Ex Vivo Models Simulating the Bone Marrow Environment and Predicting Response to Therapy in Multiple Myeloma

 , , and
, , and 
Abstract
:1. Introduction
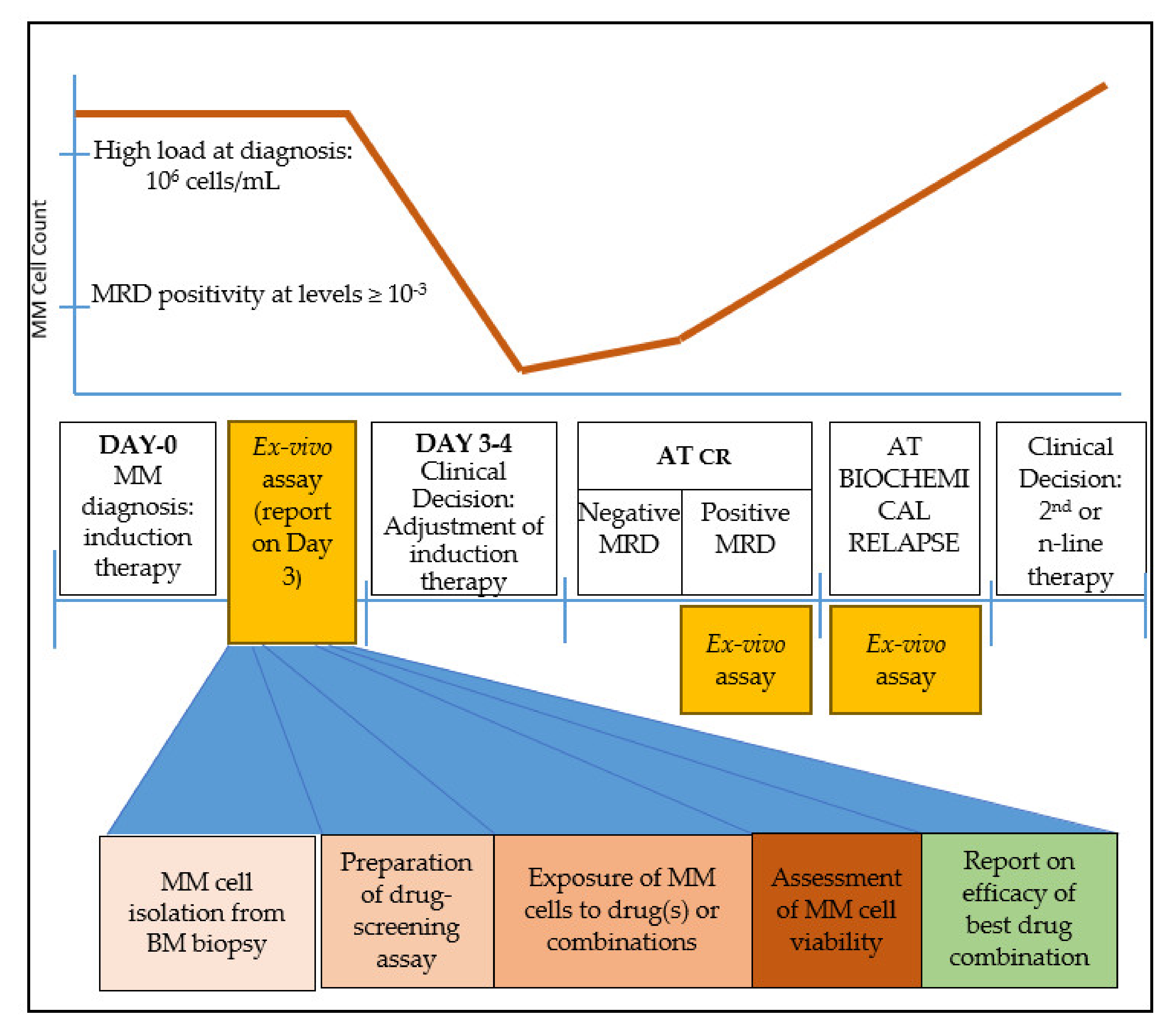
2. Study Aim and Design
3. Role of the BM Microenvironment in MM
4. The Need for Personalized Preclinical BM Models
5. Personalized Clinical Assays for MM Patients: From 2D to 3D BM Cell Cultures
6. 3D Ex Vivo Platforms Using Gel Scaffolds
7. 3D Ex Vivo Platforms Using Solid Scaffolds
8. 3D Ex Vivo Platforms Using Bioreactors
9. 3D Cultures Using Microfluidics
10. 3D Cultures Using Animal Models
11. 3D Models Partially Fulfilling the Criteria Set
12. Classification and Evaluation of the 3D MM Models
13. Time-Frame for the Application of an Ex Vivo 3D Platform in MM Patient Treatment
14. Conclusions—The Way for an Effective Ex Vivo BM Model for MM Patients
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Rajkumar, S.V.; Dimopoulos, M.A.; Palumbo, A.; Blade, J.; Merlini, G.; Mateos, M.-V.; Kumar, S.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014, 15, e538–e548. [Google Scholar] [CrossRef]
- Rajkumar, S.V. Updated Diagnostic Criteria and Staging System for Multiple Myeloma. Am. Soc. Clin. Oncol. Educ. Book 2016, 35, e418–e423. [Google Scholar] [CrossRef] [PubMed]
- Ramsenthaler, C.; Kane, P.; Gao, W.; Siegert, R.J.; Edmonds, P.M.; Schey, S.A.; Higginson, I.J. Prevalence of symptoms in patients with multiple myeloma: A systematic review and meta-analysis. Eur. J. Haematol. 2016, 97, 416–429. [Google Scholar] [CrossRef] [PubMed]
- Offidani, M.; Corvatta, L. Renal failure in multiple myeloma: Something new on the horizon. Br. J. Haematol. 2017, 176, 845–846. [Google Scholar] [CrossRef]
- Kawano, Y.; Roccaro, A.M.; Ghobrial, I.M.; Azzi, J. Multiple Myeloma and the Immune Microenvironment. Curr. Cancer Drug Targets 2017, 17, 806–818. [Google Scholar] [CrossRef]
- Reagan, M.R.; Liaw, L.; Rosen, C.J.; Ghobrial, I.M. Dynamic interplay between bone and multiple myeloma: Emerging roles of the osteoblast. Bone 2015, 75, 161–169. [Google Scholar] [CrossRef] [Green Version]
- Gupta, V.A.; Matulis, S.M.; Conage-Pough, J.E.; Nooka, A.K.; Kaufman, J.L.; Lonial, S.; Boise, L.H. Bone marrow microenvironment-derived signals induce Mcl-1 dependence in multiple myeloma. Blood 2017, 129, 1969–1979. [Google Scholar] [CrossRef] [Green Version]
- Fernández de Larrea, C.; Kyle, R.A.; Durie, B.G.M.; Ludwig, H.; Usmani, S.; Vesole, D.H.; Hajek, R.; San Miguel, J.F.; Sezer, O.; Sonneveld, P.; et al. Plasma cell leukemia: Consensus statement on diagnostic requirements, response criteria and treatment recommendations by the International Myeloma Working Group. Leukemia 2013, 27, 780–791. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.K.; Rajkumar, V.; Kyle, R.A.; van Duin, M.; Sonneveld, P.; Mateos, M.-V.; Gay, F.; Anderson, K.C. Multiple myeloma. Nat. Rev. Dis. Primer 2017, 3, 17046. [Google Scholar] [CrossRef]
- Kazandjian, D. Multiple myeloma epidemiology and survival: A unique malignancy. Semin. Oncol. 2016, 43, 676–681. [Google Scholar] [CrossRef] [Green Version]
- Röllig, C.; Knop, S.; Bornhäuser, M. Multiple myeloma. Lancet 2015, 385, 2197–2208. [Google Scholar] [CrossRef]
- Baker, H. Daratumumab improves survival in multiple myeloma. Lancet Oncol. 2016, 17, e480. [Google Scholar] [CrossRef]
- Mateos, M.-V.; Ocio, E.M.; Paiva, B.; Rosiñol, L.; Martínez-López, J.; Bladé, J.; Lahuerta, J.-J.; García-Sanz, R.; San Miguel, J.F. Treatment for patients with newly diagnosed multiple myeloma in 2015. Blood Rev. 2015, 29, 387–403. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Anderson, K.C. Immune Therapies in Multiple Myeloma. Clin. Cancer Res. 2016, 22, 5453–5460. [Google Scholar] [CrossRef] [Green Version]
- Siegel, D.S.; Dimopoulos, M.A.; Ludwig, H.; Facon, T.; Goldschmidt, H.; Jakubowiak, A.; San-Miguel, J.; Obreja, M.; Blaedel, J.; Stewart, A.K. Improvement in Overall Survival With Carfilzomib, Lenalidomide, and Dexamethasone in Patients With Relapsed or Refractory Multiple Myeloma. J. Clin. Oncol. 2018, 36, 728–734. [Google Scholar] [CrossRef]
- Di Marzo, L.; Desantis, V.; Solimando, A.G.; Ruggieri, S.; Annese, T.; Nico, B.; Fumarulo, R.; Vacca, A.; Frassanito, M.A. Microenvironment drug resistance in multiple myeloma: Emerging new players. Oncotarget 2016, 7, 60698–60711. [Google Scholar] [CrossRef] [Green Version]
- Kawano, Y.; Moschetta, M.; Manier, S.; Glavey, S.; Görgün, G.T.; Roccaro, A.M.; Anderson, K.C.; Ghobrial, I.M. Targeting the bone marrow microenvironment in multiple myeloma. Immunol. Rev. 2015, 263, 160–172. [Google Scholar] [CrossRef]
- Hynes, R.O.; Naba, A. Overview of the matrisome--an inventory of extracellular matrix constituents and functions. Cold Spring Harb. Perspect. Biol. 2012, 4, a004903. [Google Scholar] [CrossRef] [Green Version]
- Iriuchishima, H.; Takubo, K.; Miyakawa, Y.; Nakamura-Ishizu, A.; Miyauchi, Y.; Fujita, N.; Miyamoto, K.; Miyamoto, T.; Ikeda, E.; Kizaki, M.; et al. Neovascular niche for human myeloma cells in immunodeficient mouse bone. PLoS ONE 2012, 7, e30557. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Hendrix, A.; Hernot, S.; Lemaire, M.; De Bruyne, E.; Van Valckenborgh, E.; Lahoutte, T.; De Wever, O.; Vanderkerken, K.; Menu, E. Bone marrow stromal cell-derived exosomes as communicators in drug resistance in multiple myeloma cells. Blood 2014, 124, 555–566. [Google Scholar] [CrossRef] [Green Version]
- Morgan, J.J.; McAvera, R.M.; Crawford, L.J. TRAF6 Silencing Attenuates Multiple Myeloma Cell Adhesion to Bone Marrow Stromal Cells. Int. J. Mol. Sci. 2019, 20, 702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchiyama, H.; Barut, B.A.; Mohrbacher, A.F.; Chauhan, D.; Anderson, K.C. Adhesion of human myeloma-derived cell lines to bone marrow stromal cells stimulates interleukin-6 secretion. Blood 1993, 82, 3712–3720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Jiang, L.; Gao, Y.; Sun, Q.; Liu, B.; Hu, Y.; Han, X. Interaction between BMSCs and EPCs promotes IUA angiogenesis via modulating PI3K/Akt/Cox2 axis. Am. J. Transl. Res. 2018, 10, 4280–4289. [Google Scholar] [PubMed]
- Hsiao, S.T.-F.; Asgari, A.; Lokmic, Z.; Sinclair, R.; Dusting, G.J.; Lim, S.Y.; Dilley, R.J. Comparative analysis of paracrine factor expression in human adult mesenchymal stem cells derived from bone marrow, adipose, and dermal tissue. Stem Cells Dev. 2012, 21, 2189–2203. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, Y.; Kikuchi, J. Epigenetic mechanisms of cell adhesion-mediated drug resistance in multiple myeloma. Int. J. Hematol. 2016, 104, 281–292. [Google Scholar] [CrossRef]
- Abdi, J.; Chen, G.; Chang, H. Drug resistance in multiple myeloma: Latest findings and new concepts on molecular mechanisms. Oncotarget 2013, 4, 2186–2207. [Google Scholar] [CrossRef] [Green Version]
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread genetic heterogeneity in multiple myeloma: Implications for targeted therapy. Cancer Cell 2014, 25, 91–101. [Google Scholar] [CrossRef] [Green Version]
- Ledergor, G.; Weiner, A.; Zada, M.; Wang, S.-Y.; Cohen, Y.C.; Gatt, M.E.; Snir, N.; Magen, H.; Koren-Michowitz, M.; Herzog-Tzarfati, K.; et al. Single cell dissection of plasma cell heterogeneity in symptomatic and asymptomatic myeloma. Nat. Med. 2018, 24, 1867–1876. [Google Scholar] [CrossRef]
- Manier, S.; Salem, K.Z.; Park, J.; Landau, D.A.; Getz, G.; Ghobrial, I.M. Genomic complexity of multiple myeloma and its clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 100–113. [Google Scholar] [CrossRef]
- Jagannath, S.; Abonour, R.; Durie, B.G.M.; Gasparetto, C.; Hardin, J.W.; Narang, M.; Terebelo, H.R.; Toomey, K.; Wagner, L.; Srinivasan, S.; et al. Heterogeneity of Second-Line Treatment for Patients With Multiple Myeloma in the Connect MM Registry (2010-2016). Clin. Lymphoma Myeloma Leuk. 2018, 18, 480–485.e3. [Google Scholar] [CrossRef] [Green Version]
- Szalat, R.; Munshi, N.C. Genomic heterogeneity in multiple myeloma. Curr. Opin. Genet. Dev. 2015, 30, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Pawlyn, C.; Davies, F.E. Toward personalized treatment in multiple myeloma based on molecular characteristics. Blood 2019, 133, 660–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, B.; Caetano, J.; Barahona, F.; Lopes, R.; Carneiro, E.; Costa-Silva, B.; João, C. Liquid biopsies for multiple myeloma in a time of precision medicine. J. Mol. Med. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bustoros, M.; Mouhieddine, T.H.; Detappe, A.; Ghobrial, I.M. Established and Novel Prognostic Biomarkers in Multiple Myeloma. Am. Soc. Clin. Oncol. Educ. Book 2017, 37, 548–560. [Google Scholar] [CrossRef]
- Lionetti, M.; Neri, A. Utilizing next-generation sequencing in the management of multiple myeloma. Expert Rev. Mol. Diagn. 2017, 17, 653–663. [Google Scholar] [CrossRef]
- González-Calle, V.; Keane, N.; Braggio, E.; Fonseca, R. Precision Medicine in Myeloma: Challenges in Defining an Actionable Approach. Clin. Lymphoma Myeloma Leuk. 2017, 17, 621–630. [Google Scholar] [CrossRef]
- Bringhen, S.; Milan, A.; Ferri, C.; Wäsch, R.; Gay, F.; Larocca, A.; Salvini, M.; Terpos, E.; Goldschmidt, H.; Cavo, M.; et al. Cardiovascular adverse events in modern myeloma therapy - Incidence and risks. A review from the European Myeloma Network (EMN) and Italian Society of Arterial Hypertension (SIIA). Haematologica 2018, 103, 1422–1432. [Google Scholar] [CrossRef]
- Tarkun, P.; Atalay, F.; Atesoglu, E.B.; Mehtap, O.; Simsek, M.; Terzi, E.; Geduk, A.; Balli, F.; Batman, A.; Baydemir, C.; et al. Treatment of patients with multiple myeloma over 65 yr: More tolerability or better response? Eur. J. Haematol. 2015, 94, 424–430. [Google Scholar] [CrossRef]
- Bringhen, S.; De Wit, E.; Dimopoulos, M.-A. New Agents in Multiple Myeloma: An Examination of Safety Profiles. Clin. Lymphoma Myeloma Leuk. 2017, 17, 391–407.e5. [Google Scholar] [CrossRef]
- Auclair, D.; Lonial, S.; Anderson, K.C.; Kumar, S.K. Precision medicine in multiple myeloma: Are we there yet? Expert Rev. Precis. Med. Drug Dev. 2019, 4, 51–53. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Kaufman, J.L.; Gasparetto, C.; Mikhael, J.; Vij, R.; Pegourie, B.; Benboubker, L.; Facon, T.; Amiot, M.; Moreau, P.; et al. Efficacy of venetoclax as targeted therapy for relapsed/refractory t(11;14) multiple myeloma. Blood 2017, 130, 2401–2409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jovanović, K.K.; Roche-Lestienne, C.; Ghobrial, I.M.; Facon, T.; Quesnel, B.; Manier, S. Targeting MYC in multiple myeloma. Leukemia 2018, 32, 1295–1306. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Wei, R.; Qian, J.; Wang, R.; Fan, Z.; Gu, C.; Yang, Y. The impact of the bone marrow microenvironment on multiple myeloma (Review). Oncol. Rep. 2019. [Google Scholar] [CrossRef] [PubMed]
- Pampaloni, F.; Reynaud, E.G.; Stelzer, E.H.K. The third dimension bridges the gap between cell culture and live tissue. Nat. Rev. Mol. Cell Biol. 2007, 8, 839–845. [Google Scholar] [CrossRef]
- Jakubikova, J.; Adamia, S.; Kost-Alimova, M.; Klippel, S.; Cervi, D.; Daley, J.F.; Cholujova, D.; Kong, S.-Y.; Leiba, M.; Blotta, S.; et al. Lenalidomide targets clonogenic side population in multiple myeloma: Pathophysiologic and clinical implications. Blood 2011, 117, 4409–4419. [Google Scholar] [CrossRef]
- Azab, A.K.; Runnels, J.M.; Pitsillides, C.; Moreau, A.-S.; Azab, F.; Leleu, X.; Jia, X.; Wright, R.; Ospina, B.; Carlson, A.L.; et al. CXCR4 inhibitor AMD3100 disrupts the interaction of multiple myeloma cells with the bone marrow microenvironment and enhances their sensitivity to therapy. Blood 2009, 113, 4341–4351. [Google Scholar] [CrossRef] [Green Version]
- Giuliani, N.; Colla, S.; Morandi, F.; Lazzaretti, M.; Sala, R.; Bonomini, S.; Grano, M.; Colucci, S.; Svaldi, M.; Rizzoli, V. Myeloma cells block RUNX2/CBFA1 activity in human bone marrow osteoblast progenitors and inhibit osteoblast formation and differentiation. Blood 2005, 106, 2472–2483. [Google Scholar] [CrossRef]
- Edmondson, R.; Broglie, J.J.; Adcock, A.F.; Yang, L. Three-dimensional cell culture systems and their applications in drug discovery and cell-based biosensors. Assay Drug Dev. Technol. 2014, 12, 207–218. [Google Scholar] [CrossRef] [Green Version]
- Thippabhotla, S.; Zhong, C.; He, M. 3D cell culture stimulates the secretion of in vivo like extracellular vesicles. Sci. Rep. 2019, 9, 13012. [Google Scholar] [CrossRef] [Green Version]
- Asimakopoulos, F.; Hope, C.; Johnson, M.G.; Pagenkopf, A.; Gromek, K.; Nagel, B. Extracellular matrix and the myeloid-in-myeloma compartment: Balancing tolerogenic and immunogenic inflammation in the myeloma niche. J. Leukoc. Biol. 2017, 102, 265–275. [Google Scholar] [CrossRef]
- Zdzisińska, B.; Roliński, J.; Piersiak, T.; Kandefer-Szerszeń, M. A comparison of cytokine production in 2-dimensional and 3-dimensional cultures of bone marrow stromal cells of multiple myeloma patients in response to RPMI8226 myeloma cells. Folia Histochem. Cytobiol. 2009, 47, 69–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirshner, J.; Thulien, K.J.; Martin, L.D.; Debes Marun, C.; Reiman, T.; Belch, A.R.; Pilarski, L.M. A unique three-dimensional model for evaluating the impact of therapy on multiple myeloma. Blood 2008, 112, 2935–2945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parikh, M.R.; Belch, A.R.; Pilarski, L.M.; Kirshner, J. A three-dimensional tissue culture model to study primary human bone marrow and its malignancies. J. Vis. Exp. 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.-H.; Molavi, O.; Alshareef, A.; Haque, M.; Wang, Q.; Chu, M.P.; Venner, C.P.; Sandhu, I.; Peters, A.C.; Lavasanifar, A.; et al. Constitutive Activation of STAT3 in Myeloma Cells Cultured in a Three-Dimensional, Reconstructed Bone Marrow Model. Cancers 2018, 10, 206. [Google Scholar] [CrossRef] [Green Version]
- De la Puente, P.; Muz, B.; Gilson, R.C.; Azab, F.; Luderer, M.; King, J.; Achilefu, S.; Vij, R.; Azab, A.K. 3D tissue-engineered bone marrow as a novel model to study pathophysiology and drug resistance in multiple myeloma. Biomaterials 2015, 73, 70–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De la Puente, P.; Azab, A.K. 3D tissue-engineered bone marrow: What does this mean for the treatment of multiple myeloma? Future Oncol. 2016, 12, 1545–1547. [Google Scholar] [CrossRef]
- Jakubikova, J.; Cholujova, D.; Hideshima, T.; Gronesova, P.; Soltysova, A.; Harada, T.; Joo, J.; Kong, S.-Y.; Szalat, R.E.; Richardson, P.G.; et al. A novel 3D mesenchymal stem cell model of the multiple myeloma bone marrow niche: Biologic and clinical applications. Oncotarget 2016, 7, 77326–77341. [Google Scholar] [CrossRef] [Green Version]
- Arhoma, A.; Chantry, A.D.; Haywood-Small, S.L.; Cross, N.A. SAHA-induced TRAIL-sensitisation of Multiple Myeloma cells is enhanced in 3D cell culture. Exp. Cell Res. 2017, 360, 226–235. [Google Scholar] [CrossRef]
- Ji, Z.; Su, J.; Wu, D.; Peng, H.; Zhao, W.; Nlong Zhao, B.; Zhou, X. Predicting the impact of combined therapies on myeloma cell growth using a hybrid multi-scale agent-based model. Oncotarget 2017, 8, 7647–7665. [Google Scholar] [CrossRef] [Green Version]
- Braham, M.V.J.; Minnema, M.C.; Aarts, T.; Sebestyen, Z.; Straetemans, T.; Vyborova, A.; Kuball, J.; Öner, F.C.; Robin, C.; Alblas, J. Cellular immunotherapy on primary multiple myeloma expanded in a 3D bone marrow niche model. Oncoimmunology 2018, 7, e1434465. [Google Scholar] [CrossRef] [Green Version]
- Braham, M.V.; Deshantri, A.K.; Minnema, M.C.; Öner, F.C.; Schiffelers, R.M.; Fens, M.H.; Alblas, J. Liposomal drug delivery in an in vitro 3D bone marrow model for multiple myeloma. Int. J. Nanomed. 2018, 13, 8105–8118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, A.; Silva, M.C.; Sudalagunta, P.; Distler, A.; Jacobson, T.; Collins, A.; Nguyen, T.; Song, J.; Chen, D.-T.; Chen, L.; et al. An Ex Vivo Platform for the Prediction of Clinical Response in Multiple Myeloma. Cancer Res. 2017, 77, 3336–3351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reagan, M.R.; Mishima, Y.; Glavey, S.V.; Zhang, Y.; Manier, S.; Lu, Z.N.; Memarzadeh, M.; Zhang, Y.; Sacco, A.; Aljawai, Y.; et al. Investigating osteogenic differentiation in multiple myeloma using a novel 3D bone marrow niche model. Blood 2014, 124, 3250–3259. [Google Scholar] [CrossRef]
- Belloni, D.; Heltai, S.; Ponzoni, M.; Villa, A.; Vergani, B.; Pecciarini, L.; Marcatti, M.; Girlanda, S.; Tonon, G.; Ciceri, F.; et al. Modeling multiple myeloma-bone marrow interactions and response to drugs in a 3D surrogate microenvironment. Haematologica 2018, 103, 707–716. [Google Scholar] [CrossRef] [Green Version]
- Bonomi, A.; Steimberg, N.; Benetti, A.; Berenzi, A.; Alessandri, G.; Pascucci, L.; Boniotti, J.; Coccè, V.; Sordi, V.; Pessina, A.; et al. Paclitaxel-releasing mesenchymal stromal cells inhibit the growth of multiple myeloma cells in a dynamic 3D culture system. Hematol. Oncol. 2017, 35, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Khin, Z.P.; Ribeiro, M.L.C.; Jacobson, T.; Hazlehurst, L.; Perez, L.; Baz, R.; Shain, K.; Silva, A.S. A preclinical assay for chemosensitivity in multiple myeloma. Cancer Res. 2014, 74, 56–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martowicz, A.; Kern, J.; Gunsilius, E.; Untergasser, G. Establishment of a human multiple myeloma xenograft model in the chicken to study tumor growth, invasion and angiogenesis. J. Vis. Exp. 2015, e52665. [Google Scholar] [CrossRef] [Green Version]
- Calimeri, T.; Battista, E.; Conforti, F.; Neri, P.; Di Martino, M.T.; Rossi, M.; Foresta, U.; Piro, E.; Ferrara, F.; Amorosi, A.; et al. A unique three-dimensional SCID-polymeric scaffold (SCID-synth-hu) model for in vivo expansion of human primary multiple myeloma cells. Leukemia 2011, 25, 707–711. [Google Scholar] [CrossRef]
- Baker, B.M.; Chen, C.S. Deconstructing the third dimension: How 3D culture microenvironments alter cellular cues. J. Cell Sci. 2012, 125, 3015–3024. [Google Scholar] [CrossRef] [Green Version]
- Whitesides, G.M. The origins and the future of microfluidics. Nature 2006, 442, 368–373. [Google Scholar] [CrossRef]
- Lwin, S.T.; Edwards, C.M.; Silbermann, R. Preclinical animal models of multiple myeloma. Bonekey Rep. 2016, 5, 772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boulland, J.-L.; Halasi, G.; Kasumacic, N.; Glover, J.C. Xenotransplantation of human stem cells into the chicken embryo. J. Vis. Exp. 2010. [Google Scholar] [CrossRef] [PubMed]
- Fairfield, H.; Falank, C.; Farrell, M.; Vary, C.; Boucher, J.M.; Driscoll, H.; Liaw, L.; Rosen, C.J.; Reagan, M.R. Development of a 3D bone marrow adipose tissue model. Bone 2019, 118, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Cetin, A.E.; Stevens, M.M.; Calistri, N.L.; Fulciniti, M.; Olcum, S.; Kimmerling, R.J.; Munshi, N.C.; Manalis, S.R. Determining therapeutic susceptibility in multiple myeloma by single-cell mass accumulation. Nat. Commun. 2017, 8, 1613. [Google Scholar] [CrossRef] [Green Version]
- Delgado-Calle, J.; Kurihara, N.; Atkinson, E.G.; Nelson, J.; Miyagawa, K.; Galmarini, C.M.; Roodman, G.D.; Bellido, T. Aplidin (plitidepsin) is a novel anti-myeloma agent with potent anti-resorptive activity mediated by direct effects on osteoclasts. Oncotarget 2019, 10, 2709–2721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrarini, M.; Steimberg, N.; Ponzoni, M.; Belloni, D.; Berenzi, A.; Girlanda, S.; Caligaris-Cappio, F.; Mazzoleni, G.; Ferrero, E. Ex-vivo dynamic 3-D culture of human tissues in the RCCSTM bioreactor allows the study of Multiple Myeloma biology and response to therapy. PLoS ONE 2013, 8, e71613. [Google Scholar] [CrossRef]
- Moreau, P.; San Miguel, J.; Sonneveld, P.; Mateos, M.V.; Zamagni, E.; Avet-Loiseau, H.; Hajek, R.; Dimopoulos, M.A.; Ludwig, H.; Einsele, H.; et al. Multiple myeloma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2017, 28, iv52–iv61. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Year | Reference | Type of Model | Drugs Tested | Model Composition | Cell Types Cocultured (BM Cells/MM Cells) | Viability Assessment | Duration of Test | Feasibility | Cost |
|---|---|---|---|---|---|---|---|---|---|
| 2008 | Kirshner et al. [52] | Gel scaffold | MELPH, BTZ | Recombinant bone marrow; culture plates with fibronectin/collagen, ECM mixture of Matrigel/fibronectin | primary MSCs/primary MM cells | Annexin V analysis of CD38+ cells by flow cytometry; treatment efficacy evaluated using RQ-PCR to detect genomic clonotypic IgH VDJ | ⏰⏰⏰ | ★★☆ | $$ |
| 2018 | Huang et al. [54] | Gel scaffold | Stattic (a STAT3 inhibitor), BTZ | Fibronectin/Collagen scaffolds in 48-well plates seeded with cells in Matrigel | primary MSCs/U266, RPMI8226 | Cell viability measured by CellTiter 96 Assay (i.e., MTT) or trypan blue exclusion assay; apoptosis measured by an Annexin V Apoptosis Detection Kit | ⏰ | ★★★ | $$ |
| 2015 | De la Puente et al. [55,56] | Gel Scaffold | DOXO, BTZ, CFZ | 3D Tissue-Engineered BM cultures; scaffold formed by cross-linking plasma fibrinogen with CaCl2 and tranexamic acid | human umbilical vein endothelial cells, primary CD138+ and CD138- (stromal) cells/MM1s, H929, RPMI8226, MM1s-GFP-Luc | Cell survival analyzed by flow cytometry against an internal standard | ⏰⏰ | ★☆☆ | $$ |
| 2016 | Jakubikova et al. [57] | Gel Scaffold | POM, LEN, THAL, BTZ, CFZ, DOXO, DEX, MELPH | PuraMatrix hydrogel | primary MSCs/primary MM cells, OPM1, KMS11, OCIMY5 | CFSE and Annexin V-PE apoptosis assays by flow cytometry | ⏰ | ★★★ | $$ |
| 2017 | Arhoma et al. [58] | Gel Scaffold | TRAIL, HDAC inhibitors | 3D tumor spheroid cultures on alginate beads | NCI-H929, RPMI 8226, OPM-2, JJN-3, U266/one primary cell culture generated from a plasma cell leukaemia patient (ADC-1) | Caspase-3 activity measured by flow cytometry; cell viability based on CellTiter-Glo luminescent assay | ⏰⏰ | ★★☆ | $ |
| 2017 | Ji et al. [59] | Gel Scaffold | BTZ, LEN and THAL combined | Collagen gel; computational model consisting of a hybrid multi-scale agent-based model | primary MSCs, HS-5 MSC cell line/U266RPMI 8226 | Cell viability determined by MTT assay | ⏰⏰ | ★☆☆ | $ |
| 2018 | Braham et al. [60,61] | Gel Scaffold | TEGs (αβT cells engineered to express a defined γδTCR) | 3D co-cultures using Matrigel; MM cell lines and patients’ cells labelled with a Vybrant Multicolor Cell-Labeling Kit (DiO, DiI, DiD) co-cultured with MSCs | primary MSCs, embedded endothelial progenitor cells /OPM2, U266 and L363 | Viability assessed with calcein staining by confocal imaging | ⏰⏰ | ★★★ | $$$ |
| 2017 | Silva et al. [62] | Gel Scaffold | 31 drugs | 384 or 1536 multi-well plates with collagen type I and previously established MSCs | primary MSCs/primary MM cells, MM1.S | Digital Image Analysis algorithm computing differences in sequential images and identifying live cells | ⏰ | ★★☆ | $ |
| 2014 | Reagan et al. [63] | Solid scaffold | BTZ | Porous, aqueous, silk fibroin scaffolds with pores 500-600 μm cut into cylinders; fibrinogen, thrombin and Matrigel mixed with cells before seeding | primary MSCs (healthy and patient derived)/primary MM cells, MM1S, OPM2 | Calcein on a confocal microscope or LIVE/DEAD Fixable Red Dead Cell Stain kit | ⏰⏰⏰ | ★★☆ | $$ |
| 2018 | Bellloni et al. [64] | Bioreactor | ΒΤΖ, MELPH, DEX, anti-VLA-4 mAb natalizumab | Dynamic cultures in RCCS Bioreactor | primary MSCs osteoblasts, HS-5 cell line, murine L-fibroblasts, human umbilical vein endothelial cells/primary MM cells, MM1.S, U266, RPMI.8226 | Annexin V analysis of CD38+ cells by flow cytometry | n/a | ★☆☆ | $$$ |
| 2017 | Bonomi et al. [65] | Bioreactor | PTX, BTZ | Dynamic cultures in RCCS Bioreactor | MSCs (Lonza, USA)/CFPAC-1 adenocarcinoma cell line, RPMI8226 | MTT assay | ⏰⏰ | ★★☆ | $$$ |
| 2013 | Khin et al. [66] | Fluidics | MELPH, BTZ | 3D cell culture slides (µ-slide Chemotaxis 3D Ibitreat from Ibidi, LLC) with bovine collagen type I | primary MSCs/primary MM cells, RPMI-8226, HS-5, H929, 8226/LR-5 | Assessment of cell viability through membrane motion detection with ImageJ | ⏰ | ★★☆ | $$ |
| 2015 | Martowicz et al. [67] | Animal | BTZ | Spheroid grafted on chorioallantoic membrane of chicken embryos | primary MSCs/OPM-2, RPMI-8226 | Tumor cell mass measurement of eGFP contents with GFP-ELISA | ⏰⏰ | ★☆☆ | $ |
| 2011 | Calimeri et al. [68] | Animal | DEX, BTZ | PCLS scaffold cylinders surgically implanted subcutaneously into SCID mouse flank | primary MSCs (human and mouse derived)/OP9 | Detection of paraprotein levels in mouse sera; detection of MM cell apoptosis in retrieved PCLSs | ⏰⏰⏰ | ★☆☆ | $$$ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papadimitriou, K.; Kostopoulos, I.V.; Tsopanidou, A.; Orologas-Stavrou, N.; Kastritis, E.; Tsitsilonis, O.E.; Dimopoulos, M.A.; Terpos, E. Ex Vivo Models Simulating the Bone Marrow Environment and Predicting Response to Therapy in Multiple Myeloma. Cancers 2020, 12, 2006. https://doi.org/10.3390/cancers12082006
Papadimitriou K, Kostopoulos IV, Tsopanidou A, Orologas-Stavrou N, Kastritis E, Tsitsilonis OE, Dimopoulos MA, Terpos E. Ex Vivo Models Simulating the Bone Marrow Environment and Predicting Response to Therapy in Multiple Myeloma. Cancers. 2020; 12(8):2006. https://doi.org/10.3390/cancers12082006
Chicago/Turabian StylePapadimitriou, Konstantinos, Ioannis V. Kostopoulos, Anastasia Tsopanidou, Nikolaos Orologas-Stavrou, Efstathios Kastritis, Ourania E. Tsitsilonis, Meletios A. Dimopoulos, and Evangelos Terpos. 2020. "Ex Vivo Models Simulating the Bone Marrow Environment and Predicting Response to Therapy in Multiple Myeloma" Cancers 12, no. 8: 2006. https://doi.org/10.3390/cancers12082006