi, H.; Towatari, M.; Naoe, T. Lyn is an important component of the signal transduction pathway specific to FLT3/ITD and can be a therapeutic target in the treatment of AML with FLT3/ITD. Leukemia 2007, 21, 403–410. [Google Scholar] [CrossRef]
- Kim, L.C.; Song, L.; Haura, E.B. Src kinases as therapeutic targets for cancer. Nat. Rev. Clin. Oncol. 2009, 6, 587–595. [Google Scholar] [CrossRef]
- Zhang, S.; Yu, D. Targeting Src family kinases in anti-cancer therapies: Turning promise into triumph. Trends Pharmacol. Sci. 2012, 33, 122–128. [Google Scholar] [CrossRef]
- Brandvold, K.R.; Steffey, M.E.; Fox, C.C.; Soellner, M.B. Development of a highly selective c-Src kinase inhibitor. ACS Chem. Biol. 2012, 7, 1393–1398. [Google Scholar] [CrossRef]
- MacDonald, R.J.; Bunaciu, R.P.; Ip, V.; Dai, D.; Tran, D.; Varner, J.D.; Yen, A. Src family kinase inhibitor bosutinib enhances retinoic acid-induced differentiation of HL-60 leukemia cells. Leuk. Lymphoma 2018, 59, 2941–2951. [Google Scholar] [CrossRef]
- Miranda, M.B.; Redner, R.L.; Johnson, D.E. Inhibition of Src family kinases enhances retinoic acid induced gene expression and myeloid differentiation. Mol. Cancer Ther. 2007, 6, 3081–3090. [Google Scholar] [CrossRef][Green Version]
- Han, L.; Schuringa, J.J.; Mulder, A.; Vellenga, E. Dasatinib impairs long-term expansion of leukemic progenitors in a subset of acute myeloid leukemia cases. Ann. Hematol. 2010, 89, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Kropf, P.L.; Wang, L.; Zang, Y.; Redner, R.L.; Johnson, D.E. Dasatinib promotes ATRA-induced differentiation of AML cells. Leukemia 2010, 24, 663–665. [Google Scholar] [CrossRef] [PubMed]
- Paschka, P.; Schlenk, R.F.; Weber, D.; Benner, A.; Bullinger, L.; Heuser, M.; Gaidzik, V.I.; Thol, F.; Agrawal, M.; Teleanu, V.; et al. Adding dasatinib to intensive treatment in core-binding factor acute myeloid leukemia-results of the AMLSG 11-08 trial. Leukemia 2018, 32, 1621–1630. [Google Scholar] [CrossRef] [PubMed]
- Kivioja, J.L.; Thanasopoulou, A.; Kumar, A.; Kontro, M.; Yadav, B.; Majumder, M.M.; Javarappa, K.K.; Eldfors, S.; Schwaller, J.; Porkka, K.; et al. Dasatinib and navitoclax act synergistically to target NUP98-NSD1(+)/FLT3-ITD(+) acute myeloid leukemia. Leukemia 2019, 33, 1360–1372. [Google Scholar] [CrossRef]
- Saito, Y.; Yuki, H.; Kuratani, M.; Hashizume, Y.; Takagi, S.; Honma, T.; Tanaka, A.; Shirouzu, M.; Mikuni, J.; Handa, N.; et al. A pyrrolo-pyrimidine derivative targets human primary AML stem cells in vivo. Sci. Transl. Med. 2013, 5, 181ra152. [Google Scholar] [CrossRef]
- Muller, T.A.; Grundler, R.; Istvanffy, R.; Rudelius, M.; Hennighausen, L.; Illert, A.L.; Duyster, J. Lineage-specific STAT5 target gene activation in hematopoietic progenitor cells predicts the FLT3(+)-mediated leukemic phenotype. Leukemia 2016, 30, 1725–1733. [Google Scholar] [CrossRef]
- Silva, C.M. Role of STATs as downstream signal transducers in Src family kinase-mediated tumorigenesis. Oncogene 2004, 23, 8017–8023. [Google Scholar] [CrossRef]
- Choudhary, C.; Brandts, C.; Schwable, J.; Tickenbrock, L.; Sargin, B.; Ueker, A.; Bohmer, F.D.; Berdel, W.E.; Muller-Tidow, C.; Serve, H. Activation mechanisms of STAT5 by oncogenic Flt3-ITD. Blood 2007, 110, 370–374. [Google Scholar] [CrossRef]
- Leischner, H.; Albers, C.; Grundler, R.; Razumovskaya, E.; Spiekermann, K.; Bohlander, S.; Ronnstrand, L.; Gotze, K.; Peschel, C.; Duyster, J. SRC is a signaling mediator in FLT3-ITD- but not in FLT3-TKD-positive AML. Blood 2012, 119, 4026–4033. [Google Scholar] [CrossRef]
- Uras, I.Z.; Walter, G.J.; Scheicher, R.; Bellutti, F.; Prchal-Murphy, M.; Tigan, A.S.; Valent, P.; Heidel, F.H.; Kubicek, S.; Scholl, C.; et al. Palbociclib treatment of FLT3-ITD+ AML cells uncovers a kinase-dependent transcriptional regulation of FLT3 and PIM1 by CDK6. Blood 2016, 127, 2890–2902. [Google Scholar] [CrossRef]
- Liang, J.; Wu, Y.L.; Chen, B.J.; Zhang, W.; Tanaka, Y.; Sugiyama, H. The C-kit receptor-mediated signal transduction and tumor-related diseases. Int. J. Biol. Sci. 2013, 9, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Lennartsson, J.; Blume-Jensen, P.; Hermanson, M.; Ponten, E.; Carlberg, M.; Ronnstrand, L. Phosphorylation of Shc by Src family kinases is necessary for stem cell factor receptor/c-kit mediated activation of the Ras/MAP kinase pathway and c-fos induction. Oncogene 1999, 18, 5546–5553. [Google Scholar] [CrossRef] [PubMed]
- Linnekin, D.; DeBerry, C.S.; Mou, S. Lyn associates with the juxtamembrane region of c-Kit and is activated by stem cell factor in hematopoietic cell lines and normal progenitor cells. J. Biol. Chem. 1997, 272, 27450–27455. [Google Scholar] [CrossRef] [PubMed]
- Blake, R.A.; Broome, M.A.; Liu, X.; Wu, J.; Gishizky, M.; Sun, L.; Courtneidge, S.A. SU6656, a selective src family kinase inhibitor, used to probe growth factor signaling. Mol. Cell Biol. 2000, 20, 9018–9027. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Pedersen, M.; Ronnstrand, L. The D816V mutation of c-Kit circumvents a requirement for Src family kinases in c-Kit signal transduction. J. Biol. Chem. 2009, 284, 11039–11047. [Google Scholar] [CrossRef]
- Sun, J.; Pedersen, M.; Ronnstrand, L. Gab2 is involved in differential phosphoinositide 3-kinase signaling by two splice forms of c-Kit. J. Biol. Chem. 2008, 283, 27444–27451. [Google Scholar] [CrossRef]
- Timokhina, I.; Kissel, H.; Stella, G.; Besmer, P. Kit signaling through PI 3-kinase and Src kinase pathways: An essential role for Rac1 and JNK activation in mast cell proliferation. EMBO J. 1998, 17, 6250–6262. [Google Scholar] [CrossRef]
- Iwaki, S.; Spicka, J.; Tkaczyk, C.; Jensen, B.M.; Furumoto, Y.; Charles, N.; Kovarova, M.; Rivera, J.; Horejsi, V.; Metcalfe, D.D.; et al. Kit- and Fc epsilonRI-induced differential phosphorylation of the transmembrane adaptor molecule NTAL/LAB/LAT2 allows flexibility in its scaffolding function in mast cells. Cell Signal. 2008, 20, 195–205. [Google Scholar] [CrossRef][Green Version]
- Yu, M.; Luo, J.; Yang, W.; Wang, Y.; Mizuki, M.; Kanakura, Y.; Besmer, P.; Neel, B.G.; Gu, H. The scaffolding adapter Gab2, via Shp-2, regulates kit-evoked mast cell proliferation by activating the Rac/JNK pathway. J. Biol. Chem. 2006, 281, 28615–28626. [Google Scholar] [CrossRef]
- Casteran, N.; De Sepulveda, P.; Beslu, N.; Aoubala, M.; Letard, S.; Lecocq, E.; Rottapel, R.; Dubreuil, P. Signal transduction by several KIT juxtamembrane domain mutations. Oncogene 2003, 22, 4710–4722. [Google Scholar] [CrossRef]
- Chaix, A.; Arcangeli, M.L.; Lopez, S.; Voisset, E.; Yang, Y.; Vita, M.; Letard, S.; Audebert, S.; Finetti, P.; Birnbaum, D.; et al. KIT-D816V oncogenic activity is controlled by the juxtamembrane docking site Y568-Y570. Oncogene 2014, 33, 872–881. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Letard, S.; Borge, L.; Chaix, A.; Hanssens, K.; Lopez, S.; Vita, M.; Finetti, P.; Birnbaum, D.; Bertucci, F.; et al. Pediatric mastocytosis-associated KIT extracellular domain mutations exhibit different functional and signaling properties compared with KIT-phosphotransferase domain mutations. Blood 2010, 116, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- Chaix, A.; Lopez, S.; Voisset, E.; Gros, L.; Dubreuil, P.; De Sepulveda, P. Mechanisms of STAT protein activation by oncogenic KIT mutants in neoplastic mast cells. J. Biol. Chem. 2011, 286, 5956–5966. [Google Scholar] [CrossRef] [PubMed]
- Voisset, E.; Lopez, S.; Dubreuil, P.; De Sepulveda, P. The tyrosine kinase FES is an essential effector of KITD816V proliferation signal. Blood 2007, 110, 2593–2599. [Google Scholar] [CrossRef]
- Brizzi, M.F.; Dentelli, P.; Rosso, A.; Yarden, Y.; Pegoraro, L. STAT protein recruitment and activation in c-Kit deletion mutants. J. Biol. Chem. 1999, 274, 16965–16972. [Google Scholar] [CrossRef]
- Deberry, C.; Mou, S.; Linnekin, D. Stat1 associates with c-kit and is activated in response to stem cell factor. Biochem. J. 1997, 327 Pt 1, 73–80. [Google Scholar] [CrossRef]
- Gotoh, A.; Takahira, H.; Mantel, C.; Litz-Jackson, S.; Boswell, H.S.; Broxmeyer, H.E. Steel factor induces serine phosphorylation of Stat3 in human growth factor-dependent myeloid cell lines. Blood 1996, 88, 138–145. [Google Scholar] [CrossRef]
- Jacobs-Helber, S.M.; Penta, K.; Sun, Z.; Lawson, A.; Sawyer, S.T. Distinct signaling from stem cell factor and erythropoietin in HCD57 cells. J. Biol. Chem. 1997, 272, 6850–6853. [Google Scholar] [CrossRef]
- O’Farrell, A.M.; Ichihara, M.; Mui, A.L.; Miyajima, A. Signaling pathways activated in a unique mast cell line where interleukin-3 supports survival and stem cell factor is required for a proliferative response. Blood 1996, 87, 3655–3668. [Google Scholar] [CrossRef]
- Omori, I.; Yamaguchi, H.; Miyake, K.; Miyake, N.; Kitano, T.; Inokuchi, K. D816V mutation in the KIT gene activation loop has greater cell-proliferative and anti-apoptotic ability than N822K mutation in core-binding factor acute myeloid leukemia. Exp. Hematol. 2017, 52, 56–64 e54. [Google Scholar] [CrossRef]
- Paschka, P.; Marcucci, G.; Ruppert, A.S.; Mrozek, K.; Chen, H.; Kittles, R.A.; Vukosavljevic, T.; Perrotti, D.; Vardiman, J.W.; Carroll, A.J.; et al. Adverse prognostic significance of KIT mutations in adult acute myeloid leukemia with inv(16) and t(8;21): A Cancer and Leukemia Group B Study. J. Clin. Oncol. 2006, 24, 3904–3911. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.Z.; Zhu, H.H.; Jiang, Q.; Xu, L.P.; Jiang, H.; Wang, Y.; Zhao, X.S.; Liu, Y.R.; Zhang, X.H.; Liu, K.Y.; et al. Heterogeneous prognosis among KIT mutation types in adult acute myeloid leukemia patients with t(8;21). Blood Cancer J. 2018, 8, 76. [Google Scholar] [CrossRef] [PubMed]
- Yui, S.; Kurosawa, S.; Yamaguchi, H.; Kanamori, H.; Ueki, T.; Uoshima, N.; Mizuno, I.; Shono, K.; Usuki, K.; Chiba, S.; et al. D816 mutation of the KIT gene in core binding factor acute myeloid leukemia is associated with poorer prognosis than other KIT gene mutations. Ann. Hematol. 2017, 96, 1641–1652. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A. Systemic mastocytosis in adults: 2019 update on diagnosis, risk stratification and management. Am. J. Hematol. 2019, 94, 363–377. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Akin, C.; Metcalfe, D.D. Mastocytosis: 2016 updated WHO classification and novel emerging treatment concepts. Blood 2017, 129, 1420–1427. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, J.; Polivka, L.; Maouche-Chretien, L.; Frenzel, L.; Dubreuil, P.; Hermine, O. Recent advances in the understanding and therapeutic management of mastocytosis. F1000Res 2019, 8. [Google Scholar] [CrossRef]
- Komi, D.E.A.; Rambasek, T.; Wohrl, S. Mastocytosis: From a Molecular PoInt. of View. Clin. Rev. Allergy Immunol. 2018, 54, 397–411. [Google Scholar] [CrossRef]
- Draber, P.; Halova, I.; Levi-Schaffer, F.; Draberova, L. Transmembrane adaptor proteins in the high-affinity IgE receptor signaling. Front. Immunol. 2011, 2, 95. [Google Scholar] [CrossRef] [PubMed]
- Kraft, S.; Kinet, J.P. New developments in FcepsilonRI regulation, function and inhibition. Nat. Rev. Immunol. 2007, 7, 365–378. [Google Scholar] [CrossRef]
- Nagata, H.; Worobec, A.S.; Oh, C.K.; Chowdhury, B.A.; Tannenbaum, S.; Suzuki, Y.; Metcalfe, D.D. Identification of a poInt. mutation in the catalytic domain of the protooncogene c-kit in peripheral blood mononuclear cells of patients who have mastocytosis with an associated hematologic disorder. Proc. Natl. Acad. Sci. USA 1995, 92, 10560–10564. [Google Scholar] [CrossRef]
- Nishizumi, H.; Yamamoto, T. Impaired tyrosine phosphorylation and Ca2+ mobilization, but not degranulation, in lyn-deficient bone marrow-derived mast cells. J. Immunol. 1997, 158, 2350–2355. [Google Scholar] [PubMed]
- Kawakami, Y.; Kitaura, J.; Satterthwaite, A.B.; Kato, R.M.; Asai, K.; Hartman, S.E.; Maeda-Yamamoto, M.; Lowell, C.A.; Rawlings, D.J.; Witte, O.N.; et al. Redundant and opposing functions of two tyrosine kinases, Btk and Lyn, in mast cell activation. J. Immunol. 2000, 165, 1210–1219. [Google Scholar] [CrossRef] [PubMed]
- O’Laughlin-Bunner, B.; Radosevic, N.; Taylor, M.L.; Shivakrupa; DeBerry, C.; Metcalfe, D.D.; Zhou, M.; Lowell, C.; Linnekin, D. Lyn is required for normal stem cell factor-induced proliferation and chemotaxis of primary hematopoietic cells. Blood 2001, 98, 343–350. [Google Scholar] [CrossRef]
- Parravicini, V.; Gadina, M.; Kovarova, M.; Odom, S.; Gonzalez-Espinosa, C.; Furumoto, Y.; Saitoh, S.; Samelson, L.E.; O’Shea, J.J.; Rivera, J. Fyn kinase initiates complementary signals required for IgE-dependent mast cell degranulation. Nat. Immunol. 2002, 3, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Hansen, V.; Mackay, G.A.; Lowell, C.A.; Wilson, B.S.; Oliver, J.M. The Src kinase Lyn is a negative regulator of mast cell proliferation. J. Leukoc Biol. 2004, 75, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Hansen, V.; Smith, A.J.; Surviladze, Z.; Chigaev, A.; Mazel, T.; Kalesnikoff, J.; Lowell, C.A.; Krystal, G.; Sklar, L.A.; Wilson, B.S.; et al. Dysregulated FcepsilonRI signaling and altered Fyn and SHIP activities in Lyn-deficient mast cells. J. Immunol. 2004, 173, 100–112. [Google Scholar] [CrossRef]
- Odom, S.; Gomez, G.; Kovarova, M.; Furumoto, Y.; Ryan, J.J.; Wright, H.V.; Gonzalez-Espinosa, C.; Hibbs, M.L.; Harder, K.W.; Rivera, J. Negative regulation of immunoglobulin E-dependent allergic responses by Lyn kinase. J. Exp. Med. 2004, 199, 1491–1502. [Google Scholar] [CrossRef]
- Iwaki, S.; Tkaczyk, C.; Satterthwaite, A.B.; Halcomb, K.; Beaven, M.A.; Metcalfe, D.D.; Gilfillan, A.M. Btk plays a crucial role in the amplification of Fc epsilonRI-mediated mast cell activation by kit. J. Biol. Chem. 2005, 280, 40261–40270. [Google Scholar] [CrossRef]
- Kitaura, J.; Kinoshita, T.; Matsumoto, M.; Chung, S.; Kawakami, Y.; Leitges, M.; Wu, D.; Lowell, C.A.; Kawakami, T. IgE- and IgE+Ag-mediated mast cell migration in an autocrine/paracrine fashion. Blood 2005, 105, 3222–3229. [Google Scholar] [CrossRef]
- Hong, H.; Kitaura, J.; Xiao, W.; Horejsi, V.; Ra, C.; Lowell, C.A.; Kawakami, Y.; Kawakami, T. The Src family kinase Hck regulates mast cell activation by suppressing an inhibitory Src family kinase Lyn. Blood 2007, 110, 2511–2519. [Google Scholar] [CrossRef]
- Poderycki, M.; Tomimori, Y.; Ando, T.; Xiao, W.; Maeda-Yamamoto, M.; Sauer, K.; Kawakami, Y.; Kawakami, T. A minor catalytic activity of Src family kinases is sufficient for maximal activation of mast cells via the high-affinity IgE receptor. J. Immunol. 2010, 184, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.; Vemula, S.; Munugalavadla, V.; Chen, J.; Sims, E.; Borneo, J.; Kondo, T.; Ramdas, B.; Mali, R.S.; Li, S.; et al. Balanced interactions between Lyn, the p85alpha regulatory subunit of class I(A) phosphatidylinositol-3-kinase, and SHIP are essential for mast cell growth and maturation. Mol. Cell Biol. 2011, 31, 4052–4062. [Google Scholar] [CrossRef][Green Version]
- Shelburne, C.P.; McCoy, M.E.; Piekorz, R.; Sexl, V.; Roh, K.H.; Jacobs-Helber, S.M.; Gillespie, S.R.; Bailey, D.P.; Mirmonsef, P.; Mann, M.N.; et al. Stat5 expression is critical for mast cell development and survival. Blood 2003, 102, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Hundley, T.R.; Gilfillan, A.M.; Tkaczyk, C.; Andrade, M.V.; Metcalfe, D.D.; Beaven, M.A. Kit and FcepsilonRI mediate unique and convergent signals for release of inflammatory mediators from human mast cells. Blood 2004, 104, 2410–2417. [Google Scholar] [CrossRef] [PubMed]
- Morales, J.K.; Falanga, Y.T.; Depcrynski, A.; Fernando, J.; Ryan, J.J. Mast cell homeostasis and the JAK-STAT pathway. Genes Immun. 2010, 11, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Olayioye, M.A.; Beuvink, I.; Horsch, K.; Daly, J.M.; Hynes, N.E. ErbB receptor-induced activation of stat transcription factors is mediated by Src tyrosine kinases. J. Biol. Chem. 1999, 274, 17209–17218. [Google Scholar] [CrossRef] [PubMed]
- Tobio, A.; Bandara, G.; Morris, D.A.; Kim, D.K.; O’Connell, M.P.; Komarow, H.D.; Carter, M.C.; Smrz, D.; Metcalfe, D.D.; Olivera, A. Oncogenic D816V-KIT signaling in mast cells causes persistent IL-6 production. Haematologica 2020, 105, 124–135. [Google Scholar] [CrossRef]
- Baumgartner, C.; Cerny-Reiterer, S.; Sonneck, K.; Mayerhofer, M.; Gleixner, K.V.; Fritz, R.; Kerenyi, M.; Boudot, C.; Gouilleux, F.; Kornfeld, J.W.; et al. Expression of activated STAT5 in neoplastic mast cells in systemic mastocytosis: Subcellular distribution and role of the transforming oncoprotein KIT D816V. Am. J. Pathol. 2009, 175, 2416–2429. [Google Scholar] [CrossRef]
- Pan, J.; Quintas-Cardama, A.; Kantarjian, H.M.; Akin, C.; Manshouri, T.; Lamb, P.; Cortes, J.E.; Tefferi, A.; Giles, F.J.; Verstovsek, S. EXEL-0862, a novel tyrosine kinase inhibitor, induces apoptosis in vitro and ex vivo in human mast cells expressing the KIT D816V mutation. Blood 2007, 109, 315–322. [Google Scholar] [CrossRef][Green Version]
- Sibilano, R.; Frossi, B.; Pucillo, C.E. Mast cell activation: A complex interplay of positive and negative signaling pathways. Eur. J. Immunol. 2014, 44, 2558–2566. [Google Scholar] [CrossRef]
- Eiseman, E.; Bolen, J.B. Engagement of the high-affinity IgE receptor activates src protein-related tyrosine kinases. Nature 1992, 355, 78–80. [Google Scholar] [CrossRef] [PubMed]
- El-Hillal, O.; Kurosaki, T.; Yamamura, H.; Kinet, J.P.; Scharenberg, A.M. Syk kinase activation by a src kinase-initiated activation loop phosphorylation chain reaction. Proc. Natl. Acad. Sci. USA 1997, 94, 1919–1924. [Google Scholar] [CrossRef] [PubMed]
- Jouvin, M.H.; Adamczewski, M.; Numerof, R.; Letourneur, O.; Valle, A.; Kinet, J.P. Differential control of the tyrosine kinases Lyn and Syk by the two signaling chains of the high affinity immunoglobulin E receptor. J. Biol. Chem. 1994, 269, 5918–5925. [Google Scholar] [PubMed]
- Pullen, N.A.; Falanga, Y.T.; Morales, J.K.; Ryan, J.J. The Fyn-STAT5 Pathway: A New Frontier in IgE- and IgG-Mediated Mast Cell Signaling. Front. Immunol. 2012, 3, 117. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, J.W.; Kim, D.K.; Kim, H.S.; Park, H.J.; Park, D.K.; Kim, A.R.; Kim, B.; Beaven, M.A.; Park, K.L.; et al. The Src family kinase Fgr is critical for activation of mast cells and IgE-mediated anaphylaxis in mice. J. Immunol. 2011, 187, 1807–1815. [Google Scholar] [CrossRef]
- Suzuki, R.; Leach, S.; Liu, W.; Ralston, E.; Scheffel, J.; Zhang, W.; Lowell, C.A.; Rivera, J. Molecular editing of cellular responses by the high-affinity receptor for IgE. Science 2014, 343, 1021–1025. [Google Scholar] [CrossRef]
- Bischoff, S.C.; Dahinden, C.A. c-kit ligand: A unique potentiator of mediator release by human lung mast cells. J. Exp. Med. 1992, 175, 237–244. [Google Scholar] [CrossRef]
- Tkaczyk, C.; Horejsi, V.; Iwaki, S.; Draber, P.; Samelson, L.E.; Satterthwaite, A.B.; Nahm, D.H.; Metcalfe, D.D.; Gilfillan, A.M. NTAL phosphorylation is a pivotal link between the signaling cascades leading to human mast cell degranulation following Kit activation and Fc epsilon RI aggregation. Blood 2004, 104, 207–214. [Google Scholar] [CrossRef]
- Gilfillan, A.M.; Rivera, J. The tyrosine kinase network regulating mast cell activation. Immunol. Rev. 2009, 228, 149–169. [Google Scholar] [CrossRef]
- Rawlings, D.J.; Scharenberg, A.M.; Park, H.; Wahl, M.I.; Lin, S.; Kato, R.M.; Fluckiger, A.C.; Witte, O.N.; Kinet, J.P. Activation of BTK by a phosphorylation mechanism initiated by SRC family kinases. Science 1996, 271, 822–825. [Google Scholar] [CrossRef]
- Randall, N.; Courville, E.L.; Baughn, L.; Afrin, L.; Ustun, C. Bosutinib, a Lyn/Btk inhibiting tyrosine kinase inhibitor, is ineffective in advanced systemic mastocytosis. Am. J. Hematol. 2015, 90, E74. [Google Scholar] [CrossRef] [PubMed]
- Vaes, M.; Benghiat, F.S.; Hermine, O. Targeted Treatment Options in Mastocytosis. Front. Med. 2017, 4, 110. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Akin, C.; Hartmann, K.; George, T.I.; Sotlar, K.; Peter, B.; Gleixner, K.V.; Blatt, K.; Sperr, W.R.; Manley, P.W.; et al. Midostaurin: A magic bullet that blocks mast cell expansion and activation. Ann. Oncol. 2017, 28, 2367–2376. [Google Scholar] [CrossRef]
- Gotlib, J.; Kluin-Nelemans, H.C.; George, T.I.; Akin, C.; Sotlar, K.; Hermine, O.; Awan, F.T.; Hexner, E.; Mauro, M.J.; Sternberg, D.W.; et al. Efficacy and Safety of Midostaurin in Advanced Systemic Mastocytosis. N. Engl. J. Med. 2016, 374, 2530–2541. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, K.; Gotlib, J.; Akin, C.; Hermine, O.; Awan, F.T.; Hexner, E.; Mauro, M.J.; Menssen, H.D.; Redhu, S.; Knoll, S.; et al. Midostaurin improves quality of life and mediator-related symptoms in advanced systemic mastocytosis. J. Allergy Clin. Immunol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Dubreuil, P.; Letard, S.; Ciufolini, M.; Gros, L.; Humbert, M.; Casteran, N.; Borge, L.; Hajem, B.; Lermet, A.; Sippl, W.; et al. Masitinib (AB1010), a potent and selective tyrosine kinase inhibitor targeting KIT. PLoS ONE 2009, 4, e7258. [Google Scholar] [CrossRef] [PubMed]
- Lortholary, O.; Chandesris, M.O.; Bulai Livideanu, C.; Paul, C.; Guillet, G.; Jassem, E.; Niedoszytko, M.; Barete, S.; Verstovsek, S.; Grattan, C.; et al. Masitinib for treatment of severely symptomatic indolent systemic mastocytosis: A randomised, placebo-controlled, phase 3 study. Lancet 2017, 389, 612–620. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Ref. | In Response to SCF | In Response to Antigen | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cellular Functions | Signalling Pathways | Cellular Functions | Signalling Pathways | |||||||||||||||
| Proliferation (IL-3 and/or SCF) | Calcium Mobilisation | Chemotaxis | Phospho-AKT | Phospho-SHIP | Phospho-ERK1/2 | Phospho-p38 | Phospho-JNK | KIT Expression | Degranulation | Calcium Mobilisation | Cell Migration | Phospho-AKT | Phospho-SHIP | Phospho-ERK1/2 | Phospho-p38 | Phospho-JNK | FCεRI Expression | |
| Nishizumi, 1997 [81] | = | ↓ | ↓ | = | ||||||||||||||
| Kawakami, 2000 [82] | = | delayed | ↓ | ↑ | = | ↑ | = | |||||||||||
| O’Laughlin-Bunner, 2001 [83] | ↓ | ↓ | = | |||||||||||||||
| Parravicini, 2002 [84] | ↑ | ↓ | ↑ | = | ||||||||||||||
| Hernandez-Hansen, 2004 [85] | ↑ | = | = | |||||||||||||||
| Hernandez-Hansen, 2004 [86] | ↑ | delayed | ↑ | ↓ | ||||||||||||||
| Odom, 2004 [87] | ↑ | ↑ | ||||||||||||||||
| Iwaki, 2005 [88] | delayed | = | ↓ | = | = | ↓ | ↓ | ↓ | ↑ | = | ↑ | |||||||
| Kitaura, 2005 [89] | ↑ | ↓ | ||||||||||||||||
| Hong, 2007 [90] | ↑ | |||||||||||||||||
| Poderycki, 2010 [91] | ↑ | = | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | = | ||||||||
| Ma, 2011 [92] | ↑ | ↑ | ↓ | ↑ | ||||||||||||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Voisset, E.; Brenet, F.; Lopez, S.; de Sepulveda, P. SRC-Family Kinases in Acute Myeloid Leukaemia and Mastocytosis. Cancers 2020, 12, 1996. https://doi.org/10.3390/cancers12071996
Voisset E, Brenet F, Lopez S, de Sepulveda P. SRC-Family Kinases in Acute Myeloid Leukaemia and Mastocytosis. Cancers. 2020; 12(7):1996. https://doi.org/10.3390/cancers12071996
Chicago/Turabian StyleVoisset, Edwige, Fabienne Brenet, Sophie Lopez, and Paulo de Sepulveda. 2020. "SRC-Family Kinases in Acute Myeloid Leukaemia and Mastocytosis" Cancers 12, no. 7: 1996. https://doi.org/10.3390/cancers12071996
APA StyleVoisset, E., Brenet, F., Lopez, S., & de Sepulveda, P. (2020). SRC-Family Kinases in Acute Myeloid Leukaemia and Mastocytosis. Cancers, 12(7), 1996. https://doi.org/10.3390/cancers12071996